
ABSTRACT
Apoptosis plays an essential role in T cell biology. Thymocytes expressing nonfunctional or autoreactive TCRs are eliminated by apoptosis during development. Apoptosis also leads to the deletion of expanded effector T cells during immune responses. The dysregulation of apoptosis in the immune system results in autoimmunity, tumorogenesis and immunodeficiency. Two major pathways lead to apoptosis: the intrinsic cell death pathway controlled by Bcl-2 family members and the extrinsic cell death pathway controlled by death receptor signaling. These two pathways work together to regulate T lymphocyte development and function.
Similar content being viewed by others

INTRODUCTION
Apoptosis is an essential process underlying multicellular organism development and function. In the immune system, apoptosis is required for lymphocyte development and homeostasis. The dysregulation of apoptosis leads to a variety of immune disorders, including both immunodeficiency and autoimmunity. In this review, we will address three topics: i) apoptosis pathways; ii) T lymphocyte development and apoptosis; and iii) T lymphocyte function and apoptosis.
APOPTOSIS PATHWAYS
Apoptosis, a genetically conserved pathway in all metazoans, is both essential for the generation of multicellular tissues during embryonic development as well as the maintenance of cellular homeostasis 1. The typical morphological changes during apoptosis include membrane blebbing, DNA fragmentation and cellular component degradation. Notably, these changes occur without the loss of membrane integrity. During the past two decades, extensive work has been done to elucidate the molecular mechanism of apoptosis. From this large body of work it is clear that apoptosis is induced by a range of stimuli that activate two major cell death signaling pathways: the intrinsic pathway mainly controlled by Bcl-2 protein family members and the extrinsic pathway activated by death receptors of the tumor necrosis factor receptor superfamily (Fig. 1).

Apoptotic pathways. Two major pathways lead to apoptosis: the intrinsic cell death pathway controlled by Bcl-2 family members and the extrinsic cell death pathway controlled by death receptor signaling.
The intrinsic cell death pathway
The intrinsic cell death pathway is activated by a variety of apoptotic stimuli, such as genomic toxicity and cytokine withdrawal. Intrinsic cell death signals generally converge within the cell at the outer membrane of mitochondria. They result in a loss of mitochondrial membrane integrity and the subsequent activation of downstream apoptotic pathways.
Bcl-2 family members control mitochondrial membrane integrity and are the major mediators in the intrinsic cell death pathway (Fig. 1). The founding member of this family, Bcl-2, was first identified at the interchromosomal breakpoint of the t(14;18) translocation in follicular B cell lymphoma 2. Other family members were discovered based on sequence homology in four α-helical segments named Bcl-2 Homologous Domains (BH1-4) 3, 4, 5. Bcl-2 protein family members are divided into three subfamilies based on function and BH domain structure. These groupings include the anti-apoptotic BH1-4 subfamily, the pro-apoptotic BH1-3 subfamily and the pro-apoptotic BH3-only subfamily.
ANTI-APOPTOTIC BH1-4 SUBFAMILY
The members of the BH1-4 subfamily include Bcl-2, Bcl-xL, Mcl-1 and A1/Bfl-1 which all contain four BH domains. Members in this subfamily are anti-apoptotic in most experimental systems. They contain a transmembrane domain at the C-terminus and mainly localize to the outer membrane of the mitochondria where they inhibit the function of pro-apoptotic Bcl-2 family members via BH domain interactions. Bcl-2, Bcl-xL, Mcl-1 and A1 are all actively involved in T lymphocyte development and survival.
Bcl-2 protects various cell types from apoptosis. During T cell development, the expression for Bcl-2 is tightly regulated, which suggests a critical role of Bcl-2 in T lymphocytes. Indeed, Bcl-2−/− mice exhibit an intrinsic defect in T lymphocyte development probably due to increased apoptosis 6, 7. In Bcl-2−/− adult bone marrow hematopoietic stem cell reconstituted chimeric mice, donor-derived lymphocyte development is almost completely absent 8. IL-7 provides a crucial survival signal for lymphocyte precursors. It is believed that Bcl-2 plays an important anti-apoptotic role downstream of IL-7 signaling 9, 10.
A large body of work has suggested a critical role for Bcl-x in protection of different cell types from apoptosis. Five isoforms of Bcl-x (Bcl-xL, Bcl-xS, Bcl-xγ, Bcl-xβ and Bcl-xΔTM) generated by alternative splicing of mRNA have been identified 11, 12, 13, 14, 15. Bcl-x−/− mice die by embryonic day 13 due to extensive cell death in the central nerve system where Bcl-xL is highly expressed 16. Furthermore, the deficiency of Bcl-x in erythroid cells causes hemolytic anemia and profound splenomegaly 17. Recently, it has been demonstrated that Bcl-x is required for dentritic cell survival 18, 19. Therefore, Bcl-xL protects different cells from apoptosis in vivo.
Bcl-xL is the major isoform in T lymphocytes. During T lymphocyte development, the expression of Bcl-xL is tightly regulated 20, 21. Bcl-xL is not expressed at DN or SP stage. However, it is highly expressed in DP thymocytes. The high level Bcl-xL expression at DP stage is required for the survival of DP thymocytes. DP thymocytes from Bcl-x−/−Rag2−/−chimeric mice 16, 21 and RORγ−/− mice, which have a profound reduction of Bcl-xL expression at the DP stage 22, 23, have a reduced life-span. Furthermore, this defect in RORγ−/− mice can be rescued by a Bcl-xL transgene. These data demonstrated that Bcl-xL plays a non-redundant role to promote DP thymocyte survival.
Recently, Mcl-1 has also been shown to play an essential role in lymphocyte survival 24. Mcl-1 is a short-lived protein with a very fast turn-over rate. It is thought that Mcl-1 functions as a sensitive switch to decide cell fate in response to survival signals. Early deletion of Mcl-1 during lymphocyte development causes a blockade at the pro-B stage for B lineage cells and a blockade at the DN stage for T lineage cells. Moreover, the deletion of Mcl-1 in mature lymphocytes also leads to apoptosis. IL-7 signaling significantly upregulates Mcl-1 expression. Based on these data, it has been suggested that Mcl-1 is a critical pro-survival molecule downstream of IL-7 signaling 24. In addition, hematopoietic stem cells (HSCs) require Mcl-1 for their survival, and stem cell factor (SCF) signal greatly enhances Mcl-1 expression in mouse HSCs 25. Therefore, Mcl-1 is an essential anti-apoptotic protein both in lymphocytes and HSCs.
A1/Bfl-1 is a fourth member of the anti-apoptotic Bcl-2 subfamily. A1 prevents cell death as a downstream effector of NF-κB signaling 26, 27, 28, 29. A1/Bfl-1 has been identified as a transcriptional target of pre-TCR signal. The knockdown of endogenous A1/Bfl-1 expression by siRNA impairs pre-TCR+ pre-T cell line survival in response to pre-TCR signaling 30. Expression of A1/Bfl-1 rescues DN3 thymocytes from apoptosis by inhibiting the function of caspase 3 indirectly 30. Therefore, A1 plays an anti-apoptotic role in response to pre-TCR signaling during T cell development.
In summary, anti-apoptotic Bcl-2 family members play critical roles at different stages during T lymphocyte development.
PRO-APOPTOTIC BH1-3 SUBFAMILY
The pro-apoptotic BH1-3 subfamily includes the pro-apoptotic proteins Bax, Bak and Bok, which contain three BH domains (BH1-3). The expression of Bok is restricted to the reproductive system, whereas Bax and Bak have a broad expression pattern 31, 32, 33, 34, 35. Based on work done on Bax−/−Bak−/− double deficient mice, it is believed that Bax and Bak are the main cell death executioners in the intrinsic cell death pathway 36, 37, 38. Bax−/−Bak−/− cells are highly resistant to a variety of the intrinsic cell death pathway stimuli including growth factor withdrawal and DNA damage. In addition, initiator caspase activation is abolished in Bax−/−Bak−/− cells 39. It is generally thought that Bax and Bak perform their function by forming super-molecular openings in the outer membranes of mitochondria. This is supported by the fact that Bax, pro-apoptotic BH3-only protein Bid or Bid BH3 peptide and the unique mitochondrial membrane component cardiolipin form a super-molecular 'hole' in purified mitochondrial outer membranes in a cell free system. Bcl-xL inhibits the formation of the super-molecular holes by Bax, Bid and cardiolipin 40. Super-molecular openings in the outer membrane of mitochondria leads to the loss of mitochondrial integrity and the release of pro-apoptotic factors including cytochrome C, SMAC/Diablo, Omi/HtrA2, DNase endonuclease G and apoptosis inducing factor (AIF) 1.
These pro-apoptotic molecules activate downstream apoptotic machinery. Once cytochrome C enters the cytoplasm, it binds to Apaf-1 and pro-caspase 9. Cytochrome C, Apaf-1 and pro-caspase 9 then form a super-molecular complex called the apoptosome with the help of dATP. The formation of the apoptosome leads to the cleavage of pro-caspase 9 and the generation of active caspase 9 41, 42, 43. Active caspase 9 further activates downstream effector caspases, including caspase 3, 6 and 7, which in turn cleave hundreds of cellular components and result in irreversible cell death.
Caspases are a family of cytosolic cysteine proteinases that cleave their substrates after an aspartic acid residue 44. Caspases are actively involved in inflammation and apoptosis. Caspase 2, 3, 6, 7, 8, 9 and 10 are the major caspases involved in apoptosis. Most other caspases are involved in inflammation. Caspases 2, 8, 9 and 10 are initiator caspases, and caspase 3, 6 and 7 are effector caspases. Caspases are initially synthesized as an inactive zymogen, which contains an N-terminal pro-domain. For example, pro-caspase 8 contains two DED (death effector domain) domains as the pro-domain and pro-caspase 9 contains one CARD (caspase recruitment domain) domain as the pro-domain. In all pro-caspases, besides the N-terminal pro-domain, there is also one large domain of 20 kDa and one small domain of 10 kDa at the C-terminus. Cleaving the small domain and the large domain separately from the pro-domain leads to caspase activation. Caspases are the major mediators downstream of both intrinsic and extrinsic apoptotic pathways.
IAPs (inhibitor of apoptosis proteins) are a group of cytoplasmic proteins which are potent inhibitors of caspases 45, 46, 47, 48, 49. IAPs add another layer of regulation to intrinsic apoptotic pathways to ensure that unnecessary caspase activation does not occur. IAPs are regulated by two proteins released from mitochondria. One is SMAC/Diablo, the other is Omi/HtrA2. SMAC/Diablo specifically binds to IAPs and releases IAP-mediated caspase inhibition. Specific binding of SMAC/Diablo to IAPs depends on four amino acid residues at the N-terminus of SMAC/Diablo 50, 51, 52, 53. Omi/HtrA2 has a similar IAP inhibitory function as SMAC/Diablo. However, the pro-apoptotic function of Omi/HtrA2 requires its serine proteinase activity 54. Therefore, SMAC/Diablo and Omi/HtrA2 promote caspase activation by the removal of IAP inhibition.
Caspase independent mechanisms are also involved in the intrinsic death pathway downstream of Bax and Bak. Two proteins, DNase endonuclease G and AIF, mainly activate caspase independent pathways leading to apoptosis. Once released from the mitochondria, DNase endonuclease G translocates into the nucleus to initiate internucleosomal chromatin degradation and apoptosis 55, 56. AIF is also a mitochondrial protein. Once released into the cytosol, AIF translocates into the nucleus and induces chromatin condensation and large scale chromatin degradation. AIF-induced apoptosis is caspase-independent and poly (ADP-ribose) polymerase-1-dependent. Furthermore, AIF may coordinate with endonuclease G to degrade chromatin 57, 58, 59, 60.
In summary, Bax and Bak control the integrity of mitochondria and the release of many pro-apoptotic molecules from the mitochondria. Once released from the mitochondria, the pro-apoptotic molecules activate cell death via a caspase-dependent or a caspase-independent mechanism.
