
Abstract
Although we previously reported that the self-renewal of leukemia-initiating cells of B-lineage acute lymphoblastic leukemia (B-ALL LICs) was regulated by β-Arrestin1, a multiple-function protein, the cellular senescence is critical for LICs fate and leukemia progress, and worthy for further investigation. Here we found that depletion of β-Arrestin1 extended the population doubling time and the percentage of senile cells, the signatures of cellular senescence, of B-ALL LICs. Moreover, lack of β-Arrestin1 enhanced the expression of proteins (CBX, HIRA) and genes (P53, P16) related to senescence in leukemic Reh cells and B-ALL-LICs-derived leukemic mice. Further results showed that loss of β-Arrestin1 induced senescence of Reh cells through mediating hTERT-telomerase-telomere axis, which was reversed by BIBR1532, the telomerase activity inhibitor. Importantly, depletion of β-Arrestin1 decreased the binding of Sp1 to hTERT promoter at the region of −28 to −36 bp. The anti-sense oligonucleotide of this key region downregulated the transcription of hTERT and aggravated the senescence of Reh cells. Further data demonstrated that the depleted β-Arrestin1 reduced the interaction of P300 with Sp1, thus to reduce Sp1 binding to hTERT promoter, downregulate hTERT transcription, decrease telomerase activity, shorten telomere length, and promote Reh cell senescence. Interestingly, the percentage of senile cells in B-ALL LICs was decreased, which was negatively correlated to good prognosis and β-Arrestin1 mRNA expression in childhood B-ALL patients. Our study shed a light on the senescence of B-ALL LICs and is regulated by β-Arrestin1, providing the potential therapeutic target of leukemia by promoting cellular senescence with a key region of hTERT promoter.
Similar content being viewed by others


Main
Acute lymphoblastic leukemia (ALL) is the most common tumor in children under age 15. According to the affected cells, ALL is divided into B-lineage acute lymphoblastic leukemia (B-ALL) and T-lineage acute lymphoblastic leukemia (T-ALL). The long-term rates of event-free survival (EFS) for childhood B-ALL have approached close to 90%, from <10% in the 1960s, in developed countries.1, 2 However, about 10–15% of relapse and refractory B-ALL patients have still lower overall survival (OS) and EFS rates.2 The exact mechanism of relapse and refractory B-ALL is unclear. In recent years, leukemia-initiating cells (LICs), the cell population with the self-renewal capacity to initiate and maintain leukemia, have been found pivotal in relapse and drug resistance for B-ALL because of the properties LICs that share with normal hematopoietic stem cells (HSCs) such as the immunophenotyping (CD34+CD38−CD19+) and maintenance of a quiescent state that makes the cells unresponsive to cell cycle-specific cytotoxic agents.3 Besides the self-renewal ability of LICs, the cellular senescence of LICs is a critical factor for the leukemia progression,4 and aroused great concerns in researchers.
The cellular senescence means a terminal growth arrest, which includes premature senescence and replicative senescence. Premature senescence, mainly induced by stress, oncogenes, and tumor suppressors,5 has been increasingly demonstrated to be critical for the development of several kinds of leukemia.6 Replicative senescence is also called telomere-induced senescence, primarily due to shortened telomere, and the senescence is present in Ph+ CML7 and chronic lymphocytic leukemia (CLL).8 Most of the human cancers have acquired mechanisms to maintain telomeres, generally through high expression of telomerase. Telomere-induced senescence also has been shown to act as a tumor suppressor in telomerase-deficient mice.9 Therefore, telomere and telomerase are keys for cellular senescence and tumorigenesis.
Human telomerase reverse transcriptase (hTERT) is one of three telomerase main components, together with the human telomerase RNA molecules (hTR) and telomerase-associated proteins (TAP), which determines the rate of telomerase activity and expresses in most malignant tumors but not in normal tissues.10, 11 High hTERT expression was observed in some subtypes of leukemia like CLL and T-ALL.12, 13 The expression of hTERT gene is governed by its transcription through its promoter, and the transcription factor is the main regulatory aspect.14, 15 Some transcription factor-binding sites are in the region of the hTERT promoter, including Sp1, c-Myc, USF, and so on.14, 15 The Sp1 composite element centered from −1 to −110bp and with five binding sites in the proximal of hTERT promoter is particularly crucial for basal hTERT expression.14 Sp1 was identified as an activator for hTERT transcription in some tumors, including those of primary effusion lymphoma,16 prostate cancer17 and even Jurkat T cells.18 Sp1 could combine with factors like c-Myc,14 Sp3 (ref.18 to promote hTERT transcription, which also needs a permissive chromatin environment.19 For example, P300, a histone acetyltransferase, could not only bind with Sp1 (ref.20 but also be involved in the chromatin remodeling.21 Whether Sp1 binding with P300 mediates hTERT transcription and the hTERT-telomerase-telomere axis in B-ALL replicative senescence needs exploration.
β-Arrestin1, the important scaffold protein of β-Arrestin1 family, is ubiquitously distributed and of more concern regarding cancer progression, which transduce signals through and regulate the PI3K/AKT, Wnt, and Hedgehog signaling pathways to mediate cell development and differentiation, associated with the progression of malignancies.22 Both β-Arrestin1 and β-Arrestin2 could mediate the initiation and maintenance of myeloid leukemia.23, 24 In particular, β-Arrestin1 could regulate histone proteins' modification and gene transcription by coupling with CREB and YY1 to further regulate cell function.23, 24 Our previous studies showed that overexpression of β-Arrestin1 was associated with a high risk of pediatric B-ALL and promoted the self-renewal of B-ALL LICs.25, 26 Given that the cellular senescence of LICs is essential for B-ALL progress, we are interested to further explore the critical role of β-Arrestin1 in the cellular senescence of LICs and B-ALL progress.
