
Abstract
Inflammatory bowl disease (IBD) is a type 1 T helper cell (Th1)-mediated autoimmune disease. Various studies have revealed that environmental pathogens also play a significant role in the initiation and progression of this disease. Interestingly, the pathogenesis of IBD has been shown to be related to nitric oxide (NO) released from innate immune cells. Although NO is known to be highly toxic to the gut epithelia, there is very little information about the regulation of NO production, One major question in the etiology of IBD is how Th1 cells and pathogens interact in the induction of IBD. In present study, we focused on the regulation of NO. We show that macrophages require both interferon-γ (IFN-γ)-mediated and TLR4-mediated signals for the production of NO, which causes inflammation in the intestine and subsequently IBD. Thus, IBD is the result of concerted actions of innate immune signals, such as the binding of LPS to TLR-4, and adaptive immune signals, such as IFN-γ produced by Th1 cells.
Similar content being viewed by others

Introduction
Inflammatory bowl disease (IBD) is a chronic, relapsing inflammatory disorder of the digestive track with unknown etiology. Historically, IBD was believed to be an infectious disease. However, since steroid therapy causes remission of IBD, it is believed that T cells play a critical role in the progression of IBD 1. In most human cases examined, IBD is associated with a type 1 helper T cell (Th1) response 2. Most interestingly, IBD also requires the presence of phagocytic cells such as monocytes, macrophages and neutrophils 3, 4. In the chemically induced mouse model of IBD, such as that induced by acetic acid (AA), dextran sulphate (DSS), or trinitrobezene sulphonic acid (TNBS), a clear Th1 type of cytokine pattern was observed 5, 6, 7, 8. Similarly, in cell transfer studies, immunodeficient mice reconstituted with CD45RBhigh cells, which predominantly produce IFN-γ upon activation, developed IBD 9, 10, 11, 12. In addition, CD45RBhigh cells from STAT-4 deficient mice were unable to induce IBD. Since, STAT-4 is important transcription factor for IL-12 signaling, this results indicate a role for IL-12-dependent Th1 cells (IL-12 facilitates IFN-γ production in Th1 cells) in the initiation of IBD13. Anti-IFN-γ 14 or anti-IL-12 15, 16 has been shown to be effective in controlling IBD in mice, which is consistent with a role for type 1 cytokines in IBD induction. Furthermore, it was recently reported that T-bet-deficient mice, which lack Th1 cells, are resistant to IBD and also that T-bet-deficient T cells are unable to induce colitis in the T cell transfer model, suggesting a critical role for IFN-g in the induction of IBD 17.
Like other Th1 cell-induced autoimmune diseases such as experimental autoimmune encephalomyelitis and Type I diabetes mellitus, the inflammation sites of IBD contain monocytes, macrophages and neutrophils 3, 4. Upon stimulation, these cells produce reactive toxic free radicals of nitrogen and oxygen. These free radicals are normally responsible for killing phagocytosed pathogens 18. However, as a by-product of this protective effect, they also induce local tissue injury 19. Patients with both form of IBD (Crohn's disease and ulcerative colitis) show increased inducible NO synthase (iNOS) activity and citrulline production in the mucosa of colonic biopsy specimens 20. Incubation of colonic biopsy specimens from IBD patients with the NO synthase inhibitor NG-monomethyl-L-argenine (L-NMMA) inhibited citrulline production, which also suggests that NO production is associated with IBD 21. Using iNOS deficient mice, it has been shown that NO has an important role in the initiation and/or progression of DSS induced IBD 22. On the other hand, mice deficient in p47phox, an oxidase mainly present in phagocytes and required for the production of reactive oxygen intermediates, did not show any improvement in this model, which suggests that there is little or no role for toxic oxygen intermediates in inducing IBD with DSS 22. Taken together, these findings suggest that Th1 cells and their recruit and activate phagocytes are responsible for the initiation and progression of IBD in several gene different murine models.
