
Abstract
Human induced pluripotent stem (iPS) cells can be derived from lineage-restricted cells and represent an important tool to develop novel patient-specific cell therapies and research models for inherited and acquired diseases. Recently, patient-derived iPS cells, containing donor genetic background, have offered a breakthrough approach to study human genetics of neurodegenerative diseases. By offering an unlimited source of patient-specific disease-relevant cells, iPS cells hold great promise for understanding disease mechanisms, identifying molecular targets and developing phenotypic screens for drug discovery. This review will discuss the potential impact of using iPS cell-derived models in multiple sclerosis (MS) research and highlight some of the current challenges and prospective for generating novel therapeutic treatments for MS patients.
Similar content being viewed by others

Facts
-
Genetic and environmental factors are believed to be the underlying causes of the majority of autoimmune diseases such as multiple sclerosis
-
Animal models only partially recapitulate pathogenetic features of autoimmune diseases
-
iPS cells have the capability to differentiate into all cell types of the human body
-
iPS cells represent an early stage of disease development
Questions
-
The pathogenetic events involved in multiple sclerosis development and progression are still not completely understood
-
Which pathogenetic events are involved in multiple sclerosis development?
-
Are epigenetic aberrancies crucial for autoimmune diseases onset?
-
Do iPS cells have the capability to model autoimmune diseases?
-
Can iPS cells provide novel pathogenetic insights in autoimmune diseases?
Multiple sclerosis (MS) is a progressive, inflammatory, demyelinating central nervous system (CNS) disease affecting mostly young adults.1 Despite the real cause(s) remains largely unknown, MS has been conventionally classified as an autoimmune inflammatory disease affecting the white matter and only recently demonstrated to affect the grey matter as well.1
MS development has been associated with a genetic predisposition, which in concert with environmental factor exposure2 such as viral infections,3, 4 vitamin D deficiency,5 and other factors, is responsible for disease initiation.6 Initial lesions are frequently associated with a perivascular inflammation that is also considered the origin of the blood–brain barrier breakdown found in MS patients. Thus, MS is characterized by chronic leukocytes infiltration of CNS and by self-limiting attacks to glial cells, ultimately leading to a severe neuron demyelination. One of the early features of MS is the presence of neurons having few layers of myelin rather than the usual 30 layers of compact myelin with a consequent reduction of the action potential conduction along nerves. Furthermore, recovery from acute inflammation results often in ion channel damage, which in absence of defined Ranvier nodes become abnormally distributed along the axons, concurring to the failure of efficient signal conduction. Importantly, myelin destruction followed by neuronal injury is responsible for both long-term disability and cognitive impairment7 in MS patients and nowadays, all current treatments focus in reducing or blocking the autoimmune reaction.
Despite the considerable resources invested in MS research, a significant number of open questions regarding pathogenesis, disease subtypes and response to therapy are still in need to be elucidated. Animal models of autoimmune demyelinating diseases, mimicking MS phenotype, have been so far utilized with the hope to find effective treatments for MS.8 However, these animal models have failed to produce further pathogenetic insights of the disease, likely owing to the profound differences between the animal models and the human disease. The recent discovery that somatic cells can be reprogrammed to a pluripotent stem cell-like state has provided an important tool to study neurodegenerative disease in a controlled environment, including MS. Induced pluripotent stem (iPS) cells represent an early stage of disease development, and their use has the potential to identify specific disease pathways prior, during and after disease development. In addition, the possibility to obtain neurons and leukocytes with the same genetic background of MS patients can provide a deeper understanding of the genetic and epigenetic alterations contributing to the disease establishment. Recapitulating the human MS phenotype in vitro by using iPS cells might represent the rationale for the development of a drug screening approach to identify novel patient-customized targeting treatments.