PRO-APOPTOTIC BH3-ONLY SUBFAMILY
Most members in the BH3-only subfamily only contain one BH3 domain. All members are pro-apoptotic by either promoting the function of Bax and Bak or inhibiting the function of anti-apoptotic Bcl-2 family members 61, 62. BH3-only proteins include Bad, Bid, Bik, Bim, Noxa and Puma. Different BH3-only proteins have different binding affinities for different anti-apoptotic Bcl-2 family members. Bim and Puma tightly bind to all anti-apoptotic Bcl-2 family members, while Bad only binds to Bcl-2 and Bcl-xL. Bik only binds to Bcl-xL and A1, and Noxa only binds to Mcl-1 and A1 63. BH3-only proteins function as sentinels instead of direct executioners of cell death. Different cell death stimuli lead to the activation of different BH3-only proteins. Bid is activated by caspase 8 in response to death receptor signaling 64, 65, 66. Bad senses growth factor withdrawal and translocates from the cytosol to the mitochondrial outer membrane 67, 68. Bim is downstream of TCR signal induced apoptosis as Bim−/− mice have a dramatic defect in clearance of T lymphocytes after TCR stimulation 69. Noxa and Puma is mainly responsible for DNA damage induced apoptosis 70.
Bid functions as a linker between the extrinsic and intrinsic cell death pathways 64, 65, 66. Bid senses extrinsic death signals from death receptors. After death receptor activation, caspase 8 is activated. Active caspase 8 cleaves Bid and generates a smaller fragment called tBid. tBid translocates to the mitochondrial outer membrane and inhibits the function of the anti-apoptotic protein Bcl-2 and Bcl-xL, which leads to the activation of the intrinsic death pathway. Bid deficient mice are highly resistant to Fas-induced liver injury 71. Therefore, Bid functions as a critical component to amplify extrinsic death signal through intrinsic death pathway.
Bad is a sensor for growth factor withdrawal as growth factor signals hold the BH3-only protein Bad in an inactive form. Growth factor signals activate Akt kinase, which phosphorylates Bad. Phosphorylated Bad is sequestered in the cytosol by the adaptor protein 14-3–3. After growth factor withdrawal, dephosphorylated Bad translocates onto the outer membrane of mitochondria and leads to apoptosis 67, 68.
Puma and Noxa are involved in DNA damage induced cell death. The expression of Puma and Noxa is highly induced in response to irradiation in a p53-dependent manner 72, 73, 74. The deletion of Puma in human colon cancer cell lines or the knockdown of Noxa expression in MEFs (mouse embryonic fibroblasts) abolishes p53-dependent apoptosis 75, 76. The studies on Noxa−/− and Puma−/− mice have shown that Noxa deficiency partially rescues MEFs from DNA damage induced apoptosis. Puma deficiency confers thymocytes, lymphocytes and fibroblasts resistance to p53-dependent apoptotic stimuli. Interestingly, Puma−/− cells are also resistant to some p53-independent death signals, such as cytokine withdrawal, dexamethasone and phorbol ester 12-myristate 13-acetate (PMA) treatment 70. These data suggest that Noxa and Puma are the main mediators of DNA damage induced apoptosis and Puma may have a broader function than Noxa.
Another member of this subfamily, which may be related to the function of immune system, is Bik. Bik is broadly expressed in the hematopoietic system. However, Bik−/− mice do not have any significant defects in apoptosis 77, suggesting that there is a redundant or non-essential role for Bik in the immune system.
In general, different cell death stimuli activate or induce expression of different BH-3 only proteins. At the outer membrane of mitochondria, BH-3 only proteins titrate out the anti-apoptotic effect of pro-survival Bcl-2 family members. Therefore, when Bax and Bak are activated, mitochondrial integrity is disrupted and pro-apoptotic factors held inside the mitochondria are released into the cytoplasm. Apoptosis proceeds mainly through activation of downstream caspases.
The extrinsic cell death pathway
The tumor necrosis factor (TNF) superfamily and TNF receptor superfamily are evolutionarily conserved protein families involved in various biological processes 78. At least 19 members of the TNF superfamily and 32 members of the TNFR superfamily have been identified in the human genome 79. Members of the TNF superfamily and TNFR superfamily are broadly expressed in the immune system and actively involved in inflammation, adaptive immunity, lymphoid organogenesis and immune homeostasis 78. Many members of the TNF and TNFR superfamilies have been chosen as therapeutic targets or potential targets for the treatment of a variety of different human diseases, such as autoimmunity, osteoporosis and cancer.
Based on sequence homology and cellular function, the TNF receptor superfamily is classified into three groups: the first group are death receptors, which contain a death domain (DD) in their cytoplasmic tail; the second group do not contain a DD in their cytoplasmic tail, instead, they contain one or more TNF receptor associated factor (TRAF)-interacting motifs; the third group are decoy receptors which do not contain any signaling motifs in their cytoplasmic tails. Decoy receptors competitively bind to ligand and inhibit the function of the other TNFR superfamily members 78. Death receptors are initiators of the extrinsic death pathway (Fig. 1).
Death receptors (Group I) include Fas, TNFR1, death receptor 3 (DR3), tumor necrosis factor apoptosis related ligand (TRAIL) receptor1, TRAIL-R2 and DR6 80. Death receptors all contain a DD in their cytoplasmic tail 81. The death domain is an evolutionarily conserved domain composed of approximately 60 amino acids that forms a globular bundle of six α helices. After ligands bind to death receptors, the DD mediates homotypic interaction with other DD-containing adaptor proteins with high specificity. There are two major DD-containing adaptor proteins involved in death receptor signaling: Fas associated death domain (FADD) and TNF receptor associated death domain (TRADD) 82, 83. FADD binds to Fas, TRAIL-R1 and TRAIL-R2, while TRADD preferentially binds to TNFR1, DR3 and DR6 82, 83, 84, 85, 86, 87. The signal pathways for Fas and TNFR1 have been well established.
FAS MEDIATED EXTRINSIC CELL DEATH PATHWAY
Fas and FasL have been extensively studied in the immune system. It is well-known that Fas-FasL interactions play an essential role in maintaining homeostasis of the immune system. Both humans and mice carrying a mutated Fas develop a deleterious lymphoproliferative and autoimmune disease 88, 89. In vitro activation induced cell death (AICD) in T lymphocytes is mainly mediated by Fas-FasL interactions 90. Furthermore, Fas mediated killing is one of the important ways employed by cytotoxic T cells 91. Therefore, Fas delivers a critical death signal in the immune system.
Activation of Fas delivers an apoptotic signal through FADD and caspase 8/10. After FasL binds to Fas, the DD domain in the cytoplasmic tail of Fas recruits the adaptor protein FADD. FADD is a 26 kDa cytosolic adaptor protein composed of two functional domains: an N-terminal death effector domain (DED) and a C-terminal death domain (DD). The DED and the DD domain have homology at the amino acid level. Like the DD domain, the DED domain also mediates homotypic interaction between proteins containing DED domains. After the DD domain of FADD binds to Fas, the DED domain of FADD recruits pro-caspase 8 and also pro-caspase 10 in human cells, which leads to the activation of caspase 8/10. Caspase 10 is highly homologous to caspase 8 at both a sequence level and a function level. However, there is no murine homolog for caspase 10 92, 93, 94.
Active caspase 8 activates a downstream caspase cascade and leads to cell death. Because most receptors in the TNFR superfamily exist as a homotrimer on cell surface, pro-caspase 8 molecules are brought in close proximity to each other after binding to FADD, leading to auto-activation of pro-caspase 8 95, 96. Active caspase 8 cleaves and activates the downstream effector caspase 3, which cleaves cellular targets and induces apoptosis. Furthermore, caspase 8 cleaves the BH3-only protein Bid and generates a small fragment of Bid, tBid. tBid translocates to the mitochondria, inhibits the effect of anti-apoptotic Bcl-2 family members, and induces apoptosis through the intrinsic death pathway 65, 66.
c-FLIP is a critical negative regulator of the extrinsic cell death pathway. c-FLIP (also called CASH, Casper, CLARP, FLAME-1, I-FLICE and usurpin) is a homolog of pro-caspase 8. It does not have caspase activity and functions as a competitive inhibitor of caspase 8 97, 98, 99, 100, 101, 102, 103, 104. In the mouse genome, the c-FLIP gene is only 40 kb away from caspase 8. In the human genome, the c-FLIP gene is 80 kb away from caspase 8 while caspase 10 is positioned between c-FLIP and caspase 8. This genomic structure suggests a recent duplication event. Two isoforms of c-FLIP, generated by mRNA alternative splicing, have been confirmed at the protein level: 55 kDa c-FLIPL and 26 kDa c-FLIPS. Like pro-caspase 8, c-FLIPL has two DED domains at the N-terminus and a caspase-like domain at the C-terminus (Fig. 1). However, c-FLIPL does not have enzymatic activity due to the absence of several critical amino acids, including a cysteine residue within the QACXG motif and histidine residue within the HG motif 105. Both motifs are highly conserved in all caspases and essential for caspase activity. c-FLIPS contains two DED domains without any caspase-like domains 105. Both c-FLIPL and c-FLIPS are efficiently recruited to the death inducing signaling complex (DISC) upon death receptor stimulation and inhibit pro-caspase 8 activation at the DISC. However, when expressed at a low level, c-FLIPL has been shown to promote pro-caspase 8 processing 106. In the genome of some viruses, there are FLIP like genes called v-FLIP. v-FLIP genes encoded in the γ-herpesvirus genome have a similar domain structure as c-FLIPS. Like c-FLIP, v-FLIP is recruited to the DISC and inhibits apoptosis in infected cells, which may provide a protective mechanism against Fas-mediated killing 105. In general, it is believed that c-FLIP inhibits death receptor signaling and caspase 8 activation.
c-FLIP deficient mice die early during embryonic development due to impaired heart development. Experiments performed on c-FLIP−/− mouse embryonic fibroblast (MEF) cells have confirmed a critical role of c-FLIP in protection against death receptor induced apoptosis in vivo107.
TNFR1 MEDIATED EXTRINSIC CELL DEATH PATHWAY
In most cells under normal conditions, TNFR1 signaling induces an inflammatory response through NF-κB. However, when protein synthesis or NF-κB signaling is inhibited, TNFR1 ligation becomes potently apoptotic 80.
After TNFα binding, the adaptor protein TRADD is recruited to TNFR1. TRADD is a 34 kDa cytosolic adaptor protein containing one DD domain. After TRADD binds to the DD domain of TNFR1, a signaling complex forms on the cytoplasmic tail of TNFR1 83. This signaling complex is composed of TNFR1, TRADD, TNF receptor associated factor 1 (TRAF1), TRAF2 and receptor interacting protein 1 (RIP1). RIP1 is a DD domain-containing protein kinase that promotes NF-κB activation and inflammatory responses 108, 109. The main function of this TRADD, TRAF1/2 and RIP1 complex is to activate NF-κB and JNK pathways 110, 111. NF-κB is the major pro-inflammatory and pro-survival transcription factor in mammalian cells. NF-κB activates the transcription of c-FLIP, anti-apoptotic Bcl-2 family members and IAPs 26, 112, 113. Meanwhile, besides pro-inflammatory and pro-survival NF-κB activation complex, FADD and caspase 8/10 are also recruited to TRADD. FADD and caspase 8/10 form a signaling complex, activate caspase 8/10 and induce apoptosis 110, 111. How one receptor can activate two distinct signaling pathways that lead to opposite outcomes has been a puzzling question for years.
Recently, it has been shown that TNFR1 signaling leads to the sequential formation of two distinct signaling complexes in cell lines 114. After TNFα binds to TNFR1, signaling complex I forms immediately on the cytoplasmic tail of TNFR1. Signaling complex I includes TNFR1, TRADD, TRAF2, RIP1 and cIAP1. NF-κB pathways are activated by signaling complex I induced signal. Therefore, signaling complex I only delivers a pro-inflammatory signal. When signaling complex I forms on TNFR1, mono-ubiquitination or poly-ubiquitination of RIP1 and TRADD occur 115. It is speculated that these post-transcriptional modifications may cause components of signaling complex I to leave TNFR1 and the cell membrane 116.