Our data revealed that depletion of β-Arrestin1 facilitated cell senescence of B-ALL LICs in vivo and in vitro, by regulating hTERT transcription through inducing P300-Sp1 interaction at −28 to −36 bp of hTERT promoter, which was further illustrated by the data from clinical samples that decreased senile cells and elevated expression of β-Arrestin1 predicted poor prognosis in B-ALL, providing the potential therapeutic target of leukemia by promoting cellular senescence.
Results
Loss of β-Arrestin1 accelerated senescence in B-ALL LICs
On the basis of our previous report that β-Arrestin1 regulated the self-renewal of B-ALL LICs5 and the cellular senescence is another critical factor for LICs and leukemia progress;4 here we further investigate whether the cellular senescence of B-ALL LICs was mediated by β-Arrestin1. Beside isolation and identification of LICs from B-ALL as our latest report,26 we screened β-Arrestin1 expression and senescence status in different B-ALL cells to obtain the qualified cell models for B-ALL LICs. The results showed that only progenitor Reh cells had the similar pattern of cellular senescence and β-Arrestin1 expression with that of the LICs from B-ALL patients. We adopted Reh cells as the B-ALL LICs model for further exploration, and then infected Reh cells and leukemic mice injected the LICs with Si-β-Arrestin1 (Siβ1) lentivirus particles,26 named Reh-Siβ1 or Mice-Siβ1 (Supplementary Figure S2). The expression of β-Arrestin1 was then detected in Reh-Siβ1, Reh-Scram (Supplementary Figure S6D), Mice-Scram and Mice-Siβ1 (Supplementary Figure S2D) by western blot. The impaired expression of β-Arrestin1 extended the survival time of leukemic mice by Kaplan–Meier analysis (Supplementary Figure S2F). Reh-Siβ1 cells had lower population doubling times than that of Reh-Scram control cells through serial subculture (Figure 1a). Moreover, more senile cells were observed in Reh-Siβ1 cells and BM cells from Mice-Siβ1 (Figure 1b). The protein markers for senescence,5 CBX and HIRA, were then measured by western blot, and the results demonstrated that both the expression of CBX and HIRA were higher in Reh-Siβ1 cells and BM cells from Mice-Siβ1 (Figure 1c). Further, the expression of senescence-associated genes, including P16, P53, P21, and P27 were detected by real-time RT-PCR, and the data showed that the expression of these genes was higher in Reh-Siβ1 cells and BM cells from Mice-Siβ1 (Figure 1d) than those from Reh-Scram cells and BM cells from Mice-Scram, respectively. Taken together, those observations suggest that the cellular senescence of B-ALL LICs is promoted by Si-β-Arrestin1.

Loss of β-Arrestin1 promotes the senescence in B-ALL LICs. Reh cells and isolated CD34+CD38−CD19+ cells (LICs) were transiently infected with Si-β-Arrestin1 (Siβ1) or scrambled shRNA vector (Scram) lentivirus particles, named R-Siβ1, R-Scram, LICs-Siβ1, and LICs-Scram cells. 1 × 104 LICs-Siβ1 or LICs-Scram cells were injected intravenously into irradiated NSG mice (n=10), named M-Siβ1 and M-Scram mice. The mice were dead or killed 96 days after injecting cells with obvious leukemia symptoms. BM cells, PB cells, and tissues were collected. (a) R-Siβ1, R-Scram and Reh were serially subcultured in an RPMI1640 medium, and the times of doubling populations were compared. The difference between R-Siβ1 and R-Scram was performed. (b) The representative images of SA-β-gal staining (top) and the statistical data (bottom) were shown in vitro and in vivo. (c)Total protein was purified from R-Siβ1, R-Scram, and PB cells of M-Siβ1 or M-Scram mice. The expression of HIRA and CBX, the marker for senescence, was detected by Western Blot. Representative western blot images (top) and the levels of HIRA and CBX were quantified relative to their respective GAPDH level (bottom). (d) RNA was extracted from R-Siβ1, R-Scram, and PB cells of M-Siβ1 or M-Scram mice. The expression of P53, P16, P21, and P27, which is related with senescence, was detected by real-time PCR. The levels of genes were quantified relative to their respective GAPDH level. Differences among groups were calculated by t-test. *P<0.05, **P<0.001; GAP, inner control GAPDH gene; M, NSG mice; n, number; OS, overall survival; R, Reh cells;. Scale bar, 5 μm
Loss of β-Arrestin1 accelerated senescence via telomerase and telomere
To elucidate the mechanism of β-Arrestin1 regulating the senescence of B-ALL LICs, we analyzed the reactive oxygen species (ROS) for premature senescence and the telomere length for replicative senescence. The ROS level was labeled with DCFH and observed by fluorescence spectroscopy in Reh-Siβ1 cells, Reh-Scram cells, and BM cells from Mice-Siβ1 or Mice-Scram, but there was no significant difference among these groups (Supplementary Figure S4). Hence, we focused on the telomerase and telomere.