Induced Pluripotent Stem Cells
Among stem cells, human embryonic stem (ES) cells have been considered to hold greatest promises in biomedical science owing to their capability to differentiate into all the germ layer derivatives and given their potential as treatment in degenerative diseases. During the last few decades, a large amount of scientific efforts have been put in the development of functional equivalents hES-like cells for scientific and clinical purposes in order to overcome the ethical issues related to the hES use. Somatic cell nuclear transfer and cell fusion have been showed to induce reprogramming of differentiated cells to a pluripotent state; however, both techniques were highly inefficient for humans as well limited in number to be used on a large scale for disease modeling or regenerative medicine. Further, these methodologies did not solve the issues of immunological rejection of the transplanted allogeneic tissues derived from the pluripotent parental cells or the ethical issues relating to destruction of human embryos.9
The landmark discovery that pluripotent stem cells can be directly derived through the ectopic expression of defined factors opened a new frontier for regenerative medicine especially for novel disease modeling and drug screening methodologies. Overexpression of OCT4 and SOX2 in combination with KLF4 and c-MYC,10 KLF4 alone,11 LIN28 and NANOG,12 or ESRBB13 is able to induce lineage-restricted cells to reprogram to a pluripotent ES-like state. In addition, numerous reprogramming strategies have been conceived over the last few years to avoid the use of the c-Myc oncogene11, 14 and to generate safer transgene-free or integration-free iPS cells including adeno- and Sendai virus-based vectors,15, 16 episomal vectors,17 mRNA transduction,18 piggyBac transposon19 or by substituting specific reprogramming factors with chemicals.20, 21
The iPS cells have similar characteristics of blastocyst-derived ES cells such as unlimited self-renewal, gene expression profile and capacity to differentiate into all somatic cell types of the human body. Similar to hES, human iPS cells cultured in absence of FGF give rise to embryoid bodies (EBs), cell aggregates showing similar structure to the early stage of human embryos.10 EBs consist of three embryonic germ layers and represent the first step during iPS differentiation. Subsequent culture in defined conditions allows terminal differentiation of EBs to a specific cell type (Figure 1). Efficient differentiation toward selected cell phenotypes represents the greatest challenge for diseases studies. Great efforts have been made in recent years to obtain specific and functional cell types derived from pluripotent stem cells,9 that is, vascular endothelium,22 cardiomyocytes,23 most of the hematopoietic cells,24 pancreatic insulin-producing cells25 or hepatocyte-like cells,26 and various subtypes of neural cells.27, 28 Recently, it has been shown that differentiation of pluripotent stem cells is achievable in vivo by way of teratoma formation.24, 29 This innovative approach allowed the isolation of several blood elements including hematopoietic stem/progenitor cells capable of multilineage reconstitution when transplanted in immunocompromised mice. This protocol represents an alternative route to derive specific cell types from pluripotent stem cells, whereas in vitro differentiation is not a feasible approach. Another aspect to consider when handling iPS cells is the impact of potential genetic and epigenetic alterations, occurring during the reprogramming process, on the differentiation potential. Although iPS cells show a normal karyotype after the reprogramming process, it has been reported that continuous passaging of these cells is associated with the acquisition of chromosomal abnormalities (karyotype 46,XY,t(17;20) (p13;p11.2)) starting approximately at passage 13.30 Recent studies have demonstrated significant reprogramming variability among iPS cell lines, including somatic memory or aberrant reprogramming of DNA methylation.31, 32 In particular, Lister and colleagues have identified differentially methylated regions (DMRs) occurring during reprogramming by comparing iPS with ES and somatic cells. Half of the identified DMRs were related to incomplete reprogramming while the other half was deemed as epigenetic errors as they were absent in either somatic cells or ES cells. These methylation errors can be potentially inherited by the iPS-derived differentiated cell types; however, the impact of such epigenetic heterogeneity on iPS cell-differentiation capabilities as well as an approach to reduce it have not yet been fully elucidated.

Somatic cells can be reprogrammed to an ES-like state through the overexpression of four transcription factors. Pluripotent stem cells derived through this approach can differentiate to the three germ layer derivatives by the way of embryoid body formation (EBs)
iPS Cells Impact MS Genetics
The possibility to generate iPS cells from individuals affected by several diseases with a genetic component33 has attracted a world-wide attention, initiating a driving force to create real patient-customized disease models, novel drug screening platforms and eventually regenerative medicine.