Several hours after TNFα signaling, signaling complex II forms in the cytoplasm. Signaling complex II is composed of TRADD, RIP1, FADD and caspase 8/10 without TNFR1. This model explains the previous observation that although TNFR1 activates caspase 8, no direct interaction is detected between TNFR1, FADD and caspase 8 117. Apoptotic signaling is delivered by signaling complex II. It seems paradoxical that RIP1, which mainly functions as an activator of pro-inflammatory NF-κB pathway, is included in the pro-apoptotic signal complex II. However, it has been shown that RIP1 is a potential substrate for caspase 8 118. After being cleaved by caspase 8, the DD domain of RIP1 is released as a fragment called RIPc. RIPc is a dominant negative inhibitor of full-length RIP1 induced NF-κB activation 118.
Although signaling complex II is a potent apoptosis activator, the outcome of signaling complex II depends on the function of signaling complex I. If signaling complex I can successfully activate NF-κB, this transcription factor quickly induces the expression of several anti-apoptotic proteins such as c-FLIP before the formation of signaling complex II. Once the expression level of c-FLIP is high enough, c-FLIP efficiently incorporates into signaling complex II and inhibits the activation of pro-caspase 8/10. Therefore, apoptosis is inhibited. However, if NF-κB cannot be efficiently activated and the expression level of c-FLIP is not high enough to inhibit the activation of pro-caspase 8/10, apoptotic signaling complex II activates cell death pathway 114.
This two-step separated signaling complex model helps elucidate the long-standing puzzle how TNFR1 signaling can lead to both cell survival and cell death. However, this simplified model cannot incorporate all the phenomena attributed to TNFR1 signaling. A recent paper has suggested an interesting function of JNK during TNFR1 induced apoptosis 119. Transiently activated JNK is correlated with survival while sustained JNK activation leads to apoptosis. Under normal condition, JNK is transiently activated by TNFR1 signaling through TRAF2. However, the blockade of NF-κB leads to the sustained activation of JNK upon TNFR1 signaling. TNFα signaling induces the production of reactive oxygen species (ROS), which inhibits JNK-inactivating phosphatase. TNFα mediated NF-κB activation induces the antioxidant enzyme superoxide dismutase, which inhibits the accumulation of ROS. Therefore, TNFα induced NF-κB activation limits the sustained activation of JNK 120. Sustained JNK activation causes the cleavage of BH3-only protein Bid and generation of a smaller fragment called jBid. jBid translocates onto the outer membrane of mitochondria and specifically releases SMAC/Diablo from the mitochondria, which results in the promotion of TNFα induced apoptosis 119. Furthermore, JNK1−/− or JNK2−/− thymocytes are resistant to TCR/CD3 induced apoptosis, suggesting that JNK may deliver pro-apoptotic signal in vivo under certain circumstances 121.
Interestingly, another model, which is different, but not mutually exclusive from the above model, has been proposed for TNFR1 signaling 122. Based on a new technique called ligand-specific magnetic labeling and purification of TNFR1 endocytic vesicles, this model proposes that TNFR1 endocytosis is essential for TNFα induced apoptotic signaling, but not for TNFα induced NF-κB activation. After TNFα signaling, RIP1 and TRAF2 bind to TNFR1 and form a signal complex for NF-κB activation at the plasma membrane. Simultaneously, TNFR1 undergoes endocytosis after ligand binding. TRADD, FADD and pro-caspase 8 only bind to the endocytosed TNFR1 and form a death inducing signal complex (DISC) away from the plasma membrane to deliver a death signal. A TNFR1 internalization domain (TRID) has been identified. A point mutation in TRID abolishes the ability of TNFR1 to undergo endocytosis and to induce apoptosis, but still retains the ability to activate NF-κB.
In summary, a tremendous amount of work has been done to illustrate the molecular mechanism of the intrinsic and extrinsic apoptotic pathways. It is generally believed that both intrinsic and extrinsic cell death pathways are critically involved in the development and function of T lymphocytes.
T LYMPHOCYTE DEVELOPMENT AND APOPTOSIS
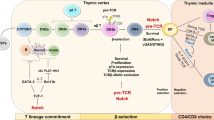
T lymphocyte development is a highly regulated process. To ensure the generation of a functional TCR repertoire, there are multiple checkpoints during thymocyte development to delete thymocytes expressing a nonfunctional or autoreactive TCR. In the thymus, T lymphocytes are divided into three major developmental stages: the CD4−CD8− double negative (DN) stage; the CD4+CD8+ double positive (DP) stage and the CD4+CD8− or CD8+CD4− single positive (SP) stage 123, 124, 125. At each stage, the major signals that trigger thymocyte apoptosis are different (Fig. 2).

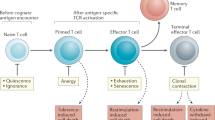
The role of apoptosis in the development and function of T lymphocytes. Major pro-apoptotic and anti-apoptotic signals/molecules are shown.
DN thymocytes, IL-7 signaling and β selection
At the DN stage, T cell precursors undergo TCRβ chain rearrangement. DN thymocytes are divided into four developmental stages DN1-DN4 by their surface expression of CD25 and CD44 (Fig. 2). In adults, thymocyte development begins with the migration of multipotent precursors from the bone marrow into the thymus. These precursor cells periodically enter the thymus through post-capillary venules in the cortex region close to the corticomedullary junction 126, 127. After entering the thymus, the precursor cells differentiate into DN1 cells (CD44+CD25−). As DN1 cells migrate towards the outer cortex, they progressively differentiate into DN2 (CD44+CD25+) and then DN3 (CD44−CD25+) cells. Chemokine receptor CCR7 signaling is critical for the migration of DN cells towards the outer cortex as DN2 cells accumulate at the corticomedullary junction in CCR7 deficient mice 128, 129. Starting from DN2 and DN3, RAG1 and RAG2 genes are expressed and the TCRβ locus undergoes vigorous genomic rearrangement. After TCRβ rearrangement, thymocytes undergo β-selection to ensure that only cells with a successfully rearranged TCRβ gene survive. These cells receive positive signals from the pre-TCR, which is composed of a TCRβ chain and an invariant pre-Tα chain. Successful β-selection leads to a series of events including proliferation and differentiation. Thymocytes that do not receive a β selection signal undergo apoptosis. After β selection, thymocytes further differentiate into the DN4 (CD44− CD25−) and immature single positive (ISP) (CD3−CD4−CD8+) cells.
During DN thymocyte development, the cytokine IL-7 has been shown to be a crucial pro-survival molecule as IL-7−/− or IL-7R−/− mice have fewer DN cells 130, 131. The intrinsic pathway anti-apoptotic Bcl-2 family members are the main effector molecules downstream of IL-7 signaling. Bcl-2 is highly expressed at the DN stage and overexpression of Bcl-2 rescues T cell development in the absence of IL-7 signal 9, 10. Recently, another anti-apoptotic Bcl-2 protein family member, Mcl-1, has been shown to be an essential pro-survival factor regulated by IL-7 signaling 24. Loss of Mcl-1 expression leads to a development blockade at the DN stage due to increased apoptosis. Furthermore, IL-7 signal induces Mcl-1 expression 24. Therefore, Bcl-2 and Mcl-1 are downstream of IL-7 signal and promote DN thymocyte survival.
β selection is a critical checkpoint at the DN3 stage mediated by pre-TCR signaling. Without a proper pre-TCR signal, DN cells undergo apoptosis, which maybe mediated by both intrinsic and extrinsic apoptotic pathways. The anti-apoptotic Bcl-2 family member A1/Bfl-1 lies downstream of the pre-TCR and protects DN thymocytes from apoptosis in response to pre-TCR signaling 30. The extrinsic cell death pathway may mediate apoptosis in DN3 cells that do not receive pre-TCR signals. It has been shown that death receptor 3 (DR3), death receptor 5 (DR5) and tumor necrosis factor receptor (TNFR)1 are expressed in DN3 132. Fas associated death domain adaptor (FADD) is an essential adaptor downstream of these death receptors. Transgenic expression of a dominant negative form of FADD (FADD-DN) rescues DN thymocytes which fail β-selection due to the inability to undergo gene rearrangement 132. This data suggests that inhibition of death receptor signaling bypasses the requirement of pre-TCR signaling to promote DN thymocyte survival.
In summary, both intrinsic and extrinsic apoptotic pathways are actively involved in IL-7 and pre-TCR mediated thymocyte survival at DN stage.
DP thymocytes and death by neglect
DP thymocytes rearrange their TCRα chain and are subjected to positive and negative selection. The majority of DP thymocytes cannot generate a functional TCR and undergo death by neglect. Activation of the intrinsic death pathway may be the major mechanism underlying death by neglect.
At the DP stage, RAG-1 and RAG-2 expression is reactivated and the TCRα loci undergo rearrangement 123, 124. Once a DP thymocyte expresses a successfully rearranged TCR molecule on its surface, it scans an array of self MHC-peptide complexes presented in the thymus. Only DP thymocytes expressing a TCR with intermediate affinity for self MHC-peptide complexes are positively selected 123, 124. Thymocytes expressing a low affinity TCR undergo death by neglect to ensure a functional TCR repertoire in the periphery 123, 124. Thymocytes expressing a high affinity TCR are subjected to negative selection to eliminate potentially autoreactive T cells 123, 124.
Death by neglect contributes to the majority of cell death during thymocyte development. It is estimated that 90% of developing thymocytes undergo death by neglect. The detailed molecular mechanism underlying death by neglect remains largely elusive. However, it is generally believed that Bcl-2 family members are the main effectors involved in death by neglect. In agreement with this notion, spontaneous thymocyte apoptosis in vitro, which may mimic in vivo death by neglect, is controlled by the intrinsic cell death pathway with little or no death receptor signaling involvement. Degradation of the anti-apoptotic proteins Bcl-2 and Bcl-xL and the release of cytochrome C into the cytoplasm are the early events of spontaneous thymocyte apoptosis. Accordingly, the pan-caspase inhibitor zVAD inhibits spontaneous thymocyte apoptosis in vitro 133.
Bcl-xL is highly expressed in DP thymocytes and promotes the life-span of DP thymocytes. In Bcl-x−/−Rag−/−chimeric mice, DP thymocytes have a decreased life-span in vitro 16. RORγ−/− mice, which have dramatically decreased Bcl-xL expression in DP thymocytes, display both a decreased life-span for DP thymocytes as well as a defect in TCRα gene segments usage. These defects are rescued by a Bcl-xL transgene 22, 23. The BH3-only protein Bim is also involved in spontaneous thymocyte apoptosis 134. During a three-day in vitro culture, sorted Bim−/− DP thymocytes survive 10-30 fold better than wild type (WT) DP thymocytes. The pro-apoptotic Bcl-2 family members Bax and Bak are the major cell death executioners in the intrinsic apoptotic pathway. Bax and Bak may participate in death by neglect as Bax−/−Bak−/− thymocytes are resistant to spontaneous apoptosis in vitro 36. After four days of in vitro culture, almost 100% Bax−/−Bak−/− thymocytes survive while less than 10% WT thymocytes survive 36. However, Apaf1−/− and caspase 9−/− thymocytes do not have any defects in death by neglect 135. These data suggest that the apoptosome is not essential for apoptosis in thymocytes. Compared with apoptosome, Bcl-2 family members have a broader function of controlling apoptosis and caspase activation 135. In summary, the Bcl-2 family members Bcl-xL, Bim, Bak and Bax control DP thymocyte survival and may participate in death by neglect process.
SP thymocytes and negative selection
Negative selection deletes thymocytes expressing high affinity TCRs for MHC/self peptide complexes to minimize the risk of autoimmunity in the periphery. A sub-population of SP thymocytes may be subjected to negative selection. While the intrinsic apoptotic pathway is the major mechanism for negative selection, the extrinsic death pathway may also be involved.
After successful positive selection, DP thymocytes further differentiate into CD4+ or CD8+ single positive thymocytes and undergo several maturation steps including the downregulation of HSA and upregulation of CCR7 136. SP thymocytes migrate from the cortex into the medulla of the thymus. CCR7 signal is required for SP thymocyte migration into the thymic medullary region as in CCR7 or CCR7 ligand deficient mice, mature SP thymocytes accumulate in the cortex instead of medulla 137. Recently, we have shown that anti-apoptotic protein c-FLIP is required for SP thymocyte maturation by providing protection against apoptosis 138. Therefore, one or more death receptors may participate in the process of SP thymocyte maturation.
