
ABSTRACT
The muscle protein myosin binding protein C (MyBPC) is a large multi-domain protein whose role in the sarcomere is complex and not yet fully understood. Mutations in MyBPC are strongly associated with the heart disease familial hypertrophic cardiomyopathy (FHC) and these experiments of nature have provided some insight into the intricate workings of this protein in the heart. While some regions of the MyBPC molecule have been assigned a function in the regulation of muscle contraction, the interaction of other regions with various parts of the myosin molecule and the sarcomeric proteins, actin and titin, remain obscure. In addition, several intra-domain interactions between adjacent MyBPC molecules have been identified. Although the basic structure of the molecule (a series of immunoglobulin and fibronectin domains) has been elucidated, the assembly of MyBPC in the sarcomere is a topic for debate. By analysing the MyBPC sequence with respect to FHC-causing mutations it is possible to identify individual residues or regions of each domain that may be important either for binding or regulation. This review looks at the current literature, in concert with alignments and the structural models of MyBPC, in an attempt to understand how FHC mutations may lead to the disease state.
Similar content being viewed by others


Introduction
Myosin binding protein C (MyBPC) was first identified as a contaminant in myosin preparations 30 years ago1, but as its assigned role in the structure and regulation of the sarcomere grows evermore complex, it has come to be of increasing interest to the scientific community. Further importance in gaining an understanding of the MyBPC molecule is illustrated by the identification of numerous disease causing mutations that results in familial hypertrophic cardiomyopathy (FHC). FHC is a serious disorder, especially in children and young adults, that can result in significant morbidity and premature death. One current challenge is to decipher the mechanism through which these mutations in the MyBPC sarcomeric protein can result in severe disease. This review aims to integrate our current understanding about the structure of cardiac MyBPC and its role in the sarcomere.
Structure and Function
MyBPC is a modular polypeptide that belongs to the intracellular immunoglobulin superfamily. It is a sarcomeric protein of approximately 137 kDa found in the thick filament of striated muscle. MyBPC is located in the central region of the A-band, known as the C-zone (Fig 1) 2. In mammalian muscle, seven to nine of the eleven structurally regular transverse C-zone stripes contain MyBPC3. The 43 nm spacing2 of these stripes dictates that only every third level of myosin heads in the C-zone is associated with a MyBPC molecule4. This, and the number of myosin heads that fall outside the C-zone, limit the number of myosin heads that can interact directly with MyBPC5. The core structure of MyBPC is comprised of seven I-class immunoglobulin (IgI) domains and three fibronectin type III (FnIII) domains, numbered from the N-terminus as Motifs 1 to 10 (Fig 1).

Position of MyBPC in the stretched sarcomere and the structure of cardiac MyBPC. The top diagram is a schematic diagram of the sarcomere indicating the location of MyBPC. MyBPC is found in transverse stripes 43 nm apart in the C-Zone, where interaction between the thick and thin filaments occurs. The bottom diagram shows that cardiac MyBPC is comprised of eight IgI domains and three FnIII domains. Motif 0 is cardiac specific as is the extra phosphorylation sites between Motifs 1 and 2. Motifs 7–10 have been found to bind to myosin and titin.
Both structural and regulatory roles have been proposed for MyBPC. It is involved in sarcomere assembly by promoting polymerization of thick filaments, via the C-terminal domains binding to specific sites on titin and light meromyosin (LMM)6. Indeed MyBPC has been demono-crossbridges, which modulates muscle contraction4.
The structural basis for the incorporation of MyBPC into the thick filament is at present poorly understood. Recent data suggest that the formation of a trimer of MyBPC, via the C-terminal half of each molecule, allows the assembly of a collar around the thick filament (Fig 2) 9. However, an alternative model for MyBPC incorporation into the thick filament has been proposed that does not involve specific interactions between MyBPC molecules (Fig 3) 10.

Schematic diagram of the trimeric collar model of cardiac MyBPC. Motifs 5–10 of three cardiac MyBPC molecules assemble in a staggered parallel array around the thick filament. The N-terminal domains (Motifs 0–4) project out from the thick filament to interact with the helical myosin S2 region and possibly the thin filament. Figure modified from Moolman-Smook et al. 20029.

Schematic diagram of the axial model of cardiac MyBPC. Motifs 7–10 are arranged axially along the myosin backbone and are able to interact with titin. The N-terminus reaches out to interact with the myosin crossbridge and/or actin. Figure modified from Squire et al. 200310.
Isoforms/alignments
Three isoforms of human MyBPC have been identified; fast skeletal, slow skeletal and cardiac8. All three isoforms map to different chromosomes indicating that they are not the result of alternative splicing. The computer program, CLUSTAL X11, was used to create multiple alignments of different isoforms of MyBPC from a range of species. These alignments identified regions of conservation and isoform divergence. The sequence identity, for domains 1–10, across human isoforms is 39.6% (Fig 4). Regions of low identity were found to occur in sequences outside of the IgI or FnIII domains, specifically the sequence that precedes Motif 1, the 1–2 linker, and, in cardiac MyBPC, an insert in Motif 5.

Multiple alignments of MyBPC isoforms from different species. hu = human, mo = mouse, ch = chick, fu = fugu, fst = fast skeletal, cc = cardiac, slw = slow skeletal. The alignment is coloured where 60% of the sequences have the same residue or the residues have similar properties (with the exception of proline and glycine which are always coloured yellow and grey, respectively); cyan = hydrophobic, red = positive, violet = negative, green = hydrophilic, blue = aromatic. Asterix above the sequence indicate residues mutated in the human MyBPC isoform that result in FHC. The alignment was built using the program CLUSTAL X11.
The sequence homology for cardiac isoforms from different species is 46.8% (Fig 4). The cardiac isoform differs from the skeletal isoforms in three major ways. Firstly, the cardiac isoform has an extra IgI motif at the N-terminus (Motif 0)12. Secondly, there are three phosphorylation sites between Motifs 1 and 2 in cardiac, compared to one in skeletal12. Finally, one of the central IgI domains (Motif 5) contains a proline/charge-rich insert12. The cardiac isoform retains most of its unique features when mapped back to the Japanese pufferfish, Fugu rubripes, genome sequence13, which also has Motif 0 and the Motif 5 insert, although one of the extra phosphorylation sites appears to be absent.