To further verify the role of β-Arrestin1 on telomere and telomerase in B-ALL LICs, we measured the telomere length, telomerase activity, and hTERT expression in Reh-Siβ1, Mice-Siβ1, and their control groups. The shortened telomere length, decreased telomerase activity, and reduced hTERT mRNA expression were observed in Reh-Siβ1 (Figures 2a, c, e, and f) and Mice-Siβ1 (Supplementary Figures S7A, S7C, S7E, and S7F) correspondingly. Moreover, BIBR1532, the inhibitor of telomerase activity, was applied to explore the specificity of telomere and telomerase in the process of β-Arrestin1 regulating the senescence of LICs, after successfully screening its best dosage (3 μmol/l), time (48 h) (Supplementary Figure S5A) and hTERT mRNA expression in Reh cells (Supplementary Figure S5B), and hTERT mRNA expression in Reh-Siβ1 and Reh-Scram (Supplementary Figure S5C). The results showed that BIBR1532 could inhibit hTERT mRNA expression and telomerase activity, with weak inhibition of telomere length (Figure 2 and Supplementary Figure S7). These results together demonstrated that β-Arrestin1 might regulate the senescence of B-ALL LICs through hTERT-telomerase-telomere axis.

Loss of β-Arrestin1 inhibits hTERT expression, decreases telomerase activity, and shortens telomere length in B-ALL LICs. DNA was extracted from R-Siβ1 cells, R-Scram cells. The length of telomere was detected by Southern blot. Calculating telomere length was performed with optical density analysis, according to the kit’s protocol. The average length of telomere (TRF) in different groups was calculated and compared with the control group. The representative images of the Southern blot (a) and the TRF (b) in Reh cells. The length of telomere was also detected by FISH in smear. The representative images of FISH (c); After calculating the fluorescence intensity of telomeres, the value was quantified relative to the level of Scram group without BIBR1532 and are presented as the relative intensity of fluorescence (bottom) in Reh cells (d). The activity of telomerase was measured by PCR-ELISA in Reh cells (e). The hTERT mRNA expression fold was detected by RT-PCR in Reh cells (f). All P-values were calculated by t-test. *P<0.05, **P<0.001; B, the inhibitor of telomerase (BIBR1532); M, marker; P, positive control; TE, the activity of telomerase; TRF, the average length of telomere. Scale bar, 10 μm
Loss of β-Arrestin1 reduced hTERT transcription by binding with Sp1
In the light of the activity of telomerase, which is positively correlated with the length of the telomere, is known to be rate-limiting regulated by hTERT gene.11 Sp1 is a transcription activator to stimulate hTERT transcription and there are five Sp1-binding sites in the proximal region of the hTERT promoter14 (Supplementary Figure S6A). Considering that the hTERT expression is governed by its transcription through its promoter, we illustrated that β-Arrestin1 didn’t affect the expression of Sp1 protein in nuclear (Supplementary Figure S6B), but β-Arrestin1 increased the binding of Sp1 to hTERT promoter by electrophoretic mobility shift assay (EMSA) in Reh cells (Figure 3a). Then, we cloned different binding fragments for Sp1 in hTERT promoter (Supplementary Figure S6C) into the PGL-3 basic reporter vector to find the most important site for hTERT transcription, and then transfected different fragments of hTERT reporter vectors with various Sp1-binding sites into Reh-Siβ1 and Reh-Scram cells. The results from luciferase reporter assay showed that luciferase was less active in Reh-Siβ1 cells than that in Reh-Scram cells, and the highest activity was detected in Reh cells transfected with the second binding fragment for Sp1 (Sp12) (− 28 to −36 bp) (Figure 3b).

Loss of β-Arrestin1 reduces the transcription of hTERT. (a) Different Reh cells were collected. We performed supershift by EMSA on purified nucleus protein to detect Sp1 binding with the hTERT promoter. (b) Reh cells were transfected by different plasmids with various cloned fragments. The luciferase activity was detected by dual-luciferase reporter assay and normalized by Renilla luciferase. The relative luciferase activity was viewed as 1.0 in Reh with a PGL-3-basic vector. (c) The anti-sense oligonucleotide was designed for the binding sites (−28 to −36 bp) of the hTERT promoter, measuring the relative luciferase activity, and comparing between Reh with or without the anti-sense oligonucleotide. R-Siβ1 cells and R-Scram cells were transfected by anti-sense oligonucleotide of Sp12 (−28 to −36 bp). The cells' total DNA and protein were collected. Then we detected the binding of the hTERT promoter with Sp1 by EMSA (d) measured the relative hTERT mRNA fold by RT-PCR (e) detected the activity of telomerase by PCR-ELISA (f) measured the length of telomere by Southern blot (g) and FISH (h), respectively, and obtained representative images through SA-β-gal staining (i). Anti-Sp1, antibody for Sp1; AS, anti-sense oligonucleotide. AS, anti-sense oligonucleotide of Sp12 (−28 to −36 bp); Ctrl, Reh cells not transfected with anti-sense oligonucleotide of Sp12 (−28 to −36 bp); E-B, E-box region; NSP, the fragment excluding the binding sites for Sp1; P-C, positive control; TE, the activity of telomerase; TRF, the average length of telomere. Scale bar, 10 μm
To study the specificity of binding sites Sp12 in affecting the transcription of the hTERT gene, we designed anti-sense oligo-DNA fragments for Sp12 (AS-Sp12) and transfected it into Reh cells. The data showed less luciferase activity in Reh cells transfected with AS-Sp12 than in those without transfection (Figure 3c). Subsequently, in Reh cells transfected with AS-Sp12, the binding of Sp1 to hTERT promoter was decreased, still measured by EMSA (Figure 3d). In addition, the reduced hTERT expression (Figure 3e), the declined telomerase activity (Figure 3f), and the shortened telomere length were consistently observed in Reh cells transfected with AS-Sp12 as well (Figures 3g and h, the statistical graphs were in Supplementary Figures S8A and S8B, respectively). Moreover, there were more senile cells than those in Reh cells without AS-Sp12 transfection (Figure 3i, statistical graphs were in Supplementary Figure S8C). These data indicated that the region (−28bp to −36bp) in the hTERT promoter was the core region for Sp1 binding, which further stimulated hTERT transcription and senescence regulated by β-Arrestin1 in B-ALL Reh cells.