Negative selection is a critical check point during thymocyte development 139, 140. In most high-affinity TCR transgenic mice that express both TCRα and TCRβ from the DN stage, massive negative selection occurs in DP cells or earlier in thymic cortex. This is seen in HY TCR transgenic mice 141. However, when the HY TCRα chain is not expressed until the DP stage, the deletion of HY thymocytes in male mice occurs at the CD8+ SP stage 142. Thus in a physiologic situation, it has been suggested that negative selection occurs during the DP to SP transition 139, 140. For MHC class II restricted thymocytes, semi-mature thymocytes (HSA+CD4+CD8−) are highly sensitive to TCR-induced apoptosis and subjected to negative selection 143. For MHC class I restricted thymocytes, whether negative selection also occurs in the HSA+ population remains to be determined.
Consistently, CD3Med SP cells are subjected to negative selection. CD3MedCD4+ or CD3MedCD8+ SP cells contain the precursors of CD3high SP cells. Sorted CD3Med SP cells differentiate into CD3high SP cells in reaggregated thymic organ culture 144. Compared with their CD3high SP counterparts, CD3Med SP cells are more sensitive to apoptotic stimuli in vitro and contain a significant higher percentage of thymocytes expressing forbidden Vβ+ TCRs reactive with endogenous superantigen 144. These data suggest that CD3Med SP thymocytes are subjected to negative selection.
Furthermore, it has been suggested that negative selection mainly occurs in thymic medulla where the two major populations of antigen presenting cells (dendritic cells and medullary thymic epithelial cells) reside 145. SP thymocytes mainly reside in thymic medulla. Therefore, SP thymocytes may be the major population subjected to negative selection.
Previous research has suggested that the extrinsic apoptotic pathway might participate in negative selection. Death receptor signaling (Fas-FasL) has been shown to play a critical role in the negative selection of semi-mature thymocytes when TCRs receive strong signals 146. However, Fas-FasL interactions are not required for the deletion of semi-mature thymocytes when TCR stimulation is moderate.
Besides Fas-FasL interactions, other death receptors may also be involved in negative selection. However, the available results are conflicting. Anti-TNFα antibody rescues CD4+ SP thymocytes which otherwise undergo negative selection in fetal thymic organ culture (FTOC) 147. TNFR1−/− and TNFR2−/− thymocytes undergo normal negative selection 148, 149. However, TNFR1−/−TNFR2−/− double deficient thymocytes exhibit a defect in negative selection in an in vitro antigen presenting cell co-culture system 150. Therefore, the in vivo role of TNFR1 and TNFR2 in negative selection is unclear at present. Studies on tumor necrosis factor related apoptosis inducing ligand (TRAIL) deficient or TRAILR−/− thymocytes have also generated contradictory conclusions 151, 152, 153, 154. In one study, TRAIL deficiency leads to a defect in anti-CD3 induced thymocyte apoptosis both in vivo and in vitro 152. On a TRAIL−/−background, DO11.10 TCR transgenic mice are resistant to OVA mediated in vivo thymocyte deletion 152. When backcrossed to BALB/C, TRAIL−/− mice also display a defect in the deletion of thymocytes expressing TCRs reactive with endogenous superantigen 152. In another study, TRAIL−/− thymocytes do not have any defects in anti-CD3 mediated thymocyte apoptosis in vitro 151. Furthermore, TRAIL−/− thymocyte display a normal deletion in response to superantigen in FTOC. When backcrossed onto a BALB/C background, TRAIL−/−mice do not have any defects in the deletion of thymocytes reactive with endogenous superantigen 151. The explanation for the discrepancy is unknown. Death receptor 3 (DR3) deficient mice have a subtle defect in thymocyte negative selection 155. DR3−/− mice display a mild defect in DP thymocyte deletion in male H-Y mice and DR3−/− thymocytes show a 10% lower apoptotic rate in response to anti-CD3 stimulation in vitro compared with WT mice 155. While these results suggest that the extrinsic death pathway may be involved in negative selection, further evidence is required.
However, there is strong evidence to support that the intrinsic apoptotic pathway is involved in thymocyte negative selection. The pro-apoptotic Bcl-2 family member Bim is required for normal negative selection in thymocytes as Bim−/− thymocytes are resistant to negative selection in several widely used negative selection models 69. Bim−/− thymocytes are resistant to anti-CD3 induced apoptosis both in vitro and in vivo 69. Furthermore, Bim−/− thymocytes are resistant to superantigen induced deletion in FTOC 69. Bim deficiency rescues OVA peptide induced thymocyte apoptosis in OT-II transgenic mice and thymocyte deletion in male H-Y transgenic mice 69. Bim deficiency rescues sorted semi-mature thymocytes from anti-CD3 induced apoptosis in vitro 156. These authors also presented data to rule out a possible role of Fas-FasL interaction in the TCR/CD3 induced apoptosis of semi-mature thymocytes in vitro 156.
The pro-apoptotic Bcl-2 family members Bax and Bak may participate in negative selection as well. Bax−/−Bak−/− thymocytes are highly resistant to anti-CD3 induced thymocyte apoptosis in vitro and endogenous superantigen induced thymocyte deletion in vivo 36. However, Apaf-1−/− thymocytes have no defect in negative selection 157. Pan-caspase inhibitor p35 T cell specific transgenic mice led to contradictory results in terms of negative selection 158, 159. In one study, the p35 transgene rescues specific peptide and superantigen induced thymocyte apoptosis in F5 TCR transgenic mice in vivo 158. p35 transgenic thymocytes are also resistant to anti-CD3 induced apoptosis in vitro 158. However, in another study, the p35 transgene could only rescue 7-10% thymocytes from anti-CD3 induced apoptosis in vitro 159. Furthermore, the p35 transgene has no effect on thymocyte deletion in H-Y and OT-1 transgenic mice 159. The reason for the discrepancy is not clear at present. In general, Bcl-2 family members Bim, Bax and Bak are required for thymocyte negative selection.
In summary, several critical check points exist during thymocyte development to ensure that only thymocytes bearing intermediate affinity TCRs towards self-MHC-peptide complexes survive and further differentiate into mature T lymphocytes. Those with useless TCRs do not receive a positive selection signal and undergo death by neglect. Those with high affinity TCRs are also eliminated by negative selection. It is estimated that about 95% T lymphocyte precursors undergo apoptosis during T cell development 5. As mentioned before, several apoptosis-related molecules play critical roles at different stages during T lymphocyte development. Therefore, apoptosis is one of the most fundamental processes underlying the development of a functional T cell repertoire.
T LYMPHOCYTE FUNCTION AND APOPTOSIS
Mounting an efficient immune response upon antigen challenge is the primary function for T lymphocytes. However, maintaining T lymphocyte homeostasis between antigenic challenges is also essential. The regulation of T cell immune responses and homeostasis has been the subject of extensive investigation. Costimulatory molecules are required for efficient T cell responses. IL-7 signal is essential for maintaining the homeostasis of both naïve and memory T cell populations. Both intrinsic and extrinsic apoptotic pathways are actively involved in the regulation of T cell responses and homeostasis. Several anti-apoptotic molecules have been identified as critical effector molecules downstream of IL-7 and costimulatory molecule signaling in mature T lymphocytes.
Maintenance of naïve T cells
Naïve T cells are long-lived resting cells, constantly circulating through blood, lymph, and secondary lymphoid organs including spleen, lymph nodes (LNs) and Peyer's Patches (PPs). In a healthy adult animal, naïve T cells do not undergo massive proliferation and T cell numbers remain constant. However, if transferred into a T cell depleted host, naïve T cells undergo homeostatic driven proliferation due to the ability of T cells to respond to empty niches in the environment. In other words, T cells can sense the extra survival signals in the host. During homeostatic driven proliferation, naïve T cells gain some phenotypic markers of effector/memory T cells, including the upregulation of CD44 and downregulation of CD62L, and they even temporarily obtain some effector functions 162.
Naïve T cells require active signals, which mainly include TCR/MHC and IL-7 signals, to survive and to undergo homeostatic driven proliferation. Molecules of the intrinsic apoptotic pathway are the major effectors downstream of TCR/MHC and IL-7 signals regulating naïve T cell survival 163, 164. Transferring naïve T cells into a MHC−/− or IL-7−/− host results in the rapid loss of transferred T cells 163, 165, 166, 167, 168, 169, 170. The MHC molecules delivering survival signals to naïve T cells are believed to be the same type of MHC/self-peptide molecules delivering positive selection signals in the thymus. Whether the peptides are identical to the peptides presented in the thymus during positive selection is still under debate 171, 172, 173. However, it is generally agreed that Bcl-2 protein family may be one of the major effector molecules downstream of TCR/MHC and IL-7 signaling to control naïve T cell survival 163, 164.
T cell immune response
When a naïve T cell encounters its cognate antigen presented by a mature dentritic cell, the T cell is activated 174, 175. Mature dentritic cells not only express high levels of MHC molecules on their surface, but also express costimulatory molecules required for T cell activation including CD80/86, OX40L, 4-1BBL and CD70. Activated T cells upregulate the expression of IL-2R and IL-2, which is the critical pro-proliferative cytokine during T cell activation. Activated T cells undergo vigorous proliferation and differentiate into effector T cells—known as T helper (TH) cells for CD4+ and cytotoxic T lymphocytes (CTLs) for CD8+ T cells. Effector CD4+ and CD8+ T cells secrete inflammatory cytokines, and CTLs directly kill infected cells. In acute infection models, the infection is usually resolved in several days. After the infection is resolved, more than 90% of the effector T cells undergo rapid apoptosis. The remaining cells further differentiate into a memory population, which provides vigorous and efficient immune protection when antigen is re-encountered.
Effector CD4+ T cells perform diverse functions during an immune response. First, effector CD4+ T cells activate antigen presenting cells (APC) by CD40-CD40L interactions. Second, effector CD4+ T cells secrete cytokines to promote a B lymphocyte response. Finally, effector CD4+ T cells provide help for CD8+ T cell responses 176, 177. It has been shown that without CD4+ T cell help, there is a defect in the maintenance of CD8+ memory T cells 178, 179, 180. Without CD4+ T cells, CD8+ memory T cells have decreased IL-7Rα expression and gradually disappear over time 181, 182. Moreover, compared with those CD8+ T cells primed with CD4+ T cell help, the CD8+ T cells primed without CD4+ T cell help (helpless CD8) express a decreased level of Bcl-2, Bcl-xL and c-FLIP, and an increased level of FasL and TRAIL upon secondary challenge 183. Specifically, TRAIL mediated death receptor signaling is critical for the elimination of helpless CD8+ T cells during secondary expansion 183.
Both CD4+ and CD8+ T cell immune responses can be divided into three phases: expansion phase, contraction phase and memory phase. Since more in vivo research has been done on the CD8+ T cell immune response, we will mainly focus on CD8+ T cells during the following discussion.
EXPANSION PHASE OF CD8 + T CELL IMMUNE RESPONSE
After CD8+ naïve T cells encounter cognate antigen presented by mature DCs, CD8+ T cells are activated and undergo up to 3-4 rounds of cell division per day. Antigen specific CD8+ T cells can amplify more than 1,000 fold in a few days 174, 175.
Multiple groups have independently discovered that the CD8+ T cell immune response is programmed and can be triggered by a brief contact with antigen. By treating animals with antibiotics after bacterial infection to limit bacteria persistence in vivo or transferring CD8+ T cells into an antigen-free host after infection, it has clearly been demonstrated that during an acute infection, 24 hours of antigen exposure in vivo is enough to initiate a CD8+ T cell immune response. After this short exposure, CD8+ T cells will undergo 7-10 rounds of cell division and generate fully functional CD8+ effector T cells in the absence of additional antigen stimulation 184, 185, 186, 187, 188. Furthermore, in vivo antigen presentation after acute bacterial infection is transient. During Listeria monocytogenes infection, in vivo antigen presentation lasts less than 3 d. If naïve CD8+ T cells are transferred more than 3 d after infection, most naïve CD8+ T cells do not undergo cell division in vivo 189.
Although a brief antigen exposure is sufficient to trigger a fully functional CD8+ T cell response, the magnitude at the peak of that response is directly correlated with the dose of initial infection. This correlation may reflect the ability of a larger infection to recruit more naïve antigen specific CD8+ T cells to the immune response 184.