Cardiac MyBPC
The cardiac isoform comprises 2% of myofibrillar protein in the heart and has been found to be particularly important during myofibrillogenesis and in regenerating muscle cells14, 15. However, despite cardiac MyBPC gene knockout mice showing that MyBPC inclusion is not necessary for the formation of the sarcomere, its absence still resulted in hypertrophy and impaired contractile function16.
The extent of phosphorylation of MyBPC correlates with increased systolic tension and occurs in response to adrenergic stimulation17. Partial extraction of MyBPC from fibres results in an increase in Ca2+-sensitivity and velocity of shortening18, 19.
The cardiac isoform of MyBPC has also been of particular interest because of its link to the heart disease, familial hypertrophic cardiomyopathy (FHC), which is caused by the expression of abnormal contractile proteins in the heart muscle. To date, mutations in ten sarcomeric proteins have been identified as causes of FHC20. The mechanism by which MyBPC mutations cause sarcomeric dysfunction at present poorly understood, but has provided a substantial stimulus to efforts to understand the basic biochemistry and physiology of this important molecule.
Familial hypertrophic cardiomyopathy
FHC is a clinically and genetically heterogeneous disorder characterised macroscopically by increased left ventricular mass in the absence of any apparent loading stress, and histologically by myofibrillar and myocyte disarray and fibrosis20. Clinically, the extent of hypertrophy determines the level of impaired cardiac function. Associated fibrosis can lead to arrythmogenesis and sudden death. Indeed FHC is the most common cause of sudden cardiac death in young athletes21. The prevalence of FHC is believed to be about 0.2%, or 1 in 50022, 23. However despite its relatively high prevalence, the mortality rate for FHC-related deaths in an unselected population was estimated to be only 1%24. This mortality rate reflects the wide range of severity associated with this disease.
The cardiac MyBPC gene (MYBPC3) was the fourth gene identified associated with FHC25. Various studies put the percentage of FHC patients with cardiac MyBPC mutations at between 20–45%26, 27, making it the second most common cause of FHC. Three types of mutations in MYBPC3 that result in FHC have been identified; truncations, point mutations and insertions.
The majority of MYBPC3 FHC mutations generate a frame-shift in the coding sequence, which results in the premature termination of translation of the C-terminus of MyBPC. Patients with these truncated forms of MyBPC usually have a mild phenotype, delayed age of onset and favourable prognosis26, 28, probably since the patients still have a normal copy of the MyBPC gene on the other allele. A similar phenotype is observed in mouse models of FHC with MyBPC C-terminal truncations29, 30, 31. Truncation mutations are thought to cause haploinsufficency, since the absence of the C-terminus of MyBPC appears to result in a failure of the mutant MyBPC to incorporate into the sarcomere32. However this has been disputed and Flavigny et al. proposed that the truncated MyBPC acts via a dominant negative mechanism, possibly as a poison peptide33.
In contrast, a number of missense mutations in MYBPC3 have been identified, some of which result in a severe FHC phenotype. Investigations of the consequences of some of these point mutations have been particularly informative in defining the structure and function of the domain in which they are found. Tab 1 lists the current FHC-causing point mutations identified in MyBPC. The precise mechanism by which many of these mutations cause FHC remains unsolved, but in some cases the degree to which these mutations may affect the structure and function of MyBPC can be inferred from sequence comparisons.
Immunoglobulin superfamily domain structure
Sequence alignments of individual modules from a number of species and proteins have identified key conserved residues for the classification of IgI and FnIII domains (Fig 5 and 6). The immunoglobulin fold is composed of two β-sheets comprising approximately 5 anti parallel β-strands in each sheet. The size and number of β-strands and the conformation of the links between each strand are used to further classify the Ig molecules into sets. The intermediate-set (I-set) of immunoglobulin domains was so named because it has characteristics of both the variable (V) and the constant (C) sets (Fig 5). In an IgI domain, one β-sheet is composed of strands A, B, E and D and the other of strands A, G, F, C and C'34. The alignment of all IgI domains within MyBPC again emphasises the presence of a cardiac-specific insert in Motif 5 of MyBPC. The domain boundaries of IgI domains are generally defined as beginning at a conserved proline residue and end approximately 100 amino acids later with a hydrophobic-X-hydrophobic sequence defining the C-terminus35.

Multiple alignments11 of IgI domains from human (hu), chick (ch), rabbit (rb) and turkey (tu) muscle proteins including; titin (ti), cardiac MyBPC (cc), skeletal MyBPC (sk), MyBPH (h) and telokin (telo). The alignment is coloured where 60% of the sequences have the same residue or the residues have similar properties (with the exception of proline and glycine which are always coloured yellow and grey, respectively); cyan = hydrophobic, red = positive, violet = negative, green = hydrophilic, blue = aromatic. The β-strands are indicated by lines across the top of the alignment and are based on the known structure of m5 from titin83 and Motif 5 from cardiac MyBPC59. The last line indicates the residues required for the classification of IgI domains. This figure highlights the cardiac-specific insert present in Motif 5 of cardiac MyBPC.

Multiple alignments 11 of FnIII domains from human (hu), and chick (ch) muscle proteins; titin (ti), cardiac MyBPC (cc), skeletal MyBPC (sk) and MyBPH (h). The alignment is coloured where 60% of the sequences have the same residue or the residues have similar properties (with the exception of proline and glycine which are always coloured yellow and grey, respectively); cyan = hydrophobic, red = positive, violet = negative, green = hydrophilic, blue = aromatic. The last line indicates the residues required for the classification of FnIII domains. The strands are indicated by lines across the top of the alignment and are based on the known structure of A71 from titin37.
The second class of protein modules found in MyBPC are fibronectin type III domains (FnIII). Similar to the IgI domain, FnIII is also composed of two β-sheets that fold into a β-sandwich, although FnIII conforms to an s-type topology, with one sheet containing strands A, B and E and the other C, C', F and G36, 37 (Fig 7). FnIII motifs are uniquely proline rich and, like most globular proteins, fold to create a hydrophobic core. Based on the alignment of multiple FnIII modules, the N-terminal boundary is defined by a PXPP motif but the C-terminal boundary is more difficult to decipher. Generally it is positioned at the residue following the last β-strand, making the domain approximately 100 amino acids long.