Loss of β-Arrestin1 reduced Sp1 interacting with P300 in hTERT promoter
To further find how β-Arrestin1 regulates Sp1 to bind with hTERT promoter, we first speculated that Sp1 is regulated by β-Arrestin1 directly. However, neither the expression of Sp1 (Figure 4a) by western blot nor the binding of β-Arrestin1 with Sp1 (Figure 4b) by Co-IP was altered in Reh cells loss of β-Arrestin1.

Loss of β-Arrestin1 decreases the formation of P300-Sp1 complex. Protein was extracted from R-Scram cells and R-Siβ1 cells, and smears were prepared. (a) Representative western blot images of Sp1 protein (top) and the levels of Sp1 protein were quantified relative to those of β-actin and are presented as folds compared with the control cells (bottom). (b) The colocalization of β-Arrestin1 and Sp1 by Co-IP. (c) Representative western blot images of P300 protein (top) and the levels of P300 protein were quantified relative to those of β-actin and are presented as folds compared with the control cells (bottom). (d) The colocalization of β-Arrestin1 and P300 by Co-IP. (e) The binding of β-Arrestin1 and P300 by immunofluorescence and confocal microscopy. (f) The colocalization of P300 and Sp1 by Co-IP. (g) The binding of P300 and Sp1 by immunofluorescence and confocal microscopy. Scale bar, 5 μm
We then assumed that β-Arrestin1 could help form a protein–protein complex to bind with hTERT promoter. In view of Sp1 is reported to work with P300 in activating gene transcription,20 we further analyzed the role of P300 in this process. Our data showed that Reh cells with β-Arrestin1-Arrestin1 depletion were not the reduced expression of total P300 protein (Figure 4c), but the loss of β-Arrestin1 reduced the interacting of β-Arrestin1 with P300 as shown by Co-IP (Figure 4d) and immunofluorescence confocal assay (Figure 4e), and the binding of Sp1 with P300 as shown by Co-IP (Figure 4f) and immunofluorescence confocal assay (Figure 4g). These findings suggest that P300, as the important mediator, could promote the formation of β-Arrestin1-P300-Sp1 complex, and that the depletion of β-Arrestin1 might decrease this complex formation.
To further testify the hypothesis, we chemically synthesized small RNA interference strands for P300 (sh-P300). Four sh-P300s targeting different fragments of the P300 gene were designed, named sh477-1, sh478-1, sh479-1, and sh480-1 (sequences of sh-P300s were listed in Supplementary Table S2). The expression of P300 was detected in Reh cells transfected with these fragments, and the sh479-1 fragment with the best inhibition was used for further research (data not shown). We found that the depletion of P300 reduced the binding of P300 with Sp1 (Figure 5a) and the binding of P300-Sp1 complex to hTERT promoter (Figure 5b). Then, the decreased hTERT transcription (Figure 5c) and the declined telomerase activity (Figure 5d), the shortened telomere length (Figure 5e), and the enhanced percentage of senile cells (Figure 5f) were observed in Reh cells knocked-down P300. Altogether, these results show that β-Arrestin1 increases the P300-Sp1 complex and the binding of P300-Sp1 to hTERT promoter, upregulates hTERT transcription, increases telomerase activity, and extends telomere length, thus, postponing cell senescence and changing the cell fate of B-ALL LICs (Figure 5g).

P300 is necessary for β-Arrestin1 regulating the senescence via the hTERT-telomerase-telomere axis in B-ALL. (a) Reh cells were transfected with shRNA-P300, and collected the cells, and then measured the binding of P300 with Sp1 by Co-IP. Then, R-Scram cells and R-Siβ1 cells were transfected with shRNA-p300. The binding of the hTERT promoter with Sp1 by EMSA (b), the relative hTERT mRNA fold by RT-PCR (c), the activity of telomerase by PCR-ELISA (d), the length of telomere by FISH (e), and the senescence by SA-β-gal staining (f) were shown. The scheme of this study, which showed that β-Arrestin1 enhances the P300-Sp1 complex and the binding of the complex to the hTERT promoter, thus regulating the senescence of B-ALL via the hTERT-telomerase-telomere axis (g). β-Arr 1, the gene of β-arrestin1; Ctrl, transfected without oligonucleotide shRNA-P300; sh-P300, transfected with oligonucleotide shRNA-P300; TE, telomerase activity; TL, telomere length. Scale bar, 5 μm
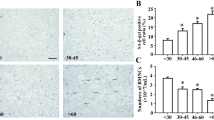
The percentage of senile cells was negatively correlated with β-Arrestin1 expression and poor prognosis in LICs from B-ALL patients
To further analyze the clinical significance of the cellular senescence of B-ALL LICs, we collected bone marrow from B-ALL patients, to isolate LICs to investigate the percentage of senile cells and the expression of β-Arrestin1 simultaneously, and further to investigate the survival time of these patients. The data showed that the ratio of senile cells is less (9 versus 15% in BM; 22 versus 30% in PB) (Figures 6a and b) through SA-β-gal staining and the population doubling time was shortened through serial subculture in B-ALL compared to their respective controls (Figure 6c). Moreover, the expression of senescence-related genes was lower in B-ALL patients than those from controls, especially the expression of P53 and P16 (Figure 6d).