During the expansion phase of a CD8+ T cell response, CD8+ T cells undergo a series of differentiation events to remodel their gene expression profiles. CD8+ T cells gain the expression of effector molecules such as interferon γ (IFNγ), perforin and granzyme B. CD8+ T cells downregulate CD62L and CCR7 while upregulating sphingosine-1-phosphate receptor 1(S1P) to facilitate migration out of secondary lymphoid organs. Furthermore, effector CD8+ T cells upregulate tissue specific chemokine receptors so that they can directly migrate into the infected tissue 175, 190.
Interestingly, the effector molecule perforin negatively regulates the expansion phase of the CD8+ T cell response 191, 192. Infection of perforin deficient mice with L. monocytogenes results in a 3-4 fold increase in CD8+ T cell expansion compared with WT mice 192. This increase of CD8+T cell expansion in perforin deficient mice is not due to an increased bacteria load in vivo as immunizing perforin deficient mice with peptide loaded dentritic cells leads to identical results 192.
In summary, a brief antigen exposure is sufficient to trigger naïve CD8+ T cells to differentiate into CD8+ effector T cells. The dose of initial infection controls the magnitude of the peak of CD8+ T cell response.
CONTRACTION PHASE OF CD8 + T CELL IMMUNE RESPONSE
After the peak of the CD8+ T cell immune response, 90-95% of the effector cells are eliminated to maintain homeostasis in the T lymphocyte compartment. It is interesting that the kinetics of contraction phase is very similar regardless of infection model and T cell antigen specificity. The size of the surviving cell population after contraction phase correlates with the size of the CD8+ T cell population at the peak of expansion 193.
The role of both the extrinsic and intrinsic apoptotic pathway in CD8+ T cell contraction phase has been investigated. For the extrinsic apoptotic pathway, it has been found that in vitro activated T cells become highly sensitive to TCR-restimulation induced cell death via Fas and TNFR1 after extended culture in IL-2 90. However, naïve T cells are highly resistant to death receptor induced apoptosis. It is now clear that IL-2 delivers a critical sensitization signal to activated T cells and downregulates the apoptosis inhibitor c-FLIP 194, 195. However, the in vivo contraction phase of the CD8+ T cell immune response after an acute infection is intact in the absence of death receptor signaling 196, 197. Moreover, in vivo administration of high doses of the pan-caspase inhibitor zVAD has little effect on the contraction phase of the CD8+ immune response, while Fas induced liver injury is largely abolished 198. These in vivo data argue against the role of the extrinsic death pathway in the contraction phase during the CD8+T cell immune response. Therefore, it is generally believed that activation induced cell death (AICD) is not involved in acute infection because most T cells will not re-encounter antigen several days after priming. Instead, AICD may be more relevant to chronic infection or autoimmunity in vivo.
The other pathway that may be involved in the contraction phase is the mitochondria-dependent intrinsic death pathway. After the peak of the CD8+ T cell response, when the infection has been resolved, growth or survival factors for effector CD8+ T cells become limited. Therefore, effector cells undergo cytokine-deprivation induced apoptosis. Bcl-2 family members are the major effectors controlling cytokine-deprivation induced apoptosis in T cells. In vitro studies have shown that a Bcl-2 transgene rescues activated T cells from cytokine deprivation induced cell death. Bax−/−Bak−/− T cells are highly resistant to multiple cell death stimuli including cytokine deprivation 36. Furthermore, in human T cells, Bim expression can be induced by TCR signaling 199. Bim-deficient T cells are resistant to cytokine deprivation134 and are defective in the CD8+contraction phase in herpes simplex virus infection200, 201. Moreover, the pro-apoptotic BH-3 only protein Noxa is induced in antigen-specific CD8+ T cells during the early period of the contraction phase 202. Overall, Bcl-2 family members may participate in the contraction phase during CD8+ T cell responses. However, a detailed mechanism is not clear at present.
Interestingly, CD8+ T cells deficient in the effector molecule IFNγ have a defect in the contraction phase following infection 192, 203. However, the mechanism underlying IFNγ mediated CD8+ T cell contraction is unclear. Recently, by treating animals with antibiotics before bacterial infection, it has been shown that the contraction phase is controlled by early inflammation. After antibiotic treatment, the initial infection and inflammation is diminished. As a result, the peak immune response is also diminished. However, a significant population of memory cells is still generated without a contraction phase 204. Whether early inflammation controls CD8+ T cell contraction phase through IFNγ is still an open question and needs further investigation.
MEMORY PHASE OF CD8 + T CELL IMMUNE RESPONSE
A small population of CD8+ T cells survive the contraction phase to differentiate into memory cells. At a cell population level, compared with antigen specific naïve T cells, the frequency of memory T cells increases. At an individual cell level, memory T cells have a lower activation threshold than naïve T cells. Therefore when antigens are reencountered, memory T cells provide more rapid and robust immune protection.
An interesting question in memory T cell development is whether there are markers to identify a subset within the effector T cell pool that will preferentially survive the contraction phase. Alternatively, apoptosis during the contraction phase may be a completely random event. Recently, IL-7R and CD8αα have been identified as markers to identify the early precursors of memory cells within the effector cell population 205, 206. IL-7R+ or CD8αα+ effector T cells have higher expression levels of anti-apoptotic Bcl-2 family members when compared with their IL-7R− or CD8αα− counterparts. Furthermore, IL-7R+ or CD8αα+ effector cells preferentially survive the contraction phase and differentiate into memory cells. In CD8αα deficient mice, CD8+ memory T cell formation after acute infection is compromised 206. These data suggest that CD8αα delivers a pro-survival signal to CD8+ memory T cell precursors. CD8αα binds to the non-classical MHC class I molecule thymic leukemia antigen (TL) 207. Surprisingly, in a mouse strain lacking non-classical MHC class I expression (β2m deficient mice crossed with single classical MHC I transgenic mice), CD8+ memory T cells develop normally. Therefore, CD8αα/TL interaction may not be required for CD8+ memory T cell survival and CD8αα may only serve as a marker for CD8+ memory T cell precursors 208. However, whether CD8αα has an alternative ligand and whether CD8αα delivers a signal without ligand binding may need further investigation.
Memory T cells need active signals to survive. Memory T cells can persist without specific antigen and have a higher turn-over rate compared to naïve T cells in a normal host. In a lymphopenic host, memory T cells undergo homeostatic driven proliferation. It is believed that CD8+ memory T cells can persist without MHC class I 209. However, CD8+ memory T cell survival depends on IL-15 in a normal host, and also depends on IL-7 in a lymphopenic host or in an IL-15 deficient host 169, 210, 211, 212, 213. CD4+ memory T cell survival and homeostatic proliferation depend on IL-7 signal and also require TCR/MHC class II interaction. The IL-7R signal and TCR signal have partial overlapping function in CD4+ memory T lymphocytes 214.
IL-7 mediated T lymphocyte survival
As discussed above, the survival of both naïve and memory CD4+and CD8+ T cells is dependent on IL-7 signaling. The IL-7R has two subunits: an IL-7Rα chain shared with thymic stromal lymphopoietin (TSLP) and a common γc, which is shared with IL-2, IL-4, IL-9, IL-15 and IL-21 164.
The expression of the IL-7R is tightly regulated during T cell development and T cell immune responses. IL-7R is expressed on the early precursors of T lymphocytes (DN stage), downregulated in ISP cells and re-expressed on mature T cells. Although after T cell activation the IL-7R is transiently downregulated, both naïve and memory T cells express IL-7R 205. Downregulation of the IL-7R at the DP stage is required to limit the competition between DN and DP thymocytes for available IL-7 215. Recently, it has been suggested that besides its role in survival and proliferation, IL-7 may have functions in thymocyte differentiation. IL-7 signaling downregulates three critical transcriptional factors involved in the ISP to DP transition: LEF, TCF-1 and RORγ 216. These data suggest that downregulation of the IL-7R may be required for the upregulation of LEF, TCF-1 and RORγ, and for the developmental transition from ISP to DP. In mature T cells, IL-7 and other pro-survival cytokine signaling downregulate the transcription of IL-7Rα. Interestingly, in CD8+ T cells, the downregulation of IL-7Rα by IL-7 signaling depends on the transcriptional factor Gfl-1, while in CD4+ T cells, it does not. It has been proposed that those T cells that have received survival signals lose the ability to compete for IL-7. Therefore, by this mechanism, the T cell pool is maximized by limited amount of IL-7 217.
The major function of IL-7 in T lymphocytes is to enhance survival. Mediators in the intrinsic apoptotic pathway are the major effectors downstream of IL-7 signaling. It is well-known that IL-7 activates a Jak/STAT signaling pathway 218. STAT5 activates Bcl-2 expression and a Bcl-2 transgene can rescue the defect in IL-7R−/− mice 9, 10. Similarly, a Bcl-2 transgene or a deficiency in Bax partially rescues the severe defects in lymphopoiesis in mice lacking Jak3, which is downstream of IL-7 signaling 219. Furthermore, IL-7 signal activates PI-3K/Akt 218. It is well established that the PI-3K/Akt pathway phosphorylates the pro-apoptotic protein Bad, which inhibits its function 68, 220. Activated Akt represses the activity of the transcription factor FOXO, which classically activates the expression of the pro-apoptotic protein Bim 221. Recently, another anti-apoptotic Bcl-2 family member, Mcl-1, has been shown to be essential for the survival of T and B lymphocytes. It is suggested that Mcl-1 may be an important downstream target of IL-7 signaling 24. Therefore, IL-7 signaling activates Bcl-2 and Mcl-1 and inhibits Bad and Bim to promote T cell survival.
Costimulatory molecules and anti-apoptotic molecules in T cells
Specific TCR/MHC-peptide interactions deliver an activation signal to T cells. However, optimal T cell activation is only achieved when costimulatory signals are present. Costimulatory signals promote T cell activation and survival. In most situations, different costimulatory molecules play functionally, spatially and temporally distinct roles in the T cell immune response. For example, compared with CD28 signals, OX40 and 4-1BB signals function in a later phase of the T cell immune response. However, different costimulatory molecules may also have overlapping functions. For instance, several costimulatory molecules, including CD28, OX40 and 4-1BB, induce the expression of pro-survival Bcl-2 family members to promote T cell survival after activation.
It is well established that CD28-CD80/CD86 signaling is crucial for T cell activation and proliferation. Because of its constitutive expression on mature T cells, CD28 is one of the most critical costimulatory molecules during the initial phase of T cell activation. In vitro studies have shown that CD28 signaling specifically upregulates the expression of the anti-apoptotic protein Bcl-xL, but not Bcl-2 222. Point mutation analysis has shown that tyrosine residue Y170 on the cytoplasmic tail of CD28 is essential for the recruitment of PI-3K, which induces the expression of Bcl-xL 223, 224, 225. CD28 carrying a mutation at Y170 can promote T cell activation, proliferation and IL-2 production. However, the mutant CD28 cannot upregulate Bcl-xL expression and cannot provide anti-apoptotic signals to T cells. CD28 deficiency has a great impact on both CD4+and CD8+ T effector cell development and function. In CD28−/− mice, basal IgG levels are greatly reduced. After infection or immunization, germinal center formation and immunoglobulin class switch are diminished 226, 227. A very similar phenotype has been observed when two of CD28's ligands are disrupted in CD80−/−CD86−/− double deficient mice 228. Effector and memory CD8+ T cell populations in CD28−/− mice are reduced by 2-3 fold in lymphocytic choriomeningitis virus (LCMV) and Listeria monocytogenes infection models 229, 230. Furthermore, the numbers of effector CD8+ T cells in secondary lymphoid organs during primary and memory responses to influenza virus infection are greatly reduced in CD28−/− mice 231. Therefore, CD28 is required for both CD4+ and CD8+ T cell responses.