Schematic diagram of IgI (top) and FnIII (bottom) domain structure. The topology (left) of these domains is represented by arrows for β-strands, with plain lines showing the connecting loops. A solved structure of an IgI domain80 and an FnIII domain83 from titin are shown to the right of the topographical diagrams. The sequential order of the strands is given by their labels: A, A', B, etc.
Interactions between β-sandwich domains are generally thought to occur via their sheets, while interactions between these domains and their ligands are thought to occur via their loops38. Additionally, often two consecutive domains are involved in ligand binding38. One study of module-module interactions, in the FnIII rich protein fibronectin, found that the highly conserved proline residues positioned at the domain-domain boundaries may be present to prevent aggregation in this multi-modular protein39. In contrast, computational modelling of FnIII domains suggests that, as proline side-chains can form low-energy interfaces for protein contacts, FnIII modules may interact via the BC and EF loops37.
Correlation between the structure of these homologous domains and the specific location of FHC mutations has allowed, in many cases, the prediction of how FHC point mutations in MyBPC may affect the structure of its motifs and thereby alter their stability, binding or function. MyBPC will be discussed in three parts; the N-terminal region containing Motifs 0–2, the central region containing Motifs 3–6 and the C-terminal region containing Motifs 7–10.
N-terminal region (Motifs 0–2)
Within Motifs 0–2 there may be up to three binding sites. The phosphorylation-dependant binding of the Motif 1–2 linker to S2 of myosin is now well established4, however recent data have suggested that Motif 0 and the Motif 0-1 linker may bind to myosin and/or actin5, 10, 33, 40. The strength of the evidence for each of these interactions is variable, and the physiological consequences remain the subject of investigation.
Despite Motif 0 being unique to the cardiac isoform of MyBPC, its function and specific role in heart muscle is still unclear. There is a surprising lack of FHC-associated mutations in this motif. Only one patient with a mild FHC phenotype has been identified. This mutation lies between strands D and E in a position of low sequence conservation26.
A possible interaction between Motif 0 and some part of the myosin crossbridge has been based on data from a mutant MyBPC knock-in mouse model5. In this mouse, MyBPC is missing both the linker between Motifs 0 and 1, and Motif 1, although the Motif 1–2 linker could still be phosphorylated. There was an increase in Ca2+ sensitivity to force production in this mutant mouse, similar to that seen previously in cardiomyocytes depleted of MyBPC 18. These data are consistent with a low affinity interaction between Motif 0 and some part of the crossbridge, or actin (discussed below). Truncation of MyBPC in the genetically manipulated mouse was thought to prevent Motif 0 from reaching out to interact with the myosin head. In contrast, sequence comparison between Motif 0 and myomesin (a myosin binding protein) suggests that Motif 0 contains a novel putative LMM binding site33. Whether this interaction occurs and what its function would be is unclear.
To further complicate interpretation of the function of this region, an actin binding site at the N-terminus of MyBPC has also been proposed. Co-sedimentation assays showed a low affinity of F-actin for all isoforms of MyBPC 8, 41, indicating that any interaction with actin is unlikely to be via a cardiac-specific region. Homology modelling with the essential light chain of myosin identified the Pro-Ala rich “linker” preceding Motif 1 as the likely candidate for binding actin10. However, no FHC mutations have been found in this linker to support its importance. Recently, fragments of MyBPC containing Motif 0 were shown to bind to actin, with the suggestion that it contributes to the weak binding state by shifting the binding of the N-terminus of MyBPC between actin and myosin40.
Motif 0 and the 0–1 linker are highly unlikely to be able to bind to the myosin crossbridge, the myosin backbone and actin simultaneously. It is possible that the N-terminus of MyBPC cycles through different binding partners, but a more detailed investigation will be required before binding partner/s or roles for Motif 0 and the 0–1 domain linker can be positively assigned.
Motifs 1 and 2 and the phosphorylatable linker that connects them, possess numerous FHC mutations of varying penetrance, stressing the importance of this region. The mutation Y237S in Motif 1 results in a mild FHC phenotype but shows a strong disease association42. This residue is highly conserved across all isoforms and species of MyBPC and is a core residue for defining both the IgI and FnIII domain folds. Located in strand F, Y237 is predicted to point into the hydrophobic core of the domain and to form stabilising hydrogen bonds with residues in strand G. Therefore, when mutated, these interactions may be disrupted, leading to a decreased stability of the domain and the resulting disease state.
The linker between Motifs 1 and 2 is of particular significance due to two additional phosphorylation sites (S284 and S304) in the cardiac isoform, including one present in a cardiac-specific LAGGGRRIS sequence (S284)12. All three phosphorylation sites can be phosphorylated by Ca2+/calmodulin-dependent (CaM-II) kinases, including the endogenous CaM-II-like kinase that co-purifies with MyBPC43, 44. The first phosphate must be added by a CaM-II kinase to residue S284, to make the other phosphorylation sites accessible45, 46. Upon adrenergic stimulation, cAMP-dependent protein kinase can phosphorylate the other two sites (S275 and S304). Interestingly, in vivo, mono- and di-phosphorylated intermediates occur, which could result in more subtle changes in MyBPC's function46.
The Motif 1–2 linker region is believed to regulate muscle contraction by specifically binding to the subfragment-2 (S2) region of myosin4, 7, 47. Phosphorylation of cardiac MyBPC releases the S2 region, which is thought to allow the myosin crossbridges to reach out and interact more efficiently with actin, increasing force generation and systolic tension48, 49, 50. Flexibility of the myosin head, both as a whole, and within its two domains, is a necessary requirement for efficient force generation51, 52, 53. The extent of phosphorylation required to abolish the interaction with S2 remains unclear4.
Several FHC mutations are found in the Motif 1–2 phosphorylatable linker. Two of these mutations, in glycine residues G278E and G279A, occur within the conserved, cardiac-specific LAGGGRRIS sequence, in close proximity to two of the phosphorylation sites27. Glycine is unique in that it has no side chain and therefore can adopt phi and psi angles in all four quadrants of the Ramachandran plot54. If it is replaced with another residue it may permit a change in the three dimensional structure of the region and/or possibly obstruct access for the kinase. Notably, the residue at position 279 is an alanine (A) in the normal murine heart.