However, in both the chemically induced and T cell transfer induced mouse models of IBD, animals raised in germ free conditions do not develop intestinal inflammation 23, 24, 25, 26. Similarly, broad-spectrum antibiotics have been shown to be effective in the prevention and resolution of IBD 27, 28. The search for specific pathogens that trigger intestinal inflammation failed to produce conclusive results 29. Instead, it has been found that reconstitution of germ-free mice with commensal bacteria can be enough to induce IBD in several gene-deficient 30, 31 as well as T cell transfer models of IBD in mice 32, 33. Therefore, instead of a specific pathogen, a broad spectrum of microbes may contribute to, or substitute for the requirement of microbes in the induction of intestinal inflammation. This makes the possibility that a common component amongst these microbes, a microbial-associated molecular pattern (MAMP), plays a role in the initiation and/or progression of IBD. Previously, it has been shown that LPS is a MAMP that signals through the TLR-4 pathway 34. Recent studies have implicated a central role for TLR-4 in the induction of a Th1 response 35, 36. Here, we show that primary peritoneal macrophages require both an innate immune signal, which is supplied by LPS, and an adaptive immune signal, which is supplied by Th1 derived IFN-γ, for the production of NO. Inhibition of NO production by using a specific iNOS inhibitor resulted in the prevention of colitis. TLR4nul/nul mice were found to be resistant to TNBS induced colitis. Therefore, IBD requires the interplay between innate and adaptive immune signals.
Materials and methods
Mice and reagents
C57BL/10 and TLR-4 mice were purchased from Jackson laboratory, (Bar Harbor, MN). They were bread and housed at our animal facility. LPS, N-(1-Naphthyl) Ethylenediamine, Sulfanilamide and Sodium Nitrite were purchased from Sigma Chemical Co, (St. Louis, MO). L-NMMA was purchased from Calbiochem, (La-Jolla, CA).
Preparation of CD45RBhi Th1 cells
RBC depleted spleen cells were stained with CD45RB, CD4 and CD8. CD4+CD45RBhigh cells were positively sorted by using a FACSvantage (Becton Dickinson, San Jose, CA). Cells were stimulated under Th1 condition in vitro with plate bound anti-CD3 (2 μg/ml), anti-CD28 (2 μg/ml), and IL-2 (10 U/ml) along with IL-12 (5 ng/ml) and anti-IL-4 (5 μg/ml) for 5 d. After 2 d in IL-2, the cells were used as Th1 for experiments.
Isolation of thioglycolate elicited macrophages and their activation
Mice were injected with 2 ml of 3% thioglycolate. Four days later elicited macrophages were harvested by injecting and aspirating 10 ml of PBS. Cells were cultured overnight in 24-well plates at a density of 1×106/ml/well in Click's EHAA with 10% FCS. Cells were washed with pre-warmed complete medium and subsequently stimulated with LPS alone, IFN-γ alone, or LPS + IFN-γ with or without the presence of L-NMMA. After 24 h, supernatants were harvested and 100 μl of each was tested for the presence of NO by using the Griess reagent.
Measurement of NO production
NO production was measured by the Griess reagent as described earlier 42. Briefly, 100 μl of culture supernatant was incubated with 100 μl Griess reagent. Griess reagent is prepared by equal volume of 1% (w/v) sulphanilamide in 5% (v/v) phosphoric acid and 0.1% N-(1-napthyl) ethylene. After 10 min incubation at room temperature, the light absorbance was measured at 550 nm. Concentrations were determined from a linear standard curve prepared by using sodium nitrite.
Histology
Small intestinal pieces were washed thoroughly in PBS/FCS and then fixed in periodate-lysin-paraformaldehyde. Tissues were paraffin embedded and 5-6 mm sections were cut using a cryostat. The sections were stained with hematoxylin and eosin and examined microscopically.
Results and discussion
Initiation of colitis require NO production
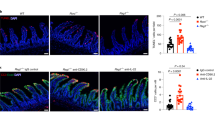
To evaluate the role of NO in a T cell transfer model, we sorted CD4+ CD45RBhigh cells from C57BL/10 mice, cultured them in Th1-polarizing conditions, and adoptively transferred these polarized cells to syngeneic RAG-1 deficient mice. These mice showed severe intestinal inflammation within three weeks after infusion of CD4, CD45RBhigh cells. A group of mice that were reconstituted with an equivalent number of Th1-polarized CD4+CD45RBhigh cells also received 500 μg/mouse/day of L-NMMA did not show signs of IBD (Figure 1). This suggests that NO is critically involved in the pathogenesis and tissue destruction of IBD. Within four weeks of reconstitution, mice received L-NMMA did not became sick were euthanized. While mice which received L-NMMA continued to be healthy and showed no signs of IBD. Clearly L-NMMA completely blocked Th1 adaptive transfer induced IBD.