OX40 is an essential costimulatory molecule for CD4+ T cell immune responses. Expression of OX40 can be detected 48 hours after T cells receive activation signals. OX40 functions after CD28 to sustain the expression of anti-apoptotic molecules in activated CD4+ T cells. OX40 signaling activates the PI-3K/Akt pathway and upregulates the anti-apoptotic proteins Bcl-xL and Bcl-2 232. OX40−/− T cells have a defect in long term survival. This survival defect is rescued by retrovirus mediated overexpression of Bcl-2, Bcl-xL or activated Akt kinase 232, 233. OX40L is detected on activated B cells, macrophages and dentritic cells 234. Interestingly, OX40L is also detected on a newly identified population of accessory cells (CD3−CD4+MHC II+ OX40L+ B220−) in the spleen in vivo 235. After immunization, OX40L+ accessory cells are in close contact with T cells 235. Furthermore, this contact correlates with the survival advantage of activated OX40+/+ T cells over OX40−/− T cells in vivo 235. The analysis of OX40−/− mice has revealed that the CD8+ T cell immune response during acute viral infection is not impaired. However, CD4+ memory T cell recall response is profoundly reduced in OX40−/− mice after immunization, although germinal center formation is intact 236. OX40−/− CD4+ T cells can produce IL-2 and expand during the initial phase of an immune response. However, OX40−/−CD4+ T cells have a survival defect in the later phase. These data suggest that OX40 is preferentially involved in CD4+ memory cell development and maintenance. Based on these observations, it has been proposed that OX40 signals function after CD28 signals to maintain the expression of anti-apoptotic proteins Bcl-xL and Bcl-2 to promote CD4+ T cell survival during immune responses.
In parallel to the function of OX40 in CD4+ T cells, the costimulatory molecule 4-1BB promotes CD8+ T cell immune response presumably by induction of anti-apoptotic molecules in activated CD8+ T cells. 4-1BB is not expressed on resting T lymphocytes. Expression of 4-1BB is induced 24-48 h after T cell activation. 4-1BBL is expressed on activated antigen presenting cells 234. 4-1BB signal also induces the anti-apoptotic proteins Bcl-xL and A1/Bfl-1 in T lymphocytes through an NF-κB dependent pathway 238, 239, 240. However, during viral infection, CD4+ T cell function is unimpaired in 4-1BB−/− mice. In contrast, effector CD8+ T cell response, especially CD8+ T cell memory response is defective in 4-1BB deficient mice 241, 242, 243. These data suggest that 4-1BB is required for CD8+ effector and memory cell development and survival during viral infection. 4-1BB may function after CD28 to promote the expression of anti-apoptotic molecules in activated CD8+ T cells.
CD27 is another critical costimulatory molecule required for memory T cell development. CD27 has a low to moderate expression level on naïve CD4+ and CD8+ T lymphocytes 234. After T cell activation, CD27 is quickly upregulated and downregulated again during the expansion phase of the T cell response 234. During viral infection in both mice and humans, a CD27−CD8+ cell population has been identified as the most efficient effector cell population 244, 245. CD27 signal not only promotes T cell division, but also T cell survival after activation in vitro 231. Whether CD27 signals promote T cell survival by induction of anti-apoptotic Bcl-2 family members needs further investigation. In CD27−/− mice, after intranasal influenza infection, there is a dramatic reduction of effector CD4+ and CD8+ cells in the lung and draining lymph nodes (DLN) and a mild reduction of effector T cells in the spleen. This is true both in primary and memory responses. In contrast, in the same infection model the major defect for CD28−/− mice lies in the accumulation of effector T cells in the spleen 231, 246.
In summary, costimulatory molecules are essential for T cell function. Anti-apoptotic proteins are important mediators downstream of costimulatory signals. Interestingly, although Bcl-xL is one of the most important effector molecules downstream of CD28, OX40 and 4-1BB signals, Bcl-xL is dispensable for the development of effector and memory T lymphocytes 247. This result may be explained by the redundancy between different anti-apoptotic Bcl-2 family members or different anti-apoptotic pathways.
CONCLUSION
The function of apoptosis in the development and function of T lymphocytes has been an exciting field for years. However, many issues remain to be addressed. For examples, what are the detailed molecular mechanisms of thymocyte apoptosis in vivo? Which molecules are essential for effector and memory T lymphocyte survival? Which molecules are involved in the contraction phase of T cell immune responses? The answers to these questions will not only further our knowledge of T cell biology, but will have great implications in vaccine design and immunotherapy as well.
References
Danial NN, Korsmeyer SJ . Cell death: critical control points. Cell 2004; 116:205–19.
Bakhshi A, Jensen JP, Goldman P, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 1985; 41:899–906.
Vander Heiden MG, Thompson CB . Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nat Cell Biol 1999; 1:E209–16.
Gross A, McDonnell JM, Korsmeyer SJ . BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999; 13:1899–911.
Chao DT, Korsmeyer SJ . BCL-2 family: regulators of cell death. Annu Rev Immunol 1998; 16:395–419.
Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ . Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993; 75:229–40.
Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY . Targeted disruption of Bcl-2 alpha beta in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci U S A 1994; 91:3700–4.
Matsuzaki Y, Nakayama K, Tomita T, et al. Role of bcl-2 in the development of lymphoid cells from the hematopoietic stem cell. Blood 1997; 89:853–62.
Akashi K, Kondo M, von Freeden-Jeffry U, Murray R, Weissman IL . Bcl-2 rescues T lymphopoiesis in interleukin-7 receptor-deficient mice. Cell 1997; 89:1033–41.
Maraskovsky E, O'Reilly LA, Teepe M, et al. Bcl-2 can rescue T lymphocyte development in interleukin-7 receptor-deficient mice but not in mutant rag-1-/- mice. Cell 1997; 89:1011–9.
Boise LH, Gonzalez-Garcia M, Postema CE, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993; 74:597–608.
Gonzalez-Garcia M, Perez-Ballestero R, Ding L, et al. bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development 1994; 120:3033–42.
Fang W, Rivard JJ, Mueller DL, Behrens TW . Cloning and molecular characterization of mouse bcl-x in B and T lymphocytes. J Immunol 1994; 153:4388–98.
Shiraiwa N, Inohara N, Okada S, et al. An additional form of rat Bcl-x, Bcl-xbeta, generated by an unspliced RNA promotes apoptosis in promyeloid cells. J Biol Chem 1996; 271:13258–65.
Yang XF, Weber GF, Cantor H . A novel Bcl-x isoform connected to the T cell receptor regulates apoptosis in T cells. Immunity 1997; 7:629–39.
Motoyama N, Wang F, Roth KA, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 1995; 267:1506–10.
Wagner KU, Claudio E, Rucker EB 3rd, et al. Conditional deletion of the Bcl-x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Development 2000; 127:4949–58.
Hon H, Rucker EB 3rd, Hennighausen L, Jacob J . bcl-xL is critical for dendritic cell survival in vivo. J Immunol 2004; 173:4425–32.
Hou WS, Van Parijs L . A Bcl-2-dependent molecular timer regulates the lifespan and immunogenicity of dendritic cells. Nat Immunol 2004; 5:583–9.
Grillot DA, Merino R, Nunez G . Bcl-XL displays restricted distribution during T cell development and inhibits multiple forms of apoptosis but not clonal deletion in transgenic mice. J Exp Med 1995; 182:1973–83.
Ma A, Pena JC, Chang B, et al. Bclx regulates the survival of double-positive thymocytes. Proc Natl Acad Sci U S A 1995; 92:4763–7.
Guo J, Hawwari A, Li H, et al. Regulation of the TCRalpha repertoire by the survival window of CD4(+)CD8(+) thymocytes. Nat Immunol 2002; 3:469–76.
Sun Z, Unutmaz D, Zou YR, et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 2000; 288:2369–73.
Opferman JT, Letai A, Beard C, et al. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003; 426:671–6.
Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005; 307:1101–4.
Chen C, Edelstein LC, Gelinas C . The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol Cell Biol 2000; 20:2687–95.
Duriez PJ, Wong F, Dorovini-Zis K, Shahidi R, Karsan A . A1 functions at the mitochondria to delay endothelial apoptosis in response to tumor necrosis factor. J Biol Chem 2000; 275:18099–107.
D'Sa-Eipper C, Chinnadurai G . Functional dissection of Bfl-1, a Bcl-2 homolog: anti-apoptosis, oncogene-cooperation and cell proliferation activities. Oncogene 1998; 16:3105–14.
Craxton A, Chuang PI, Shu G, Harlan JM, Clark EA . The CD40-inducible Bcl-2 family member A1 protects B cells from antigen receptor-mediated apoptosis. Cell Immunol 2000; 200:56–62.
Mandal M, Borowski C, Palomero T, et al. The BCL2A1 gene as a pre-T cell receptor-induced regulator of thymocyte survival. J Exp Med 2005; 201:603–14.
Farrow SN, White JH, Martinou I, et al. Cloning of a bcl-2 homologue by interaction with adenovirus E1B 19K. Nature 1995; 374:731–3.
Chittenden T, Harrington EA, O'Connor R, et al. Induction of apoptosis by the Bcl-2 homologue Bak. Nature 1995; 374:733–6.
Kiefer MC, Brauer MJ, Powers VC, et al. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature 1995; 374:736–9.
Oltvai ZN, Milliman CL, Korsmeyer SJ . Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993; 74:609–19.
Hsu SY, Hsueh AJ . A splicing variant of the Bcl-2 member Bok with a truncated BH3 domain induces apoptosis but does not dimerize with antiapoptotic Bcl-2 proteins in vitro. J Biol Chem 1998; 273:30139–46.
Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB . Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol 2002; 3:932–9.
Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 2000; 6:1389–99.
Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292:727–30.
Ruiz-Vela A, Opferman JT, Cheng EH, Korsmeyer SJ . Proapoptotic BAX and BAK control multiple initiator caspases. EMBO Rep 2005; 6:379–85.
Kuwana T, Mackey MR, Perkins G, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002; 111:331–42.
Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91:479–89.
Liu X, Kim CN, Yang J, Jemmerson R, Wang X . Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996; 86:147–57.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997; 90:405–13.
Degterev A, Boyce M, Yuan J . A decade of caspases. Oncogene 2003; 22:8543–67.
Deveraux QL, Takahashi R, Salvesen GS, Reed JC . X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997; 388:300–4.
Chai J, Shiozaki E, Srinivasula SM, et al. Structural basis of caspase-7 inhibition by XIAP. Cell 2001; 104:769–80.
Huang Y, Park YC, Rich RL, et al. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 2001; 104:781–90.
Riedl SJ, Renatus M, Schwarzenbacher R, et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell 2001; 104:791–800.
Salvesen GS, Duckett CS . IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 2002; 3:401–10.
Chai J, Du C, Wu JW, et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000; 406:855–62.
Du C, Fang M, Li Y, Li L, Wang X . Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102:33–42.
Liu Z, Sun C, Olejniczak ET, et al. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 2000; 408:1004–8.
Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102:43–53.
Suzuki Y, Imai Y, Nakayama H, et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 2001; 8:613–21.
Li LY, Luo X, Wang X . Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001; 412:95–9.
Parrish J, Li L, Klotz K, et al. Mitochondrial endonuclease G is important for apoptosis in C elegans. Nature 2001; 412:90–4.
Joza N, Susin SA, Daugas E, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001; 410:549–54.
Susin SA, Lorenzo HK, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999; 397:441–6.
Wang X, Yang C, Chai J, Shi Y, Xue D . Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 2002; 298:1587–92.
Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297:259–63.
Huang DC, Strasser A . BH3-Only proteins-essential initiators of apoptotic cell death. Cell 2000; 103:839–42.
Marsden VS, Strasser A . Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol 2003; 21:71–105.
Chen L, Willis SN, Wei A, et al. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol Cell 2005; 17:393–403.
Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ . BID: a novel BH3 domain-only death agonist. Genes Dev 1996; 10:2859–69.
Li H, Zhu H, Xu CJ, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94:491–501.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94:481–90.
Yang E, Zha J, Jockel J, et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995; 80:285–91.
Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ . Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996; 87:619–28.
Bouillet P, Purton JF, Godfrey DI, et al. BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 2002; 415:922–6.
Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003; 302:1036–8.
Yin XM, Wang K, Gross A, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999; 400:886–91.
Nakano K, Vousden KH . PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683–94.
Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288:1053–8.
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B . PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7:673–82.
Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L . PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A 2003; 100:1931–6.
Schuler M, Maurer U, Goldstein JC, et al. p53 triggers apoptosis in oncogene-expressing fibroblasts by the induction of Noxa and mitochondrial Bax translocation. Cell Death Differ 2003; 10:451–60.