Although there are high expectations for future applications of iPS cells to repair damaged organs including the CNS, the most compelling and important concerns/limitations remain the feasibility to produce ‘large-scale’ patient-derived cells carrying specific genetic susceptibility and to subsequently analyze these cells under controlled conditions in the laboratory. Recently, fibroblasts from MS patients have been reprogrammed to pluripotency34 making an important step toward MS research. Indeed, multiple sclerosis iPS cells are able to differentiate to all germ layer derivatives, including oligodendrocytes, astrocytes and functional neurons. Many iPS lines generated from several pathological conditions are characterized by specific genetic driving mutation.33 In contrast, MS is a multifactorial disease caused by a complex interaction between environment and genetic susceptibility thereby contributing to the pathological heterogeneity.35 A growing body of evidences suggests that the genetic component has a crucial role in the disease development36 (Table 1). Despite the MHC loci still represent the major dominant risk region for MS development,37, 38 many other non-MHC genetic variants involved in MS pathogenesis have been recently identified.39 Interestingly, the majority of the immunological genes identified to have a role in the disease onset are known to be important for T-cell responses, including cytokine receptors for interleukin-7 (IL-7) and IL-2,40, 41 and signal transducers such as tyrosine kinase 2 (TYK2) for type I interferons, IL-10 and IL-12.7 Genetic variants of adhesion molecules such as CD58 and CD226 are also demonstrated to be associated with MS development because of its involvement in abnormal stimulating signals.42 Interestingly, neurological gene variants have also been found associated with MS, including amiloride-sensitive cation channel 1, neuronal (ACCN1)43 and kinesin family member 1B (KIF1B).44 Although the role of the ACNN1 channel is poorly understood, KIF1B encodes a protein involved in axonal transport of mitochondria and synaptic vesicle precursors. Although it is reasonable to hypothesize a contribution of these genes to the inflammatory process and neuronal degeneration observed in MS, the functional role of these factors during the disease development still requires to be specifically investigated. The advent of iPS cells makes it now possible to investigate the impact of genetic variants on specific cell population behavior and survival. In particular, generation of several neural population with a defined genetic MS background may allow functional evaluation of specific genetic variants and polymorphisms by unraveling their role in the disease establishment. A similar approach has the potential to shed light on the participation of several environmental insults, such as reactive oxygen species or abnormal cytokine levels, on apoptosis or cell death of specific neural population and to recapitulate at least partially the inflammatory environment of MS. Further, by enabling studies on the differentiation capability of MS-iPS45-derived neural stem/precursors to terminally differentiated neurons and glial cells, this strategy will allow us to understand whether specific genetic variants interfere with self-renewal and regeneration of damaged CNS tissues in MS patients.
In summary, the use of iPS cells might unravel specific pathogenetic aspects of MS initiation and development that, up to date, remain largely unknown. Understanding the function of specific genetic variants, detected in neural and immunological population of MS patients, is critical to pave the way for the discovery of novel therapeutic targets to tackle this complex debilitating disease.
iPS Cells Impact MS Epigenetics
In the last few years, a large amount of studies showed that the interplay between environmental factors and individual genetic susceptibility is likely to produce a pathogenetic predisposition to MS.6 Currently, numerous studies point at the role of epigenetic aberrancies to both disease development and progression.46
Epigenetic modifications are heritable changes affecting gene expression without altering the DNA sequence. The molecular changes underlying epigenetics modifications include DNA methylation and histone modifications. The epigenetic modifications are important not only for tissue development but also for homeostasis and for the establishment of cellular identity. Transcriptional activation is associated with the presence of both lineage-specific transcription factors and of activation marks on lysine and arginine residues of histone tails. Gene repression instead is achieved by repressive marks on amino-acid residues of histone tails.47
DNA methylation is linked to gene silencing and it is considered more likely an irreversible phenomenon.48 Non-coding RNAs operate at both transcriptional49 and post-transcriptional50 level by finely regulating expression of specific target genes. All the epigenetic modifications are affected by environmental signals and their dysregulation can lead to many diseases including MS.51 Patients with MS have been found to share specific epigenetic modifications (Table 2), yet the functional role of such aberrancies in the disease development is not completely understood.