Nitric oxide is primarily responsible for the tissue destruction in Th1 induced IBD. Th1 polarized CD4+CD45RBhigh cells were adoptively transferred into RAG-1-deficient mice. These mice were divided into two groups. One group received 500 μg/mouse/d of L-NMMA in PBS, the other group received PBS alone for 10 d. Animals were killed and their intestines were fixed and embedded in paraffin. The sections were stained with hematoxylin and eosin and examined microscopically (10 ×).
Primary macrophages require both innate and adaptive immune signals for the production of NO
It is now evident that IFN-γ, which is a product of Th1 cells, and MAMPs, which are products of microbes, are essential components for the induction of IBD. Therefore, we evaluated the role of IFN-γ and of MAMPs for the induction of NO. To do so, we activated thioglycolate elicited peritoneal macrophages with different doses of IFN-γ and LPS, a well known MAMP, either individually or in combination. After 24 h, supernatants were tested for the presence of NO. It was found that neither LPS nor IFN-γ alone could induce NO production at detectable levels. However, the combination of these two ligands dramatically increased NO production in culture supernatants (Figure 2). Importantly, the elevated NO production was inhibited by the addition of 0.5 mM L-NMMA (Figure 2). This demonstrates that iNOS was elevated only when both innate immune and adaptive immune signals were provided. It is important to note that LPS alone can induce NO production in some macrophage tumor cell lines such as RAW. Therefore, our experiments used freshly isolated macrophage. Thus, this renders the possibility that upon LPS and IFN-γ activation macrophages induce expression of an oncogene(s), which is already expressed in these tumor cell lines, and this gene(s) is required for the NO production. Other Th1 induced autoimmune diseases, such as experimental autoimmune encephalomyelitis (EAE) are also associated with NO production 37, and introduction of iNOS inhibitors have been found to partially protect from experimental autoimmune neuritis (EAN) 38.

Macrophages require two signals for the production of nitric oxide. Macrophages were cultured in Click's EHAA medium overnight and subsequently stimulated with LPS, IFN-γ or a combination of both. L-NMMA at a concentration of 0.5 mM was added to respective wells. After 24 h, supernatants were harvested and tested for NO production by using Greiss reagent. Neither LPS nor IFN-γ (200 IU/ml alone was sufficient for inducing the production of NO. However, their combination showed dramatically increased NO production.
IFN-γ and LPS induced NO production is defective in TLR4nul/nul- macrophages.
Macrophage activated by an LPS signal occurs mainly through TLR4, and TLR4-deficient mice show reduced Th1 induction 35, 36, 39. Therefore, we tested the NO producing capability of macrophages from TLR4nul/nul mice. Peritoneal macrophages derived from these mice were activated with IFN-g and LPS for 24 h, after which their supernatants were tested for the content of NO. It was found that macrophages derived from TLR4-deficient mice produce significantly less NO (Figure 3). This observation demonstrates that TLR4 recognition of LPS has a significant role in the production of inducible NO (Figure 3). Taken together with the earlier data showing in vivo treatment with L-NMMA prevents IBD, these observations suggest that NO is required for the initiation of IBD. These results suggest that IBD is induced by monocytes or macrophages if they are simultaneously activated with IFN-γ and LPS.

LPS and IFN-γ induced nitric oxide in macrophages require TLR-4. Macrophages were isolated from the peritoneal cavity from wild type or TLR-4nul/nul mice. Cells were stimulated with LPS and IFN-γ (200 IU/ml) for 24 h. The production of NO was assessed by the Greiss reagent.
TLR4nul/nul- mice are resistant to TNBS induced colitis
In the preceding experiments, we showed that TLR4nul/nul mice have little capacity to produce NO because they fail to receive LPS-mediated signals. We therefore thought that these mice would be resistant to Th1 induced IBD. Because of the integral homeostatic regulatory mechanisms, adoptively transferred T cells into TLR4nul/nul mice will not be engrafted normally.
Thus, we chose TNBS induced IBD as an alternative for Th1 induced IBD. Therefore, to examine our hypothesis, we performed TNBS-induced colitis in TLR4nul/nul mice. We determined the weight loss as the parameter for the status of inflammation. Mice were sacrificed after four weeks and examined macroscopically for the status of their colon. Weight loss and macroscopical examinations of the intestine of these mice suggested that TLR4nul/nul mice are resistant to TNBS induced colitis (data not shown). When the intestine was examined histologically, massive leukocyte infiltration was observed in C57BL/6 mice, but not in TLR4nul/nul mice (Figure 4), suggesting inflammation of progressive IBD is only in wild type mice.