Coultas L, Bouillet P, Stanley EG, et al. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol Cell Biol 2004; 24:1570–81.
Locksley RM, Killeen N, Lenardo MJ . The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 2001; 104:487–501.
Muppidi JR, Tschopp J, Siegel RM . Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 2004; 21:461–5.
Dempsey PW, Doyle SE, He JQ, Cheng G . The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev 2003; 14:193–209.
Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E . The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci 1995; 20:342–4.
Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM . FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995; 81:505–12.
Hsu H, Xiong J, Goeddel DV . The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995; 81:495–504.
Chaudhary PM, Eby M, Jasmin A, et al. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997; 7:821–30.
Schneider P, Thome M, Burns K, et al. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997; 7:831–6.
Chinnaiyan AM, O'Rourke K, Yu GL, et al. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science 1996; 274:990–2.
Pan G, Bauer JH, Haridas V, et al. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett 1998; 431:351–6.
Rieux-Laucat F, Le Deist F, Hivroz C, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 1995; 268:1347–9.
Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S . Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992; 356:314–7.
Lenardo M, Chan KM, Hornung F, et al. Mature T lymphocyte apoptosis—immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol 1999; 17:221–53.
Nagata S, Suda T . Fas and Fas ligand: lpr and gld mutations. Immunol Today 1995; 16:39–43.
Kischkel FC, Hellbardt S, Behrmann I, et al. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. Embo J 1995; 14:5579–88.
Boldin MP, Goncharov TM, Goltsev YV, Wallach D . Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996; 85:803–15.
Muzio M, Chinnaiyan AM, Kischkel FC, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death—inducing signaling complex. Cell 1996; 85:817–27.
Medema JP, Scaffidi C, Kischkel FC, et al. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). Embo J 1997; 16:2794–804.
Yang X, Chang HY, Baltimore D . Autoproteolytic activation of pro-caspases by oligomerization. Mol Cell 1998; 1:319–25.
Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388:190–5.
Shu HB, Halpin DR, Goeddel DV . Casper is a FADD- and caspase-related inducer of apoptosis. Immunity 1997; 6:751–63.
Hu S, Vincenz C, Ni J, Gentz R, Dixit VM . I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J Biol Chem 1997; 272:17255–7.
Goltsev YV, Kovalenko AV, Arnold E, et al. CASH, a novel caspase homologue with death effector domains. J Biol Chem 1997; 272:19641–4.
Srinivasula SM, Ahmad M, Ottilie S, et al. FLAME-1, a novel FADD-like anti-apoptotic molecule that regulates Fas/TNFR1-induced apoptosis. J Biol Chem 1997; 272:18542–5.
Han DK, Chaudhary PM, Wright ME, et al. MRIT, a novel death-effector domain-containing protein, interacts with caspases and BclXL and initiates cell death. Proc Natl Acad Sci U S A 1997; 94:11333–8.
Inohara N, Koseki T, Hu Y, Chen S, Nunez G . CLARP, a death effector domain-containing protein interacts with caspase-8 and regulates apoptosis. Proc Natl Acad Sci U S A 1997; 94:10717–22.
Rasper DM, Vaillancourt JP, Hadano S, et al. Cell death attenuation by 'Usurpin', a mammalian DED-caspase homologue that precludes caspase-8 recruitment and activation by the CD-95 (Fas, APO-1) receptor complex. Cell Death Differ 1998; 5:271–88.
Thome M, Tschopp J . Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol 2001; 1:50–8.
Chang DW, Xing Z, Pan Y, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. Embo J 2002; 21:3704–14.
Yeh WC, Itie A, Elia AJ, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 2000; 12:633–42.
Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV . TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996; 4:387–96.
Kelliher MA, Grimm S, Ishida Y, et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998; 8:297–303.
Hsu H, Shu HB, Pan MG, Goeddel DV . TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996; 84:299–308.
Liu ZG, Hsu H, Goeddel DV, Karin M . Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 1996; 87:565–76.
Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J . NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol 2001; 21:5299–305.
Wang Q, Wang X, Evers BM . Induction of cIAP-2 in human colon cancer cells through PKC delta/NF-kappa B. J Biol Chem 2003; 278:51091–9.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114:181–90.
Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C . Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 2003; 18:655–64.
Barnhart BC, Peter ME . The TNF receptor 1: a split personality complex. Cell 2003; 114:148–50.
Harper N, Hughes M, MacFarlane M, Cohen GM . Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem 2003; 278:25534–41.
Lin Y, Devin A, Rodriguez Y, Liu ZG . Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 1999; 13:2514–26.
Deng Y, Ren X, Yang L, Lin Y, Wu X . A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003; 115:61–70.
Kamata H, Honda SI, Maeda S, et al. Reactive Oxygen Species Promote TNFalpha-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell 2005; 120:649–661.
Sabapathy K, Kallunki T, David JP, et al. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J Exp Med 2001; 193:317–28.
Schneider-Brachert W, Tchikov V, Neumeyer J, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 2004; 21:415–28.
Goldrath AW, Bevan MJ . Selecting and maintaining a diverse T-cell repertoire. Nature 1999; 402:255–62.
Starr TK, Jameson SC, Hogquist KA . Positive and negative selection of T cells. Annu Rev Immunol 2003; 21:139–76.
Godfrey DI, Zlotnik A . Control points in early T-cell development. Immunol Today 1993; 14:547–53.
Donskoy E, Foss D, Goldschneider I . Gated importation of prothymocytes by adult mouse thymus is coordinated with their periodic mobilization from bone marrow. J Immunol 2003; 171:3568–75.
Foss DL, Donskoy E, Goldschneider I . The importation of hematogenous precursors by the thymus is a gated phenomenon in normal adult mice. J Exp Med 2001; 193:365–74.
Witt CM, Robey EA . The ins and outs of CCR7 in the thymus. J Exp Med 2004; 200:405–9.
Misslitz A, Pabst O, Hintzen G, et al. Thymic T cell development and progenitor localization depend on CCR7. J Exp Med 2004; 200:481–91.
Peschon JJ, Morrissey PJ, Grabstein KH, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med 1994; 180:1955–60.
von Freeden-Jeffry U, Vieira P, Lucian LA, et al. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med 1995; 181:1519–26.
Newton K, Harris AW, Strasser A . FADD/MORT1 regulates the pre-TCR checkpoint and can function as a tumour suppressor. Embo J 2000; 19:931–41.
Zhang J, Mikecz K, Finnegan A, Glant TT . Spontaneous thymocyte apoptosis is regulated by a mitochondrion-mediated signaling pathway. J Immunol 2000; 165:2970–4.
Bouillet P, Metcalf D, Huang DC, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 1999; 286:1735–8.
Marsden VS, O'Connor L, O'Reilly LA, et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 2002; 419:634–7.
Rosen H, Alfonso C, Surh CD, McHeyzer-Williams MG . Rapid induction of medullary thymocyte phenotypic maturation and egress inhibition by nanomolar sphingosine 1-phosphate receptor agonist. Proc Natl Acad Sci U S A 2003; 100:10907–12.
Ueno T, Saito F, Gray DH, et al. CCR7 signals are essential for cortex-medulla migration of developing thymocytes. J Exp Med 2004; 200:493–505.
Zhang N, He YW . An essential role for c-FLIP in the efficient development of mature T lymphocytes. J Exp Med 2005; 202:395–404.
Palmer E . Negative selection—clearing out the bad apples from the T-cell repertoire. Nat Rev Immunol 2003; 3:383–91.
Sprent J, Kishimoto H . The thymus and negative selection. Immunol Rev 2002; 185:126–35.
von Boehmer H . Developmental biology of T cells in T cell-receptor transgenic mice. Annu Rev Immunol 1990; 8:531–56.
Baldwin TA, Sandau MM, Jameson SC, Hogquist KA . The timing of TCR{alpha} expression critically influences T cell development and selection. J Exp Med 2005; 202:111–21.
Kishimoto H, Sprent J . Negative selection in the thymus includes semimature T cells. J Exp Med 1997; 185:263–71.
Sheard MA, Liu C, Takahama Y . Developmental status of CD4-CD8+ and CD4+CD8- thymocytes with medium expression of CD3. Eur J Immunol 2004; 34:25–35.
Kyewski B, Derbinski J . Self-representation in the thymus: an extended view. Nat Rev Immunol 2004; 4:688–98.
Kishimoto H, Surh CD, Sprent J . A role for Fas in negative selection of thymocytes in vivo. J Exp Med 1998; 187:1427–38.
Page DM . Cutting edge: thymic selection and autoreactivity are regulated by multiple coreceptors involved in T cell activation. J Immunol 1999; 163:3577–81.
Pfeffer K, Matsuyama T, Kundig TM, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993; 73:457–67.
Erickson SL, de Sauvage FJ, Kikly K, et al. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature 1994; 372:560–3.
Page DM, Roberts EM, Peschon JJ, Hedrick SM . TNF receptor-deficient mice reveal striking differences between several models of thymocyte negative selection. J Immunol 1998; 160:120–33.
Cretney E, Uldrich AP, Berzins SP, et al. Normal thymocyte negative selection in TRAIL-deficient mice. J Exp Med 2003; 198:491–6.
Lamhamedi-Cherradi SE, Zheng SJ, Maguschak KA, Peschon J, Chen YH . Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL-/- mice. Nat Immunol 2003; 4:255–60.
Diehl GE, Yue HH, Hsieh K, et al. TRAIL-R as a negative regulator of innate immune cell responses. Immunity 2004; 21:877–89.
Corazza N, Brumatti G, Jakob S, Villunger A, Brunner T . TRAIL and thymocyte apoptosis: not so deadly? Cell Death Differ 2004; 11 Suppl 2:S213–5.
Wang EC, Thern A, Denzel A, et al. DR3 regulates negative selection during thymocyte development. Mol Cell Biol 2001; 21:3451–61.
Villunger A, Marsden VS, Zhan Y, et al. Negative selection of semimature CD4(+)8(-)HSA+ thymocytes requires the BH3-only protein Bim but is independent of death receptor signaling. Proc Natl Acad Sci U S A 2004; 101:7052–7.
Hara H, Takeda A, Takeuchi M, et al. The apoptotic protease-activating factor 1-mediated pathway of apoptosis is dispensable for negative selection of thymocytes. J Immunol 2002; 168:2288–95.
Izquierdo M, Grandien A, Criado LM, et al. Blocked negative selection of developing T cells in mice expressing the baculovirus p35 caspase inhibitor. Embo J 1999; 18:156–66.
Doerfler P, Forbush KA, Perlmutter RM . Caspase enzyme activity is not essential for apoptosis during thymocyte development. J Immunol 2000; 164:4071–9.
Marrack P, Bender J, Hildeman D, et al. Homeostasis of alpha beta TCR+ T cells. Nat Immunol 2000; 1:107–11.
Freitas AA, Rocha B . Population biology of lymphocytes: the flight for survival. Annu Rev Immunol 2000; 18:83–111.
Goldrath AW, Bogatzki LY, Bevan MJ . Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med 2000; 192:557–64.
Kirberg J, Berns A, von Boehmer H . Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med 1997; 186:1269–75.
Fry TJ, Mackall CL . Interleukin-7: master regulator of peripheral T-cell homeostasis? Trends Immunol 2001; 22:564–71.
Nesic D, Vukmanovic S . MHC class I is required for peripheral accumulation of CD8+ thymic emigrants. J Immunol 1998; 160:3705–12.
Viret C, Wong FS, Janeway CA Jr . Designing and maintaining the mature TCR repertoire: the continuum of self-peptide:self-MHC complex recognition. Immunity 1999; 10:559–68.
Rooke R, Waltzinger C, Benoist C, Mathis D . Targeted complementation of MHC class II deficiency by intrathymic delivery of recombinant adenoviruses. Immunity 1997; 7:123–34.
Takeda S, Rodewald HR, Arakawa H, Bluethmann H, Shimizu T . MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity 1996; 5:217–28.
Schluns KS, Kieper WC, Jameson SC, Lefrancois L . Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol 2000; 1:426–32.
Tan JT, Dudl E, LeRoy E, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A 2001; 98:8732–7.