Recently, DNA methylation has been investigated in CD4+ peripheral blood lymphocytes from three twin pairs discordant for MS.52 Two genes TMEM and PEX14 were found to be differentially methylated. Furthermore, Liggett et al.53 analyzed the DNA methylation pattern of 56 genes previously shown to be associated with cancer development using cell-free plasma DNA of MS patients. Interestingly, 15 out of 56 genes including PAX5, TP73 and FAS were found to be differentially methylated suggesting the potential use of different T-cell subpopulations to further investigate the role of DNA methylation in MS.
Nucleosomal histone tails are target of many enzymatic processes including acetylation, methylation, phosphorylation, sumoylation, citrullination and ubiquitination. Acetylation of lysine in position 9 and 14 and tri-methylation of lysine in position 4, 36 and 79 of histone H3 are commonly associated with active chromatin and gene expression. Inversely, acetylation and tri-methyation loss at those levels tend to correspond to inactive chromatin and gene repression.51 Recently, it has been showed an increased deamination of arginine residues H3 histone tails in MS patients. This phenomenon also known as citrullination has been associated with protein instability and might be contributing to oligodendrocytes apoptosis54 and T-cell activation.55
MicroRNAs have been also found differentially expressed between MS patients and healthy individuals. Using high-throughput miRNA profiling, key miRNAs involved in MS development have been identified in several tissues including whole blood, peripheral blood mononuclear cells and lymphocytes.56 Interestingly, few miRNAs involved in repression of genes associated with T-cell activation such as miR-17 and miR-20a57 were found significantly decreased (Table 2). Therefore, downregulation or upregulation of RNA molecules involved in the immune response could represent a key second hit during MS development.
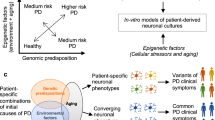
Epigenetic changes have a pivotal role during nuclear reprogramming and are fundamental for pluripotency acquisition.58 From this prospective, nuclear reprogramming offers an ideal model to study the role of specific epigenetic abnormalities in MS development and progression. The generation of genetically matched but epigenetically distinct cells might help understanding the involvement of epigenetic alterations in the disease pathogenesis. Recently, it has been shown that the possibility to reset epigenetic abnormalities in cancer by nuclear reprogramming,59, 60 leading the cancer epigenome to an ES-like ground state.
The same approach can be exploited in MS in order to reactivate immune response key factors poisoned by DNA methylation. This approach might allow the functional evaluation of epigenetic alteration role in MS development. Reactivation of critical genes of immune response regulation such as FAS and TP73 might lead to understand whether ‘apoptosis escaping’ driven by epigenetic mutations concurs to generate self-reactive clones after inflammation and whether those types of epigenetic abnormalities represent a precipitating pathogenetic event during autoimmune disease development (Figure 2).

T cells isolated from MS patients might present an aberrant epigenetic state of key immunological genes. These cells can be reprogrammed to an induced pluripotent-like state with reactivation of several immune response-related genes. iPS cells can then differentiate to T-cell lineages with an epigenetic profile resembling the ES ground state and subsequently used for functional experiments
Toward Pharmaceutical Applications of iPS Cells
The comprehension of how genetic variation contributes to disease pathogenesis is inevitable for the development of preventive strategies and new generation treatments. Historically, gain- and loss-of-function animal models have been used for this purpose; however, several disease phenotypes failed to be reproduced in these models. Furthermore, the limitation in obtaining human tissues, other than blood, has been so far an obstacle for conducting analysis and relevant research for specific diseases. Thus, the advent of iPS cells has represented a turning point to overcome the mentioned limitations. The possibility to use pluripotent cells derived from patients able to differentiate into cells or tissues not accessible otherwise (e.g., neurons, glial cells, etc) has initiated a new research field with the potential to unravel specific pathogenetic mechanisms, which are still largely unknown. In addition, owing to the unlimited self-renewal capabilities, iPS cells become a suitable tool for drug screening or toxicology studies. Furthermore, iPS cells can mimick disease models in so-called ‘disease in a dish’ system. In order to obtain a specific ‘disease in a dish’ phenotype, two aspects must be satisfied: (1) to derive pathological relevant cell populations and (2) to recapitulate key aspects of the disease onset. This approach might even more benefit from the application of three-dimensional (3D) cell culture model system.61 Traditionally, two-dimensional (2D) cell culture systems have been considered the chosen and most simplistic tools to reproduce ‘disease in a dish’ for drug discovery and screening purposes whereas ‘in vivo’ models have been preferred for the efficacy and safety assessment before proceeding to clinical trials. Unfortunately, both approaches display several limitations: 2D cultures are unable to mirror the physiological condition of the native tissue of study, thus impairing therapeutics screening as shown by the high drug failure rate (~95%);62 the use of animal models, when available for specific disease testing, not always translates into therapies able to improve the outcome of human disease and often for reasons not clearly understood.63 In order to bridge the gap between in vitro and in vivo experiments, the 3D tissue culture system has been recently developed.