Another mutation in this region, R326Q, is more controversial. It is a highly conserved residue and is associated with incomplete penetrance and is associated with onset FHC42, 55, 56. However, it has also been found in healthy controls57, 58, suggesting that it may be a neutral polymorphism. These data suggest that any sarcomeric dysfunction associated with this mutation may be minimal.
Central region (Motifs 3–6)
Both IgI Motifs 3 and 4 have, as of yet, no experimentally defined roles, but may be required for the flexibility of the N-terminal region for its interactions with either the S2 region of myosin or the actin filament. Three FHC mutations have been identified in arginine residues in Motif 3 (R495Q, R502Q and R502W). Despite being highly conserved in MyBPC, they are not essential residues for defining the IgI fold and mutation results in a favourable disease prognosis26, 27, 55.
The first detailed structural study of an isolated domain of cardiac MyBPC, and two FHC related point mutations, was performed by NMR and circular dichroism on the Motif 5 domain59. In addition to possessing the key characteristics of the IgI set, the structure exhibited a novel feature. An additional ten amino acids beyond the predicted N-terminus of Motif 5 were required for the stability of the isolated domain, with these 10 residues forming an integral part of the isolated Motif 5 fold. It is not clear whether this unusual packing occurs in the full length MyBPC, or if these 10 residues actually form a short linker between domains 4 and 5, as the sequence alignments would predict. Electron micrographs of isolated cardiac MyBPC revealed over half of the protein molecules were V-shaped, with arms of 22±4.5 nm60, implying a point of flexibility occurs between Motifs 4 and 59. Electron micrographs of skeletal MyBPC also showed V-shaped molecules61. An atomic resolution structure of Motifs 4 and 5 together is needed to determine the true nature of this unique “linker”.
The second feature of the NMR structure of isolated Motif 5 is the 28 residue cardiac-specific insertion in the CD loop. This loop was unstructured, highly dynamic and pointing away from the domain's surface59. This proline/charge rich insert, while always present in the cardiac isoform, varies greatly in its sequence and length (Fig 5), and has been identified as a possible target binding region for an, as yet, unidentified ligand. One suggestion is that this insert forms an SH3 domain recognition sequence, perhaps binding the Calmodulin class -II (CaM-II) like kinase that co-purifies with cardiac MyBPC12, 43, 62. However, only the human cardiac isoform conforms to the PXXP–PXXP sequence usually required for SH3 target recognition. Additionally, the proposed target CaM-II kinase does not contain an identifiable SH3 domain63. Nevertheless, the Motif 5 insert could bind a different class of kinase.
Recently, a structural model of the MyBPC molecule has been developed, based on data from a yeast two-hybrid assay, using the Motif 5 sequence as “bait”. Motif 5 was chosen due to its unique cardiac insert and the identification of several FHC related mutations. In conjunction with deletion mapping studies, the yeast two hybrid assay surprisingly identified Motif 8 as a preferential binding partner in a screen of >7×106 clones9. Additional yeast two-hybrid assays revealed an interaction between Motifs 7 with 10. An interaction between Motifs 6 and 9 is also predicted to occur. As a result of these studies, a model of the myosin filament was developed, in which three MyBPC molecules formed a 'collar' around the myosin thick filament, stabilized by intermolecular interaction between Motifs 5–7 of each molecule and Motifs 8–10 of the next molecule, thus forming a staggered parallel arrangement (Fig 2)9.
To date, three FHC-associated mutations have been identified in Motif 5. The N755K point mutation in Motif 5 exhibits a severe phenotype and lies in the highly conserved position 1 of a Type 1 β-turn connecting the F and G strands of the IgI domain. It was predicted that this highly conserved β-turn is stabilized by hydrogen bonding between N755 and G758 at position 4 of this β-turn 64. Circular dichroism and NMR have confirmed that this N755K mutant is unstable and largely unfolded compared to the wild-type Motif 5, due to the loss of several key interactions59, 65. The NMR study suggested that tight packing by adjacent domains may partially stabilize the mutant Motif 5 in a folded conformation, to allow it to withstand modest mechanical stress. However, the severe phenotype of this mutation suggests significant stress within the sarcomere is likely to lead to partial or complete loss of structure and, thus, function. Furthermore, the N755K mutation leads to a weakened interaction with its binding partner Motif 8, where a 10 fold decrease in the affinity of Motif 5 for 8 was measured in the presence of the mutation9.
A second FHC mutation in Motif 5, R654H, exhibits a milder phenotype, and results in a smaller decrease in the binding affinity between Motif 5 and Motif 8 (2 fold decrease)9. The R654H mutation did not appear to affect the stability or overall fold of the module, in good agreement with the location on an exposed protein surface 56. Residue R654 is located on the CFGA' face of Motif 5. CFGA' together with the negatively charged cardiac-specific insert, results in a highly negative surface59. This surface on Motif 5 may be the binding site for the positively charged surface on Motif 8. Thus, the R654H mutation, although it does not affect the protein fold or stability, is likely to affect an interaction with Motif 8.
A third mutation in Motif 5, R668H, was recently found 42. This residue, located in strand B on the surface of Motif 5, is not a key residue for the IgI fold, although it is a highly conserved residue in MyBPC. Substitution of a polar positive histidine for a polar positive arginine would not be predicted to cause significant structural instability of the fold of the domain, but it may impact on Motif 5's ability to bind a ligand. However, this mutation is not located on the negatively charged surface proposed to be a target for Motif 8, and is thus unlikely to interfere with Motif 8 binding.
No specific functions or sarcomeric interactions have so far been assigned to the Motif 6 fibronectin domain. Based on its location between Motifs 5 and 7, the trimeric collar model of MyBPC9 suggests that Motif 6 may bind to Motif 9. This specific interaction has not been directly tested, although a construct of Motifs 5–7 binds to a construct of Motifs 8–10 with higher affinity than Motif 5 alone9. The demonstration of a specific interaction between Motifs 6 and 9 and a definition of their binding interface will be important for interpretation of FHC mutations in Motif 6.
FnIII modules Motifs 6 and 7 are reasonably homologous to similar FnIII domain pairs found in titin and fibronectin63, 64, 65, 66, 67, 68, 69. This homology includes a short linker region between the domains. FnIII domain pairs from titin have been shown to possess several highly conserved residues that are found in loops, in particular the BC loop. These residues point towards the module-module interface and interact via electrostatic charges37. The EF loop at the “bottom” of one domain is predicted to form a salt bridge and hydrogen bonds with the BC loop at the “top” of the following domain (Fig 7 and 8). Residue A833 is a mutational hot spot at the end of strand E in Motif 6 (Fig 8)27, 42. It is possible that mutations in this conserved amino acid may disrupt the bonds between the EF loop of Motif 6 and the BC loop of Motif 7, resulting in incorrect packing and assembly of MyBPC.