Less senile cells are correlated with high expression of β-Arrestin1 in B-ALL. BM cells and PB cells were collected from 20 B-ALL patients (P-AL from PB cells, B-AL from BM cells) and 20 non-leukemic patients as a control (P-C from PB cells, B-C from BM cells). BM smears were prepared. RNA and protein were purified from PB cells. The β-Arrestin1, P53, P16, P21 and P27 mRNA were detected by real-time RT-PCR, and the data are presented as folds using GAPDH as the reference gene, and the relative β-Arrestin1/GAPDH expression in the P-C was viewed as 1.0-fold. (a) PB cells and BM cells were stained by SA-β-gal, the stained cells were prepared on smear. The representative images were shown. Both P-C and B-C were from patients #4, #5, #6; both P-AL and B-AL were from person No. 1, No. 2, No. 3. (b) The statistical graphs for SA-β-gal staining of all specimens. The data are represented as the means±S.E.M and P-value is calculated by t-test. (c) B-C and B-AL cells were serially cultured in an RPMI1640 medium, and the times of doubling populations were compared by t-test. (d) The expression of P53, P21, P16 and P27 mRNA were detected by real-time RT-PCR. The statistics for all specimens were shown with the means±SEM and P-value is calculated by t-test. (e) Overall survival of B-ALL based on the K value (k value=the percentage of senile cells in P-AL cells subtract the average percentage of senile cells in P-C cells), was analyzed by the Kaplan–Meier method, with K=−5% as the cutoff value in P-AL cells. (f) The correlation analysis of the percentage of senile cells with mRNA folds of β-Arrestin1 gene. (g) Representative western blot images of β-Arrestin1 (top) and the relative mRNA expression of β-Arrestin1 (bottom) in patients. (h) Representative immunofluorescence images of β-Arrestin1 expression in patients. (i) the overall survival of B-ALL based on β-Arrestin1 mRNA expression was analyzed by Kaplan–Meier method, with 2.5-fold β-Arrestin1 mRNA expression as the cutoff value. *P<0.05, **P<0.001; P-C, PB cells in non-leukemic patients; P-AL, PB cells in B-ALL patients; B-C, BM cells in non-leukemic patients; B-AL, BM cells in B-ALL patients; β-Arr 1, the gene of β-arrestin1; GAP, inner control GAPDH gene; n, the number of patients; OS, overall survival; Scale bar, 5 μm
Overall survival (OS) of B-ALL based on the K-value (K=percentage of senile cells in PB-ALL (P-AL) cells subtract the average percentage of senile cells in PB-Control (P-C) cells), was analyzed by the Kaplan–Meier method. K<−5% could distinguish B-ALL patients' with a poor prognosis from those with a good prognosis (Figure 6e), suggesting that a low percentage of senile cells in PB cells were positively associated with an unfavorable prognosis in B-ALL.
On the basis of our previous studies,25, 26 here we further investigated whether β-Arrestin1 affected the OS of leukemia patients with detailed listed information (Supplementary Table S1). Enhanced expression of β-Arrestin1 was observed (Figures 6g and h) and the median value of β-Arrestin1 mRNA was about 2.2-fold in B-ALL patients compared with corresponding controls. We then chose 2.5-fold as a cutoff value for the survival analysis. Leukemic patients (Figure 6i) with ≥2.5-fold of β-Arrestin1 mRNA expression level had poor prognosis (patients: 2.8 years versus 3.9 years), indicating that a high expression of β-Arrestin1 is positively correlated with an unfavorable prognosis for B-ALL patients. Interestingly, we found that the mRNA expression of β-Arrestin1 was negatively correlated with senescence in B-ALL patients (P<0.001, R2=0.55) (Figure 6f).
In order to further investigate the relationship of senescence with β-Arrestin1 on LICs, we isolated different fractions of LICs from BM cells of 20 newly diagnosed pediatric B-ALL patients, using MACS and identified them using FACS (Supplementary Figure S1A). The least senile cells were observed on the LICs (Supplementary Figures S1B and S1C), and the highest expression of β-Arrestin1-Arrestin1 was also demonstrated on the LICs, as shown in our latest report.26 All four fractions showed that a smaller percentage of senile cells was correlated with higher expression of β-Arrestin1, but significant correlation was observed only in LICs (P<0.001, R2=0.91) (Supplementary Figures S1D–S1G).
After isolation and identification, the LICs and other fractions as control from newly diagnosed pediatric B-ALL patients were injected into mice, respectively, we then collected PB and BM specimens after identifying leukemia (Supplementary Figure S2), and found that less senile cells (Supplementary Figure S3D) and higher expression of β-Arrestin1 were shown in leukemic mice derived from LICs fraction (Supplementary Figures S3A and S3B). To explore the relationship of survival with senescence and the expression of β-Arrestin1 in leukemic mice, we got the similar survival results with leukemic patients (Supplementary Figures S3E and S3C). The mRNA expression of β-Arrestin1 was also negatively correlated with senescence in the leukemic mice (P<0.001, R2=0.67) (Supplementary Figure S3F). Altogether, our data indicate that the role of LICs’ senescence of related to β-Arrestin1 on predictive ability for B-ALL.
Discussion
Both cellular senescence and β-Arrestin1 play critical roles in the survival of pediatric B-ALL patients. Here we uncovered that depletion of β-Arrestin1 enhanced cell senescence of B-ALL LICs in vivo and in vitro by regulating the hTERT-telomerase-telomere axis through inducing P300-Sp1 interaction at the −28 to −36 bp region of hTERT promoter, and decreased senile cells and elevated expression of β-Arrestin1 predicted poor prognosis in B-ALL. Collectively, our data shed light on a previously unexpected role of β-Arrestin1 in the cellular senescence of B-ALL LICs, and showed that β-Arrestin1, this protein working with P300-Sp1 is a key regulator for hTERT transcription. Combining with our previous reports of the role of β-Arrestin1 on the self-renewal5 and proliferation4 of LICs, here we extend the critical role of β-Arrestin1 on cellular senescence of LICs.