TLR-4nul/nul mice were resistant to TNBS induced colitis. TLR-4nul/nul, and wild type mice were induced by TNBS for the development of colitis as described. Mice were observed for weight loss. After three weeks mice were sacrificed and their intestines were examined for inflammation. Histological examinations show that TLR-4nul/nul mice are resistant to TNBS induced colitis, whereas wild type mice show considerable inflammation.
Apart from Th1-induced IBD, there are few mouse models of IBD that show Th2 mediated inflammation 40. In these models of IBD, it is possible that NO does not play a role in tissue destruction because Th2 cytokines, namely IL-4 and IL-13, are potent inhibitors of iNOS 41. However, there are no data available yet that suggest this is the mechanism. Therefore, in the Th2-induced IBD model, the toxic materials that cause tissue destruction remain to be demonstrated. In the case of human IBD, involvement of Th2 cytokines has not been demonstrated in either form of IBD.
In summary, it has been a matter of debate whether IBD is an autoimmune or an infectious disease. Here, we show that microbial-derived LPS and a Th1 derived cytokine (IFN-γ) delivered simultaneously are both necessary for the induction of iNOS in macrophages which are in mucosal tissue, which leads to IBD. Upon signaling with both LPS and IFN-γ, macrophages produce reactive NO. As reactive NO is a major component leading to tissue destruction in IBD, therefore, we conclude that inhibition of iNOS by L-NMMA largely prevents the tissue destruction commonly seen in IBD. The iNOS activation that leads to tissue damage was due to signals from both types of immune system, the innate immune system in LPS triggering of TLR-4, and the adaptive immune system in which CD4+, CD45RBhigh cells produce IFN-γ. Either signal by itself was not sufficient to induce iNOS or the tissue destruction seen in IBD. Therefore, IBD required the interaction of both innate immune mechanisms and those of adaptive immunity.
References
Boismenu R, Chen Y . Insights from mouse models of colitis. J Leukoc Biol 2000; 67:267–78.
Blumberg RS, Saubermann LJ, Strober W . Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr Opin Immunol 1999; 11:648–56.
Caradonna L, Amati L, Lella P, Jirillo E, Caccavo D . Phagocytosis, killing, lymphocyte-mediated antibacterial activity, serum autoantibodies, and plasma endotoxins in inflammatory bowel disease. Am J Gastroenterol 2000; 95:1495–502.
Vainer B, Nielsen OH, Horn T . Comparative studies of the colonic in situ expression of intercellular adhesion molecules (ICAM-1, -2, and -3), beta2 integrins (LFA-1, Mac-1, and p150, 95), and PECAM-1 in ulcerative colitis and Crohn's disease. Am J Surg Pathol 2000; 24:1115–24.
MacPherson BR, Pfeiffer CJ . Experimental production of diffuse colitis in rats. Digestion 1978; 17:135–50.
Dieleman LA, Elson CO, Tennyson GS, Beagley KW . Kinetics of cytokine expression during healing of acute colitis in mice. Am J Physiol 1996; 271:G130–6.
Hans W, Scholmerich J, Gross V, Falk W . Interleukin-12 induced interferon-gamma increases inflammation in acute dextran sulfate sodium induced colitis in mice. Eur Cytokine Netw 2000; 11:67–74.
Romagnani S . Th1/Th2 cells. Inflamm Bowel Dis 1999; 5:285–94.
Powrie F, Correa-Oliveira R, Mauze S, Coffman RL . Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell- mediated immunity. J Exp Med 1994; 179:589–600.
Morrissey PJ, Charrier K, Braddy S, Liggitt D, Watson JD . CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med 1993; 178:237–44.
Claesson MH, Whiting CV, Bonhagen K, et al. Colitis-inducing potency of CD4+ T cells in immunodeficient, adoptive hosts depends on their state of activation, IL-12 responsiveness, and CD45RB surface phenotype. J Immunol 1999; 162:3702–10.
Mason D, Powrie F . Control of immune pathology by regulatory T cells. Curr Opin Immunol 1998; 10:649–55.
Simpson SJ, Shah S, Comiskey M, et al. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon gamma expression by T cells. J Exp Med 1998; 187:1225–34.