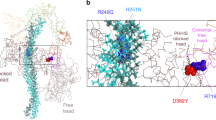
Solved structure of a FnIII pair from human fibronectin69. The homologous positions of FHC mutations found in Motifs 6 (left domain) and 7 (right domain) are superimposed as spheres. The sequential order of the strands is given by their labels: A, A', B, etc.
Another region of mutational activity reported in 3 patients occurs in strand C at the conserved residues R810 and K811 (Fig 8)27, 70. However, the reported mutations (R810H and K811R) do not alter the charge of the residue and are thus less likely to affect the stability of the FnIII fold. Of relevance is the clinical observation that these mutations appear to result in a mild phenotype, since one heterozygote had only moderate hypertrophy, and severe hypertrophy was only seen in a very unusual homozygote70.
The proposed mutation R820Q (Fig 8) in Motif 6 has led to some uncertainty concerning the wildtype amino acid sequence of human cardiac MyBPC. Numerous entries in the NCBI database for wild-type human cardiac MyBPC show a glutamine at position 820, compared to one entry submitted by Niimura et al. in 1997 (number AAC04620) which has an arginine. On the other hand, MyBPC sequence alignments show that all isoforms and species other than human cardiac have either an arginine or a lysine residue at position 820, suggesting that the glutamine in the wild-type human cardiac sequence may not be correct. The proposed mutation R820Q is located in strand C' in Motif 6 and is not predicted to be part of the Motif 6–7 interface. The associated FHC phenotype appears to be mild, usually with first presentation being late in life67, 68. The confusion in defining R820Q as a FHC mutation or a neutral polymorphism makes the prediction of possible structural defects difficult.
C-terminal region (Motifs 7–10)
Motifs 7 to 10 of the C-terminal region of MyBPC bind the backbone region of the myosin thick filament47, 72. The primary myosin and titin binding regions of MyBPC are localised to Motif 10 and Motifs 8-10, respectively 73, 74. These three C-terminal domains are the minimal requirement for incorporation into the A-band of the sarcomere6, with Motif 7 improving the targeting of MyBPC to the C-Zone75. The trimeric collar model9 also proposes specific interactions between C-terminal domains of adjacent MyBPC molecules.
A large majority of FHC mutations result in the premature termination of translation of the C-terminus of MyBPC, thus eliminating the titin and/or myosin binding sites26, 32, 64, 76, 77. This probably results in minimal or no incorporation of these truncated mutant proteins into the sarcomere32.
Three missense mutations are located in Motif 7 (Fig 8). The first, P873H60, changes a proline that is highly conserved across species and isoforms of MyBPC and is at the position that defines the N-terminal boundary in FnIII domains. The mutation P873H is unique, being found in only one patient with a mild phenotype. Modelling predicts that this mutation may affect the formation of strand A or the stability of the linker between Motifs 6 and 7.
It is unclear whether a second mutation, V896M (Fig 8)42, 78, located in strand B of Motif 7 is disease causing, disease modifying or represents a neutral polymorphism 27, 57. Patients identified with the V896M mutation in several studies were found to be double heterozygous, with these patients also possessing known FHC mutations in the myosin heavy chain gene27, 42. Furthermore, a clinically unaffected relation was found to be homozygous for the mutation. The V896M variant has also been found in control subjects57. Thus this mutation may not cause disease, but it may increase the severity of the disease in the presence of other FHC mutations. V896 is a conserved residue, predicted to point into the core of the FnIII domain. The replacement of the valine residue by the longer methionine may affect domain stability, but, as they are both non-polar residues, it is unlikely to result in incorrect folding.
The final mutation found in Motif 7 is N948T (Fig 8) 56. This mutation, found in only one patient, occurs in a conserved position and results in a severe disease phenotype. From homology modelling this mutation is expected to be located in a non-classical β-type turn connecting strands F and G37. This is in a similar position to the N755K mutation found in Motif 5, although the asparagine in Motif 5 forms part of a tight, type-1 β turn. Therefore, N948 is likely to be critical for domain folding and its loss could result in a poorly folded domain unable to help target MyBPC to the A band of the sarcomere. Additionally, the mutation may disrupt the interaction between Motif 7 and its proposed binding partner, Motif 10.
Only one FHC mutation has been identified in Motif 8. The mutation R1002Q58 is located in strand C, but, while it is conserved across species and isoforms of MyBPC, it is not a key IgI folding residue. Motif 8 is the potential binding partner of Motif 5 in the trimeric collar model, in which a negative face on Motif 5 is thought to bind to a positive region on Motif 89. This mutation results in the loss of a positively charged amino acid but this loss probably has little impact on the Motifs 5/8 interaction, consistent with the disease phenotype for this mutation being mild. The binding faces for this proposed interaction require elucidation before any definite conclusions can be made.
Motif 9 contains no known FHC mutations. The trimeric collar model predicts that it may interact with Motif 6, although this has not been directly tested9. Its contribution to myosin and titin binding has only been evaluated as part of the C-terminal 3 or 4 domains6, 75. Thus, the functional importance of Motif 9 remains unclear and may be clarified by future structural studies.
The binding of the C-terminal Motif 10 domain to the myosin filament has been extensively studied and nine key residues involved in binding to the filament have been identified by mutagenesis/sedimentation assays6, 73, 79. When these residues were positioned onto a model of cardiac Motif 10, the myosin binding faces were found to be located on two surfaces, the first formed by strands B and E and the second by strand C of the IgI fold. A unique insertion-mutation associated with FHC is a 6 amino acid duplication of residues 1248-125377. Functional and spectroscopic characterization of the isolated wildtype and the insertion-mutant of Motif 10, using a myosin binding sedimentation assay, circular dichroism spectroscopy and molecular modeling, revealed that the structure of Motif 10 is minimally perturbed and the mutation does not interfere with myosin binding80. Molecular modelling positioned the six amino acid duplication-mutation on the surface shown to not be involved in myosin binding80. In addition, the close location of the single FHC point mutation in a conserved hydrophobic residue, A1255T27, is also unlikely to interfere with myosin binding. These results suggest that both the unique 6 amino acid 1248-1253 duplication mutation and the A1255T mutation may affect some other function of Motif 10, possibly its binding to titin78 or an alteration in an interaction that may occur with Motif 7 or the adjacent Motif 9.