The conventional idea is that tumor cells lost the ability to senescence. Thus, an increasing number of studies clarified that inducible senescence is one of the mechanisms for chemical drug,7 and is even responsible for prognosis in leukemia and some solid tumors.27 Recently, Collado proposed that tumors might still undergo senescence.28 In our study, the senescence was present in leukemic patients and mice, but the percentage of senile cells was less than those in non-leukemic patients and mice, respectively. And our data first proved that the percentage of senile cells may be a novel factor for B-ALL prognosis. The reasons for this are as follows: On one hand, not only senescence could cause tumor cells to be cleared by immune cells, possibly, resulting in efficient tumor regression,29 but also senescence could reflect the limit of stemness.27 On the other hand, both tumor regression and limited stemness are markers for excellent prognosis of patients with tumors.30, 31 Therefore, we deduce that cell senescence is critical for B-ALL prognosis.
We detected the ROS level for stress-induced senescence, hypothesizing that there is a difference between a patient with leukemia and a healthy person, but the effect of β-Arrestin1 on ROS was rare in leukemia. Moreover, the senescence-related oncogenes or tumor suppressors, including P16, P53, P21, and P27 were investigated, and the expression of P16 was significantly different. P16 is involved in premature senescence as a tumor suppressor,32 which is also involved in replicative senescence by the P16-RB pathway.33 Thus, we explored the role of β-Arrestin1 on telomere-related replicative senescence, and found that β-Arrestin1 regulated the senescence via the hTERT-telomerase-telomere axis. However, the clear role of P16 gene on this process still need further study.
Abundant evidence has shown that the length of the telomere depended on the activity of telomerase. Regulation of telomerase is multifactorial involving hTERT expression, post-translational protein–protein interactions, and protein phosphorylation.34 In our study, we found that the alteration of hTERT mRNA expression was positively correlated with the alteration of telomerase activity in the Reh cells' presence or absence of β-Arrestin1, so we concentrated on studying the expression of the hTERT gene. The effect of other factors on telomerase needs further investigation.
Many ways for regulating hTERT expression, including miRNA,35 tumor suppressor or oncogene pathways,36 epigenetic mechanisms,37 and transcriptional regulation.38 β-Arrestin1, as a possible oncogene in B-ALL, could be involved in these pathways to regulate the epigenetic and gene transcription. Thus, based on transcriptional regulation of the hTERT gene being the major mechanism for cancer-specific activation of telomerase, an activator of transcription was applied to study the possible mechanism for β-Arrestin1 regulating hTERT expression. We cannot rule out other possible factors and potential mechanisms, especially miRNA and other epigenetic influence for hTERT expression.
A number of factors have been directly or indirectly identified as regulating the hTERT promoter, including cellular transcriptional activators (c-Myc, Sp1, HIF-1, AP2, ER, Ets, etc.) as well as repressors.14, 39 Recent research showed, however, that no reasons could clearly account for the cancer specificity of hTERT expression; the chromatin structure has been suggested as an important factor for the binding of transcriptional activators with the hTERT promoter.37 It has been reported that P300, histone acetyltransferase, mediated by β-Arrestin1 (ref. 40 and binding with Sp1 (ref. 20 regulates the gene transcription. Although we demonstrated that β-Arrestin1 increased the binding of P300 with Sp1, thus promoting hTERT transcription in this study, the change of the chromatin structure needs further exploration.
An increasing number of potential drugs targeting the telomere and telomerase have been designed to treat tumors, and most of strategies have been proposed for telomerase inhibition such as BIBR1532 (ref. 41 or telomere uncapping like 6-thio-dG42 and Ber8 (ref. 43 at present. In our study, we designed the anti-sense nucleotide for the fragment (−28 to −36 bp) of hTERT promoter, and identified the effect. The chemical drug with the anti-sense oligo-DNA for −28 to −36 bp in hTERT promoter is promising and needs to further study. Collectively, our results have provided novel insights into the β-Arrestin1-mediated regulation of the senescence in leukemic cells, which may benefit for pediatric B-ALL patients’ treatment.
Materials and methods
Isolated and identified different cell fractions
We collected the LICs and control cells from BM of B-ALL patients using the magnetic-activated cell sorting (MACS; Miltenyi, Bergisch Gladbach, Germany) system with anti-CD34, anti-CD38, and anti-CD19 antibodies. The purity of the cell fractions was identified by flow cytometry as previous report.26
Senescence-associated beta-galactosidase (SA-β-gal) activity assay
SA-β-gal activity was measured as mentioned previously with minor modifications. Bone marrow cells from pediatric B-ALL patients or leukemic mice, or Reh cells, were washed with PBS, fixed in 3% formaldehyde for 20 min at room temperature, washed, and incubated with 1 ml staining solution (Beyotime Biotech., Shanghai, China) overnight at 37 °C, 5% CO2. We counted the blue-stained cells in at least 200 cells, and calculated the percentage of positive cells.
TRAP and ELISA
A quantitative measurement of telomerase activity was conducted one of the two ways: either using PCR-based telomeric repeat amplification protocol (TRAP assay) (Roche, Mannheim, Germany) or using the PCR-ELISA (Roche), with the absorbance read at OD450 nm applying an ELISA reader.