Bender J, Mitchell T, Kappler J, Marrack P . CD4+ T cell division in irradiated mice requires peptides distinct from those responsible for thymic selection. J Exp Med 1999; 190:367–74.
Ernst B, Lee DS, Chang JM, Sprent J, Surh CD . The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity 1999; 11:173–81.
Goldrath AW, Bevan MJ . Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity 1999; 11:183–90.
Badovinac VP, Harty JT . CD8(+) T-cell homeostasis after infection: setting the 'curve'. Microbes Infect 2002; 4:441–7.
Kaech SM, Wherry EJ, Ahmed R . Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol 2002; 2:251–62.
Ridge JP, Di Rosa F, Matzinger P . A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998; 393:474–8.
Bennett SR, Carbone FR, Karamalis F, et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998; 393:478–80.
Janssen EM, Lemmens EE, Wolfe T, et al. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003; 421:852–6.
Sun JC, Bevan MJ . Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003; 300:339–42.
Shedlock DJ, Shen H . Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003; 300:337–9.
Sun JC, Williams MA, Bevan MJ . CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol 2004; 5:927–33.
Bevan MJ . Helping the CD8(+) T-cell response. Nat Rev Immunol 2004; 4:595–602.
Janssen EM, Droin NM, Lemmens EE, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 2005; 434:88–93.
Kaech SM, Ahmed R . Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol 2001; 2:415–22.
Bevan MJ, Fink PJ . The CD8 response on autopilot. Nat Immunol 2001; 2:381–2.
van Stipdonk MJ, Lemmens EE, Schoenberger SP . Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol 2001; 2:423–9.
Mercado R, Vijh S, Allen SE, et al. Early programming of T cell populations responding to bacterial infection. J Immunol 2000; 165:6833–9.
Wong P, Pamer EG . Cutting edge: antigen-independent CD8 T cell proliferation. J Immunol 2001; 166:5864–8.
Wong P, Pamer EG . Feedback regulation of pathogen-specific T cell priming. Immunity 2003; 18:499–511.
Cyster JG . Chemokines, Sphingosine-1-Phosphate, and Cell Migration in Secondary Lymphoid Organs. Annu Rev Immunol 2004.
Badovinac VP, Hamilton SE, Harty JT . Viral infection results in massive CD8+ T cell expansion and mortality in vaccinated perforin-deficient mice. Immunity 2003; 18:463–74.
Badovinac VP, Tvinnereim AR, Harty JT . Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science 2000; 290:1354–8.
Badovinac VP, Porter BB, Harty JT . Programmed contraction of CD8(+) T cells after infection. Nat Immunol 2002; 3:619–26.
Algeciras-Schimnich A, Griffith TS, Lynch DH, Paya CV . Cell cycle-dependent regulation of FLIP levels and susceptibility to Fas-mediated apoptosis. J Immunol 1999; 162:5205–11.
Schmitz I, Weyd H, Krueger A, et al. Resistance of short term activated T cells to CD95-mediated apoptosis correlates with de novo protein synthesis of c-FLIPshort. J Immunol 2004; 172:2194–200.
Nguyen LT, McKall-Faienza K, Zakarian A, et al. TNF receptor 1 (TNFR1) and CD95 are not required for T cell deletion after virus infection but contribute to peptide-induced deletion under limited conditions. Eur J Immunol 2000; 30:683–8.
Reich A, Korner H, Sedgwick JD, Pircher H . Immune down-regulation and peripheral deletion of CD8 T cells does not require TNF receptor-ligand interactions nor CD95 (Fas, APO-1). Eur J Immunol 2000; 30:678–82.
Nussbaum AK, Whitton JL . The contraction phase of virus-specific CD8+ T cells is unaffected by a pan-caspase inhibitor. J Immunol 2004; 173:6611–8.
Sandalova E, Wei CH, Masucci MG, Levitsky V . Regulation of expression of Bcl-2 protein family member Bim by T cell receptor triggering. Proc Natl Acad Sci U S A 2004; 101:3011–6.
Pellegrini M, Belz G, Bouillet P, Strasser A . Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci U S A 2003; 100:14175–80.
Pellegrini M, Bouillet P, Robati M, et al. Loss of Bim increases T cell production and function in interleukin 7 receptor-deficient mice. J Exp Med 2004; 200:1189–95.
Haring JS, Corbin GA, Harty JT . Dynamic regulation of IFN-gamma signaling in antigen-specific CD8+ T cells responding to infection. J Immunol 2005; 174:6791–802.
Badovinac VP, Harty JT . Adaptive immunity and enhanced CD8+ T cell response to Listeria monocytogenes in the absence of perforin and IFN-gamma. J Immunol 2000; 164:6444–52.
Badovinac VP, Porter BB, Harty JT . CD8+ T cell contraction is controlled by early inflammation. Nat Immunol 2004; 5:809–17.
Kaech SM, Tan JT, Wherry EJ, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 2003; 4:1191–8.
Madakamutil LT, Christen U, Lena CJ, et al. CD8alphaalpha-mediated survival and differentiation of CD8 memory T cell precursors. Science 2004; 304:590–3.
Leishman AJ, Naidenko OV, Attinger A, et al. T cell responses modulated through interaction between CD8alphaalpha and the nonclassical MHC class I molecule, TL. Science 2001; 294:1936–9.
Williams MA, Bevan MJ . Cutting edge: a single MHC class ia is sufficient for CD8 memory T cell differentiation. J Immunol 2005; 175:2066–9.
Murali-Krishna K, Lau LL, Sambhara S, et al. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science 1999; 286:1377–81.
Marks-Konczalik J, Dubois S, Losi JM, et al. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A 2000; 97:11445–50.
Yajima T, Nishimura H, Ishimitsu R, et al. Overexpression of IL-15 in vivo increases antigen-driven memory CD8+ T cells following a microbe exposure. J Immunol 2002; 168:1198–203.
Schluns KS, Williams K, Ma A, Zheng XX, Lefrancois L . Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol 2002; 168:4827–31.
Becker TC, Wherry EJ, Boone D, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med 2002; 195:1541–8.
Seddon B, Tomlinson P, Zamoyska R . Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat Immunol 2003; 4:680–6.
Munitic I, Williams JA, Yang Y, et al. Dynamic regulation of IL-7 receptor expression is required for normal thymopoiesis. Blood 2004; 104:4165–72.
Yu Q, Erman B, Park JH, Feigenbaum L, Singer A . IL-7 receptor signals inhibit expression of transcription factors TCF-1, LEF-1, and RORgammat: impact on thymocyte development. J Exp Med 2004; 200:797–803.
Park JH, Yu Q, Erman B, et al. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity 2004; 21:289–302.
Khaled AR, Durum SK . Death and Baxes: mechanisms of lymphotrophic cytokines. Immunol Rev 2003; 193:48–57.
Wen R, Wang D, McKay C, et al. Jak3 selectively regulates Bax and Bcl-2 expression to promote T-cell development. Mol Cell Biol 2001; 21:678–89.
Harada H, Becknell B, Wilm M, et al. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell 1999; 3:413–22.
Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ . Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 2000; 10:1201–4.
Boise LH, Minn AJ, Noel PJ, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 1995; 3:87–98.
Okkenhaug K, Wu L, Garza KM, et al. A point mutation in CD28 distinguishes proliferative signals from survival signals. Nat Immunol 2001; 2:325–32.
Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL . CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol 2003; 171:166–74.
Burr JS, Savage ND, Messah GE, et al. Cutting edge: distinct motifs within CD28 regulate T cell proliferation and induction of Bcl-XL. J Immunol 2001; 166:5331–5.
Shahinian A, Pfeffer K, Lee KP, et al. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993; 261:609–12.
Lee BJ, Reiter SK, Anderson M, Sarawar SR . CD28(-/-) mice show defects in cellular and humoral immunity but are able to control infection with murine gammaherpesvirus 68. J Virol 2002; 76:3049–53.
McAdam AJ, Farkash EA, Gewurz BE, Sharpe AH . B7 costimulation is critical for antibody class switching and CD8(+) cytotoxic T-lymphocyte generation in the host response to vesicular stomatitis virus. J Virol 2000; 74:203–8.
Suresh M, Whitmire JK, Harrington LE, et al. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J Immunol 2001; 167:5565–73.
Mittrucker HW, Kursar M, Kohler A, Hurwitz R, Kaufmann SH . Role of CD28 for the generation and expansion of antigen-specific CD8(+) T lymphocytes during infection with Listeria monocytogenes. J Immunol 2001; 167:5620–7.
Hendriks J, Xiao Y, Borst J . CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med 2003; 198:1369–80.
Rogers PR, Song J, Gramaglia I, Killeen N, Croft M . OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001; 15:445–55.
Song J, Salek-Ardakani S, Rogers PR, et al. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat Immunol 2004; 5:150–8.
Croft M . Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol 2003; 3:609–20.
Kim MY, Gaspal FM, Wiggett HE, et al. CD4(+)CD3(-) accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collaborate with B cells. Immunity 2003; 18:643–54.
Kopf M, Ruedl C, Schmitz N, et al. OX40-deficient mice are defective in Th cell proliferation but are competent in generating B cell and CTL Responses after virus infection. Immunity 1999; 11:699–708.
Gramaglia I, Jember A, Pippig SD, et al. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol 2000; 165:3043–50.
Lee HW, Park SJ, Choi BK, et al. 4-1BB promotes the survival of CD8+ T lymphocytes by increasing expression of Bcl-xL and Bfl-1. J Immunol 2002; 169:4882–8.
Laderach D, Movassagh M, Johnson A, Mittler RS, Galy A . 4-1BB co-stimulation enhances human CD8(+) T cell priming by augmenting the proliferation and survival of effector CD8(+) T cells. Int Immunol 2002; 14:1155–67.
Bukczynski J, Wen T, Ellefsen K, Gauldie J, Watts TH . Costimulatory ligand 4-1BBL (CD137L) as an efficient adjuvant for human antiviral cytotoxic T cell responses. Proc Natl Acad Sci U S A 2004; 101:1291–6.
DeBenedette MA, Wen T, Bachmann MF, et al. Analysis of 4-1BB ligand (4-1BBL)-deficient mice and of mice lacking both 4-1BBL and CD28 reveals a role for 4-1BBL in skin allograft rejection and in the cytotoxic T cell response to influenza virus. J Immunol 1999; 163:4833–41.
Tan JT, Whitmire JK, Ahmed R, Pearson TC, Larsen CP . 4-1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T cell responses. J Immunol 1999; 163:4859–68.
Kwon BS, Hurtado JC, Lee ZH, et al. Immune responses in 4-1BB (CD137)-deficient mice. J Immunol 2002; 168:5483–90.
Hamann D, Kostense S, Wolthers KC, et al. Evidence that human CD8+CD45RA+CD27- cells are induced by antigen and evolve through extensive rounds of division. Int Immunol 1999; 11:1027–33.
Wherry EJ, Teichgraber V, Becker TC, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 2003; 4:225–34.
Hendriks J, Gravestein LA, Tesselaar K, et al. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol 2000; 1:433–40.
Zhang N, He YW . The antiapoptotic protein Bcl-xL is dispensable for the development of effector and memory T lymphocytes. J Immunol 2005; 174:6967–73.
Acknowledgements
This work was supported by National Institutes of Health grant CA92123 and American Cancer Society grant RSG-0125201 (to YW HE).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
ZHANG, N., HARTIG, H., DZHAGALOV, I. et al. The role of apoptosis in the development and function of T lymphocytes. Cell Res 15, 749–769 (2005). https://doi.org/10.1038/sj.cr.7290345
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290345
Keywords
This article is cited by
-
Apoptosis-mediated ADAM10 activation removes a mucin barrier promoting T cell efferocytosis
Nature Communications (2024)
-
Selenium–GPX4 axis protects follicular helper T cells from ferroptosis
Nature Immunology (2021)
-
Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development
Experimental & Molecular Medicine (2020)
-
Blockade of miR-142-3p promotes anti-apoptotic and suppressive function by inducing KDM6A-mediated H3K27me3 demethylation in induced regulatory T cells
Cell Death & Disease (2019)
-
Lymphocyte Apoptosis and FAS Expression in Patients with 22q11.2 Deletion Syndrome
Journal of Clinical Immunology (2019)