A second FHC point mutation in Motif 10, also resulting in a change from a hydrophobic alanine to a polar threonine at position 119427, is located at the end of strand A preceding the A-B loop. Modelling suggests that replacement of this residue with the more bulky threonine could alter the interaction with strand F. This residue is also located close to two identified key myosin-binding residues79. Therefore, this FHC mutation, unlike the others found in Motif 10, may disrupt binding to the myosin filament.
Models for the assembly of MyBP-C onto the thick filament
There are currently two models for the arrangement of MyBPC in the sarcomere. The trimeric collar model proposed by Moolman-Smook et al. (2002)9, has three staggered MyBPC molecules forming a ring around the thick filament. The collar is thought to be stabilized by specific interactions that have been demonstrated between Motifs 5 and 8, and Motifs 7 and 10 (Fig 2). Motifs 0 to 4, forming the N-terminal half of MyBP-C are predicted to have sufficient length to reach out from the thick filament and interact with the myosin crossbridge and/or the actin filament5, 10.
This model raises several issues. Firstly, the geometry of this model needs further verification. The diameter of the myosin thick filament is 13–15 nm82, resulting in a circumference of approximately 41–47 nm. The trimeric collar model proposes that the length of the ring that wraps around the myosin thick filament is 9 immunoglobulin domains. The longest diameter of these domains is 3.4–3.9 nm83, 84, resulting in the largest ring possible being 35–36 nm, 5–12 nm shorter than the measured circumference of the myosin thick filament. No current model of MyBPC function suggests that the C-terminal motifs are subjected to high degrees of stretch. Alternatively, the MyBPC linker regions between domains may provide additional length to the collar-forming C-terminal half of the model. However, the only linker of significant length in this half of MyBPC is located between Motifs 9 and 10, and stretching of this linker may result in mismatch between interacting domains in the trimeric collar model (Fig 2).
Secondly, how does the titin molecule fit into this model? Titin is the molecular ruler of the sarcomere85 and three strands are bound axially to the thick filament (Fig 2). The C-terminal Motifs 7–10 of MyBPC bind to both titin and the LMM region of the myosin thick filament. It is unclear how titin interacts with Motifs 7–10 in this model, given that the trimeric collar model would orient them as being close to mutually perpendicular74.
An alternative model of MyBPC has the C-terminal motifs running parallel to the myosin backbone (Fig 3)10. This more recent model was developed primarily to explain differences between the observed length of MyBPC in experimental X-ray diffraction patterns and the predicted spatial arrangement of MyBPC in the sarcomere. MyBPC has a longer periodicity than the myosin filament repeat 86, 87. This model accounts for this discrepancy by allowing the N-terminal domain of MyBPC to interact with neighbouring actin filaments in defined muscle states, thereby shortening the overall length of MyBPC running parallel with the myosin filament. Additionally, with the MyBPC molecules arranged axially they could more readily interact with titin, in contrast to the trimeric collar model. However, apart from the X-ray diffraction patterns, there is no direct experimental evidence that currently supports this model and no explanation for the experimental findings of Motif 5 binding to 8 and Motif 7 binding to 10. Clearly, more experimental data is required to clarify the manner in which MyBPC binds to the thick filament.
Conclusion
Analysing the effect of a mutant protein on muscle cell architecture, physiology, and biochemistry should provide an insight into the FHC disease process. A first step in this process is a careful examination of the sequence of MyBPC, with an extensive comparison of both the sequence and the predicted individual domain structures. The analysis presented here has clarified possible structural consequences of FHC mutations, laying a framework for the design of future structural and physiological studies to directly test these predictions. Most notably the predictions presented here emphasise the correlation between disease phenotype and the extent of conservation of the associated mutant residue/s.
References
Starr R, Offer G . Polypepetide chains of intermediate molecular weight in myosin preparations. FEBS Lett 1971; 15(1):40–4.
Craig R, Offer G . The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond [Biol] 1976; 192(1109):451–61.
Bennett P, Craig R, Starr R, Offer G . The ultrastructural location of C-protein, X-protein and H-protein in rabbit muscle. J Muscle Res Cell Motil 1986; 7(6):550–67.
Gruen M, Prinz H, Gautel M . cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett 1999; 453(3):254–9.
Witt CC, Gerull B, Davies MJ, Centner T, Linke WA, Thierfelder L . Hypercontractile properties of cardiac muscle fibers in a knock-in mouse model of cardiac myosin-binding protein-C. J Biol Chem 2001; 276(7):5353–9.
Gilbert R, Kelly MG, Mikawa T, Fischman DA . The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. J Cell Sci 1996; 109(Pt 1):101–11.
Gruen M, Gautel M . Mutations in β-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol 1999; 286(3):933–49.
Yamamoto K, Moos C . The C-proteins of rabbit red, white, and cardiac muscles. J Biol Chem 1983; 258(13):8395–401.
Moolman-Smook J, Flashman E, de Lange W, et al. Identification of novel interactions between domains of Myosin binding protein-C that are modulated by hypertrophic cardiomyopathy missense mutations. Circ Res 2002; 91(8):704–11.
Squire JM, Luther PK, Knupp C . Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J Mol Biol 2003; 331(3):713–24.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG . The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997; 25(24):4876–82.
Gautel M, Zuffardi O, Freiburg A, Labeit S . Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J 1995; 14(9):1952–60.
Aparicio S, Chapman J, Stupka E, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 2002; 297(5585):1301–10.
Offer G, Moos C, Starr R . A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification and characterization. J Mol Biol 1973; 74(4):653–76.
Saad AD, Obinata T, Fischman DA . Immunochemical analysis of protein isoforms in thick myofilaments of regenerating skeletal muscle. Dev Biol 1987; 119(2):336–49.
Harris SP, Bartley CR, Hacker TA, et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res 2002; 90(5):594–601.
Winegrad S . Cardiac myosin binding protein C. Circ Res 1999; 84(10):1117–26.
Hofmann PA, Hartzell HC, Moss RL . Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J Gen Physiol 1991; 97(6):1141–63.