Powrie F, Leach MW, Mauze S, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994; 1:553–62.
Liu Z, Geboes K, Heremans H, et al. Role of interleukin-12 in the induction of mucosal inflammation and abrogation of regulatory T cell function in chronic experimental colitis. Eur J Immunol 2001; 31:1550–60.
Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W . Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med 1995; 182:1281–90.
Neurath MF, Weigmann B, Finotto S, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med 2002; 195:1129–43.
Raupach B, Kaufmann SH . Immune responses to intracellular bacteria. Curr Opin Immunol 2001; 13:417–28.
Whittle BJ . Nitric oxide—a mediator of inflammation or mucosal defence. Eur J Gastroenterol Hepatol 1997; 9:1026–32.
Boughton-Smith NK, Evans SM, Hawkey CJ, et al. Nitric oxide synthase activity in ulcerative colitis and Crohn's disease. Lancet 1993; 342:338–40.
Middleton SJ, Shorthouse M, Hunter JO . Relaxation of distal colonic circular smooth muscle by nitric oxide derived from human leucocytes. Gut 1993; 34:814–7.
Krieglstein CF, Cerwinka WH, Laroux FS, et al. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J Exp Med 2001; 194:1207–18.
Matsumoto S, Okabe Y, Setoyama H, et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut 1998; 43:71–8.
Cahill RJ, Foltz CJ, Fox JG, et al. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun 1997; 65:3126–31.
Dianda L, Hanby AM, Wright NA, et al. T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol 1997; 150:91–7.
Sellon RK, Tonkonogy S, Schultz M, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 1998; 66:5224–31.
Panwala CM, Jones JC, Viney JL . A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol 1998; 161:5733–44.
Videla S, Vilaseca J, Guarner F, et al. Role of intestinal microflora in chronic inflammation and ulceration of the rat colon. Gut 1994; 35:1090–7.
Fiocchi C . Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology 1998; 115:182–205.
Kullberg MC, Ward JM, Gorelick PL, et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect Immun 1998; 66:5157–66.
Foltz CJ, Fox JG, Cahill R, et al. Spontaneous inflammatory bowel disease in multiple mutant mouse lines: association with colonization by Helicobacter hepaticus. Helicobacter 1998; 3:69–78.
Aranda R, Sydora BC, McAllister PL, et al. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J Immunol 1997; 158:3464–73.
Camerini V, Sydora BC, Aranda R, et al. Generation of intestinal mucosal lymphocytes in SCID mice reconstituted with mature, thymus-derived T cells. J Immunol 1998; 160:2608–18.
Guha M, Mackman N . LPS induction of gene expression in human monocytes. Cell Signal 2001; 13:85–94.
Okamoto M, Oshikawa T, Ohe G, et al. Severe impairment of anti-cancer effect of lipoteichoic acid-related molecule isolated from a penicillin-killed Streptococcus pyogenes in toll-like receptor 4-deficient mice. Int Immunopharmacol 2001; 1:1789–95.
Pulendran B, Kumar P, Cutler CW, et al. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol 2001; 167:5067–76.
Willenborg DO, Staykova MA, Cowden WB . Our shifting understanding of the role of nitric oxide in autoimmune encephalomyelitis: a review. J Neuroimmunol 1999; 100:21–35.
Zielasek J, Jung S, Gold R, et al. Administration of nitric oxide synthase inhibitors in experimental autoimmune neuritis and experimental autoimmune encephalomyelitis. J Neuroimmunol 1995; 58:81–8.
Schnare M, Barton GM, Holt AC, et al. Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2001; 2:947–50.
Iqbal N, Oliver JR, Wagner FH, et al. T helper 1 and T helper 2 cells are pathogenic in an antigen-specific model of colitis. J Exp Med 2002; 195:71–84.
Paludan SR, Lovmand J, Ellermann-Eriksen S, Mogensen SC . Effect of IL-4 and IL-13 on IFN-gamma-induced production of nitric oxide in mouse macrophages infected with herpes simplex virus type 2. FEBS Lett 1997; 414:61–4.
Ding AH, Nathan CF, Stuehr DJ . Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol 1988; 141:2407–12.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shi, D., Das, J. & Das, G. Inflammatory bowel disease requires the interplay between innate and adaptive immune signals. Cell Res 16, 70–74 (2006). https://doi.org/10.1038/sj.cr.7310009
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310009