Southern blot
The measurements of terminal restriction fragment (TRF) length in tumor and normal samples were applied using the TeloTAGGG telomere length assay kit (Roche), according to the manufacturer's protocols. The intensity of the hybridization was evaluated by densitometric analysis with Quantity One software (Bio Rad, Hercules, USA) and mean TRF length was estimated according to the formula as described.44
FISH
The slides were processed with the Q-FISH method, as reported previously. In brief, the telomeres were labeled with a Cy3-labeled 5′-CCCTAACCCTAACCCTAA-3′ probe (Fasmac, Atsugi, Japan) and the centromeres were labeled with an FITC-labeled 5′-CTTCGTTGGAAACGGGGT-3′ probe (CENP1: a non-specific centromere probe, Fasmac). Microscopic images were captured with the Image-Pro Plus software package (version 5.0, Media Cybernetics, MD, USA), and analyzed using the public domain NIH image program. We calculated the average fluorescence intensity of telomeres within each nucleus in the FISH images.45
Electrophoretic Mobility Shift Assay (EMSA)
Binding of the Sp1 protein with gene was detected by using LightShift Chemiluminescent EMSA Kit (Thermo, Rockford, USA), according to the manufacturer’s protocol. The Sp1 probe was labeled by biotin and Sp1 consensus oligo was 5′-ATTCGATCGGGGCGGGGCGAGC-3′, 3′-TAAGCTAGCCCCGCCCCGCTCG-5′ (Beyotime Biotech). The supershift was performed with the anti-Sp1 antibody (Abcam, Cambridge, USA).
Luciferase reporter assay
Luciferase reporter plasmids were kindly provided by Professor Weihui Zhou.46 Reh cells were incubated in 10 cm plates (1 × 106 cells per plate) for 24 h, and then transfected with the constructed reporter plasmids (10 μg) and pRL-SV40 (0.01 μg) according to the handbook of effect transfection reagent (Qiagen, Hilden, Germany). Luciferase activity was measured with a dual-luciferase reporter assay system (Promega, Madison, USA) and normalized against the Renilla luciferase activity. All experiments were performed at least three times in each plasmid and represented as the average relative luciferase activity.
References
Hunger SP, Lu X, Decidas M, Camitta BM, Gaynon PS, Winick NJ et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 2012; 30: 1663–1669.
Ghaderi S, Lie RT, Moster D, Ruud E, Syse A, Wesenberg F et al. Cancer in childhood, adolescence, and young adults: a population-based study of changes in risk of cancer death during four decades in Norway. Cancer Causes Control 2012; 23: 1297–1305.
Lang F, Wojcik B, Rieger MA . Stem cell hierarchy and clonal evolution in acute lymphoblastic leukemia. Stem Cells Int 2015; 2015: 137164.
Park IK, Morrison SJ, Clarke MF . Bmi1, stem cell, and senescence regulation. J Clin Invest 2004; 113: 175–179.
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS . The essence of senescence. Genes Dev 2010; 24: 2463–2479.
Lee SH, Um SJ, Kim EJ . CBX8 antagonizes the effect of Sirtinol on premature senescence through the AKT-RB-E2F1 pathway in K562 leukemia cells. Biochem Biophys Res Commun 2016; 469: 884–890.
Grandjenette C, Schnekenburger M, Karius T, Ghelfi J, Gaigneaux A, Henery E et al. 5-aza-2'-deoxycytidine-mediated c-myc down-regulation triggers telomere-dependent senescence by regulating human telomerase reverse transcriptase in chronic myeloid leukemia. Neoplasia 2014; 16: 511–528.
Nunes C, Wong R, Mason M, Fegan C, Man S, Pepper C . Expansion of a CD8 (+) PD-1(+) replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin Cancer Res 2012; 18: 678–687.
Feldser DM, Greider CW . Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 2007; 11: 461–469.
Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biol Ther 2015; 16: 420–429.
Hasegawa H, Yamada Y, Iha H, Tsukasaki K, Nagai K, Atogami S et al. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia 2009; 23: 2090–2101.
Tchirkov A, Chaleteix C, Magnac C, Vasconcelos Y, Davi F, Michel A et al. hTERT expression and prognosis in B-chronic lymphocytic leukemia. Ann Oncol 2004; 15: 1476–1480.
Cogulu O, Kosova B, Karaca E, Gunduz C, Ozkinay F, Aksoylar S et al. Evaluation of telomerase mRNA (hTERT) in childhood acute leukemia. Leuk Lymphoma 2004; 45: 2477–2480.
Takakura M, Kyo S, Kanaya T, Hirano H, Takeda J, Yutsudo M et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res 1999; 59: 551–557.
Kyo S, Takakura M, Taira T, Kanaya T, Itoh H, Yutsudo M et al. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res 2000; 28: 669–677.
Long C, Wang J, Guo W, Wang H, Wang C, Liu Y et al. Triptolide inhibits transcription of hTERT through down-regulation of transcription factor specificity protein 1 in primary effusion lymphoma cells. Biochem Biophy Res Commun 2016; 469: 87–93.
Liu YB, Gao X, Deeb D, Pindolia K, Gautam SC . Role of telomerase in anticancer activity of pristimerin in prostate cancer cells. J Exp Ther Oncol 2015; 11: 41–49.
Pang JX, Cheng XY, Xu W, Wu SG . Antisense Sp1 oligodeoxynucleotide decreases telomerase activity by inhibiting hTERT mRNA expression in Jurkat T cells. Acta Pharmacol Sin 2003; 24: 91–96.
Cheng D, Zhao Y, Wang S, Jia W, Kang J, Zhu J . Human telomerase reverse transcriptase (hTERT) transcription requires sp1/sp3 binding to the promoter and a permissive chromatin environment. J Biol Chem 2015; 290: 30193–30203.
Hung JJ, Wang YT, Chang WC . Sp1 deacetylation induced by phorbol ester recruits p300 to activate 12(S)-lipoxygenase gene transcription. Mol Cell Biol 2006; 26: 1770–1785.