Hofmann PA, Greaser ML, Moss RL . C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J Physiol 1991; 439:701–15.
Chung MW, Tsoutsman T, Semsarian C . Hypertrophic cardiomyopathy: from gene defect to clinical disease. Cell Res 2003; 13(1):9–20.
Maron BJ . Triggers for sudden cardiac death in the athlete. Cardiol Clin 1996; 14(2):195–210.
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE . Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92(4):785–9.
Miura K, Nakagawa H, Morikawa Y, et al. Epidemiology of idiopathic cardiomyopathy in Japan: results from a nationwide survey. Heart 2002; 87(2):126–30.
Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–64.
Carrier L, Hengstenberg C, Beckmann JS, et al. Mapping of a novel gene for familial hypertrophic cardiomyopathy to chromosome 11. Nat Genet 1993; 4(3):311–3.
Niimura H . Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998; 338(18):1248–57.
Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107(17):2227–32.
Charron P, Dubourg O, Desnos M, et al. Genotype-phenotype correlations in familial hypertrophic cardiomyopathy. A comparison between mutations in the cardiac protein-C and the β-myosin heavy chain genes. Eur Heart J 1998; 19(1):139–45.
Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J . A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest 1998; 102(7):1292–300.
McConnell BK, Jones KA, Fatkin D, et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest 1999; 104(9):1235–44.
Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J . In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res 1999; 85(9):841–7.
Rottbauer W, Gautel M, Zehelein J, et al. Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization Of cardiac transcript and protein. J Clin Invest 1997; 100(2):475–82.
Flavigny J, Souchet M, Sebillon P, et al. COOH-terminal truncated cardiac myosin-binding protein C mutants resulting from familial hypertrophic cardiomyopathy mutations exhibit altered expression and/or incorporation in fetal rat cardiomyocytes. J Mol Biol 1999; 294(2):443–56.
Harpaz Y, Chothia C . Many of the immunoglobulin superfamily domains in cell adhesion molecules and surface receptors belong to a new structural set which is close to that containing variable domains. J Mol Biol 1994; 238(4):528–39.
Politou AS, Gautel M, Joseph C, Pastore A . Immunoglobulin-type domains of titin are stabilized by amino-terminal extension. FEBS Lett 1994; 352(1):27–31.
Main AL, Harvey TS, Baron M, Boyd J, Campbell ID . The three-dimensional structure of the tenth type III module of fibronectin: an insight into RGD-mediated interactions. Cell 1992; 71(4):671–8.
Goll CM, Pastore A, Nilges M . The three-dimensional structure of a type I module from titin: a prototype of intracellular fibronectin type III domains. Structure 1998; 6(10):1291–302.
Bork P, Holm L, Sander C . The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol 1994; 242(4):309–20.
Steward A, Adhya S, Clarke J . Sequence conservation in Ig-like domains: the role of highly conserved proline residues in the fibronectin type III superfamily. J Mol Biol 2002; 318(4):935–40.
Kulikovskaya I, McClellan G, Flavigny J, Carrier L, Winegrad S . Effect of MyBP-C Binding to Actin on Contractility in Heart Muscle. J Gen Physiol 2003; 122(6):761–74.
Moos C, Mason CM, Besterman JM, Feng IN, Dubin JH . The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1. J Mol Biol 1978; 124(4):571–86.
Morner S, Richard P, Kazzam E, et al. Identification of the genotypes causing hypertrophic cardiomyopathy in northern Sweden. J Mol Cell Cardiol 2003; 35(7):841–9.
Hartzell HC, Glass DB . Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. J Biol Chem 1984; 259(24):15587–96.
Schlender KK, Bean LJ . Phosphorylation of chicken cardiac C-protein by calcium/calmodulin-dependent protein kinase II. J Biol Chem 1991; 266(5):2811–7.
Mohamed AS, Dignam JD, Schlender KK . Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys 1998; 358(2):313–9.
McClellan G, Kulikovskaya I, Winegrad S . Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys J 2001; 81(2):1083–92.
Starr R, Offer G . The interaction of C-protein with heavy meromyosin and subfragment-2. Biochem J 1978; 171(3):813–6
Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH . Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res 2000; 86(1):51–8.
Weisberg A, Winegrad S . Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc Natl Acad Sci USA 1996; 93(17):8999–9003.
Kulikovskaya I, McClellan G, Levine R, Winegrad S . Effect of extraction of myosin binding protein C on contractility of rat heart. Am J Physiol 2003; 285(2):H857–65.
Brown LJ, Klonis N, Sawyer WH, Fajer PG, Hambly BD . Independent movement of the regulatory and catalytic domains of myosin heads revealed by phosphorescence anisotropy. Biochemistry 2001; 40(28):8283–91
Baumann BA, Hambly BD, Hideg K, Fajer PG . The regulatory domain of the myosin head behaves as a rigid lever. Biochemistry 2001; 40(26):7868–73
Palm T, Sale K, Brown L, Li H, Hambly B, Fajer PG . Intradomain distances in regulatory domain of myosin head in prepower and postpower stroke states: fluorescence energy transfer. Biochemistry 1999; 38(40):13026–34
Hovmoller S, Zhou T, Ohlson T . Conformations of amino acids in proteins. Acta Crystallogr D Biol Crystallogr 2002; 58(Pt 5):768–76.
Maron BJ, Niimura H, Casey SA, et al. Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J Am Coll Cardiol 2001; 38(2):315–21.
Daehmlow S, Erdmann J, Knueppel T, et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun 2002; 298(1):116–20.
Jaaskelainen P, Kuusisto J, Miettinen R, et al. Mutations in the cardiac myosin-binding protein C gene are the predominant cause of familial hypertrophic cardiomyopathy in eastern Finland. J Mol Med 2002; 80(7):412–22.
Niimura H, Patton KK, McKenna WJ, et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002; 105(4):446–51.
Idowu SM, Gautel M, Perkins SJ, Pfuhl M . Structure, stability and dynamics of the central domain of cardiac myosin binding protein C (MyBP-C): implications for multidomain assembly and causes for cardiomyopathy. J Mol Biol 2003; 329(4):745–61.
Hartzell HC, Sale WS . Structure of C protein purified from cardiac muscle. J Cell Biol 1985; 100(1):208–15.
Swan RC, Fischman DA . Electron microscopy of C-protein molecules from chicken skeletal muscle. J Muscle Res Cell Motil 1986; 7(2):160–6.
Redwood CS, Moolman-Smook JC, Watkins H . Properties of mutant contractile proteins that cause hypertrophic cardiomyopathy. Cardiovasc Res 1999; 44(1):20–36.
Hudmon A, Schulman H . Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J 2002; 364(Pt 3):593–611.
Yu B, French JA, Carrier L, et al. Molecular pathology of familial hypertrophic cardiomyopathy caused by mutations in the cardiac myosin binding protein C gene. J Med Genet 1998; 35(3):205–10.
Singh L, Oakley C, Brown LJ, Hambly B . Structural effects of hypertrophic cardiomyopathy mutations on sarcomeric proteins. Biophys J 2003; 84:452.
Leahy DJ, Aukhil I, Erickson HP . 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 1996; 84(1):155–64.
de Pereda JM, Wiche G, Liddington RC . Crystal structure of a tandem pair of fibronectin type III domains from the cytoplasmic tail of integrin α6β4. EMBO J 1999; 18(15):4087–95.
Sharma A, Askari JA, Humphries MJ, Jones EY, Stuart DI . Crystal structure of a heparin- and integrin-binding segment of human fibronectin. EMBO J 1999; 18(6):1468–79.
Leahy DJ, Erickson HP, Aukhil I, Joshi P, Hendrickson WA . Crystallization of a fragment of human fibronectin: introduction of methionine by site-directed mutagenesis to allow phasing via selenomethionine. Proteins 1994; 19(1):48–54.
Nanni L, Pieroni M, Chimenti C, et al. Hypertrophic cardiomyopathy: two homozygous cases with 'typical' hypertrophic cardiomyopathy and three new mutations in cases with progression to dilated cardiomyopathy. Biochem Biophys Res Commun 2003; 309(2):391–8.
Konno T, Shimizu M, Ino H, et al. A novel missense mutation in the myosin binding protein-C gene is responsible for hypertrophic cardiomyopathy with left ventricular dysfunction and dilation in elderly patients. J Am Coll Cardiol 2003; 41(5):781–6.
Moos C, Offer G, Starr R, Bennett P . Interaction of C-protein with myosin, myosin rod and light meromyosin. J Mol Biol 1975; 97(1):1–9.
Okagaki T, Weber FE, Fischman DA, Vaughan KT, Mikawa T, Reinach FC . The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J Cell Biol 1993; 123(3):619–26.
Freiburg A, Gautel M . A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur J Biochem 1996; 235(1–2):317–23.
Gilbert R, Cohen JA, Pardo S, Basu A, Fischman DA . Identification of the A-band localization domain of myosin binding proteins C and H (MyBP-C, MyBP-H) in skeletal muscle. J Cell Sci 1999; 112(Pt 1):69–79.
Bonne G, Carrier L, Bercovici J, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet 1995; 11(4):438–40.
Watkins H, Conner D, Thierfelder L, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 1995; 11(4):434–7.
Moolman-Smook JC, De Lange WJ, Bruwer EC, Brink PA, Corfield VA . The origins of hypertrophic cardiomyopathy-causing mutations in two South African subpopulations: a unique profile of both independent and founder events. Am J Hum Genet 1999; 65(5):1308–20.
Miyamoto CA, Fischman DA, Reinach FC . The interface between MyBP-C and myosin: site-directed mutagenesis of the CX myosin-binding domain of MyBP-C. J Muscle Res Cell Motil 1999; 20(7):703–15.
Brown LJ, Singh L, Sale KL, et al. Functional and spectroscopic studies of a familial hypertrophic cardiomyopathy mutation in Motif X of cardiac myosin binding protein-C. Eur Biophys J 2002; 31(5):400–8.
Furst DO, Vinkemeier U, Weber K . Mammalian skeletal muscle C-protein: purification from bovine muscle, binding to titin and the characterization of a full-length human cDNA. J Cell Sci 1992; 102(Pt 4):769–78.
Squire J, Cantino M, Chew M, et al. Myosin rod-packing schemes in vertebrate muscle thick filaments. J Struct Biol 1998; 122(1–2):128–38.
Pfuhl M, Pastore A . Tertiary structure of an immunoglobulin-like domain from the giant muscle protein titin: a new member of the I set. Structure 1995; 3(4):391–401.
Improta S, Politou AS, Pastore A . Immunoglobulin-like modules from titin I-band: extensible components of muscle elasticity. Structure 1996; 4(3):323–37.
Whiting A, Wardale J, Trinick J . Does titin regulate the length of muscle thick filaments? J Mol Biol 1989; 205(1):263–8.
Huxley HE, Brown W . The low-angle x-ray diagram of vertebrate striated muscle and its behaviour during contraction and rigor. J Mol Biol 1967; 30(2):383–434.
Sjostrom M, Squire JM . Cryo-ultramicrotomy and myofibrillar fine structure: a review. J Microsc 1977; 111(3):239–78.
Andersen PS, Havndrup O, Bundgaard H, et al. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations. J Med Genet 2001; 38(12):E43.
Moolman-Smook JC, Mayosi B, Brink P, Corfield VA . Identification of a new missense mutation in MyBP-C associated with hypertrophic cardiomyopathy. J Med Genet 1998; 35(3):253–4.
Acknowledgements
CEO is a recipient of an Australian Postgraduate Award. This work was supported by a National Heart Foundation of Australia Grant-in-Aid.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
OAKLEY, C., HAMBLY, B., CURMI, P. et al. Myosin binding protein C: Structural abnormalities in familial hypertrophic cardiomyopathy. Cell Res 14, 95–110 (2004). https://doi.org/10.1038/sj.cr.7290208
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290208
Keywords
This article is cited by
-
Genetic, clinical, molecular, and pathogenic aspects of the South Asian–specific polymorphic MYBPC3Δ25bp variant
Biophysical Reviews (2020)
-
The effect of disease on human cardiac protein expression profiles in paired samples from right and left ventricles
Clinical Proteomics (2014)
-
Cardiac myosin binding protein-C as a central target of cardiac sarcomere signaling: a special mini review series
Pflügers Archiv - European Journal of Physiology (2014)
-
Myosin binding protein-C: an essential protein in skeletal and cardiac muscle
Journal of Muscle Research and Cell Motility (2011)