Rahman I, Marwick J, Kirkham P . Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem Pharmacol. 2004; 68: 1255–1267.
Palmitessa A, Hess HA, Bany IA, Kim YM, Koelle MR, Benovic JL . Caenorhabditus elegans arrestin regulates neural G protein signaling and olfactory adaptation and recovery. J Biol Chem 2005; 280: 24649–24662.
Fereshteh M, Ito T, Kovacs JJ, Zhao C, Kwon HY, Tornini V et al. β-Arrestin2 mediates the initiation and progression of myeloid leukemia. Proc Natl Acad Sci USA 2012; 109: 12532–12537.
Qin R, Li K, Qi X, Zhou X, Wang L, Zhang P et al. β-Arrestin1 promotes the progression of chronic myeloid leukaemia by regulating BCR/ABL H4 acetylation. Br J Cancer 2014; 111: 568–576.
Liu H, Long J, Zhang PH, Li K, Tan JJ, Sun B et al. Elevated beta-arrestin1 expression correlated with risk stratification in acute lymphoblastic leukemia. Int J Hematol 2011; 93: 494–501.
Shu Y, Zhou X, Qi X, Liu S, Li K, Tan J et al. beta-Arrestin1 promotes the self-renewal of the leukemia-initiating cell-enriched subpopulation in B-lineage acute lymphoblastic leukemia related to DNMT1 activity. Cancer Lett 2015; 357: 170–178.
Kirsch DG, Kastan MB . Tumor-suppressor p53: implications for tumor development and prognosis. J Clin Oncol 1998; 16: 3158–3168.
Collado M, Serrano M . Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10: 51–57.
Galderisi U, Helmbold H, Squillaro T, Alessio N, Komm N, Khadang B et al. in vitro senescence of rat mesenchymal stem cells is accompanied by downregulation of stemness-related and DNA damage repair genes. Stem Cells Dev 2009; 18: 1033–1042.
Rödel C, Martus P, Papadoupolos T, Fuzesi L, Klimpfinger M, Fietkau R et al. Prognostic significance of tumor regression after preoperative chemoradiotherapy for rectal cancer. J Clin Oncol 2005; 23: 8688–96v.
Beier D, Wischhusen J, Dietmaier W, Hau P, Proescholdt M, Brawanski A et al. CD133 expression and cancer stem cells predict prognosis in high-grade oligodendroglial tumors. Brain Pathol 2008; 18: 370–377.
Mirzayans R, Andrais B, Hansen G, Murray D . Role of p16(INK4A) in replicative senescence and DNA damage-induced premature senescence in p53-deficient human cells. Biochem Res Int 2012; 2012: 951574.
Macha MA, Rachagani S, Pai P, Gupta S, Lydiatt WM, Smith RB et al. MUC4 regulates cellular senescence in head and neck squamous cell carcinoma through p16/Rb pathway. Oncogene 2015; 34: 1698–1708.
Liu JP . Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J 1999; 13: 2091–104v.
Zhu HY, Li C, Bai WD, Su LL, Liu JQ, Li Y et al. MicroRNA-21 regulates hTERT via PTEN in hypertrophic scar fibroblasts. PLoS ONE 2014; 9: e97114v.
Lin SY, Elledge SJ . Multiple tumor suppressor pathways negatively regulate telomerase. Cell 2003; 113: 881–889.
Kharbanda S, Kumar V, Dhar S, Pandey P, Chen C, Majumder P et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr Biol 2000; 10: 568–575.
Horikawa I, Barrett JC . Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis 2003; 24: 1167–1176.
Nishi H, Nakada T, Kyo S, Inoue M, Shay JW, Isaka K . Hypoxia-inducible factor 1 mediates upregulation of telomerase (hTERT). Mol Cell Biol 2004; 24: 6076–6083.
Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M et al. A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell 2005; 123: 833–847.
Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I et al. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J Biol Chem 2002; 277: 15566–15572.
Mender I, Gryaznov S, Dikmen ZG, Wright WE, Shay JW . Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2'-deoxyguanvosine. Cancer Discov 2015; 5: 82–95.
Xiong YX, Su HF, Lv P, Ma Y, Wang SK, Miao H et al. A newly identified berberine derivative induces cancer cell senescence by stabilizing endogenous G-quadruplexes and sparking a DNA damage response at the telomere region. Oncotarget 2015; 6: 35625–35635.
Hiraishi Y, Kato S, Ishihara T, Takano T . Quantitative Southern blot analysis in the dystrophin gene of Japanese patients with Duchenne or Becker muscular dystrophy: a high frequency of duplications. J Med Genet 1992; 29: 897–901.
Tanemura K, Ogura A, Cheong C, Gotoh H, Matsumoto K, Sato E et al. Dynamic rearrangement of telomeres during spermatogenesis in mice. Dev Biol 2005; 281: 196–207.
Xiang Y, Meng S, Wang J, Li S, Liu J, Li H et al. Two novel DNA motifs are essential for BACE1 gene transcription. Sci Rep 2014; 4: 6864.
Acknowledgements
We thank Professor Gang Pei for lentiviral vectors of β-Arrestin1, and Professor Weihui Zhou for Luciferase reporter plasmids. This work was supported in part by research grants from the National Natural Science Foundation of China (81373444, 81570142), Natural Science Foundation of Chongqing (Cstc2015shmszx120012).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U Simon
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, S., Liu, H., Qin, R. et al. The cellular senescence of leukemia-initiating cells from acute lymphoblastic leukemia is postponed by β-Arrestin1 binding with P300-Sp1 to regulate hTERT transcription. Cell Death Dis 8, e2756 (2017). https://doi.org/10.1038/cddis.2017.164
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2017.164