Although nuclear reprogramming appears to be a very inefficient process, to date many iPS lines from patients affected by genetic disease including Duchenne muscular dystrophy, Down’s syndrome, Parkinson’s and so on have been derived.33 Among genetic diseases from which iPS cells have been derived, spinal muscular athrophy (SMA) looks to be one of the most promising to generate a valuable disease modeling.64 SMA is a neurodegenerative genetic disease in which motor neurons degenerate bringing the young patients to paralysis and often death. iPS cells derived from SMA fibroblasts were capable to differentiate normally to neural tissues and motor neurons. However, motor neurons were selectively lost in the neural culture recapitulating the patient-specific SMA phenotype.64 Interestingly, valproic acid or tobramycin treatments were capable to increase the levels of SMN protective protein. These findings prove that generation of a valuable disease in a dish model might aid the identification of novel compounds and pathogenetic mechanisms in the disease initiation and development.
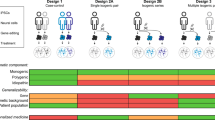
Heterogeneity of human diseases has never been recapitulated in animal models. Generation of iPS lines from patients’ samples with same disease but different outcome has the potential to shed light on the mechanisms driving the severity of a specific disease. For MS, unraveling the pathogenetic events behind the disease subtypes represents a major goal for understanding and generating the most appropriate and efficacious treatment for that disease phenotype. In particular, derivation of iPS cells from patients affected by different types of MS might facilitate identification of genetically distinct disease subtypes (Figure 3).

Somatic cells from skin, blood or other tissues can be obtained from patients with different MS disease subtypes and reprogrammed to iPS cells. The approach allows the establishment of subtype disease in a dish has the potential to identify novel pathogenetic mechanisms in MS as well as produce specific broad drug or toxicology screening during a drug development
Understanding drug toxicity before starting a clinical trial is one of the greatest challenges in pharmaceutical research. A reliable and accessible platform to predict toxicity in vivo will offer two major advantages: (1) it will lead to more efficient generation of safer compounds and (2) it will decrease the costs for drug development. Animal models have been extensively used for toxicity purpose, however, differences between human and murine tissue entail a poor accuracy in toxicity prediction. iPS cells capable to differentiate to tissues often target of drug toxicity (e.g., cardiac, neural and hepatic tissues) represent now an important tool to dissect pre-clinical drug toxicity. Pluripotent stem cell libraries derived from skin of patients for whom that drug is being developed represent an important resource to predict accurately toxicity in a specific cohort of patients. Finally, the possibility to derive disease subtype iPS cells will help in developing patient-customized treatments. For MS case, this approach will allow for a timely identification of patients that would response adversely or favorably to a specific compound improving and accelerating the drug development process.
Concluding Remarks
The major finding that somatic cells can artificially reach a pluripotent stem cell-like state holds new promises for discovery of novel therapeutic treatments. Recent studies warn some caution for the use of iPS cells for clinical applications due to both the use of exogenous DNA and for the presence of important epigenetic alterations found in several iPS lines.31 On the other hand, it is clear that the most promising application of iPS cells resides in disease modeling and drug screening. Establishment of the ‘disease in a dish’ using disease-specific iPS cells will lead to the identification of pathogenetic mechanisms that are still poorly understood. iPS cells promise to overcome all the limitations associated with animal models used to recapitulate specific disease phenotypes, offering an unlimited source of pathological tissues involved in specific disease development. Combination of exploratory genomic approaches such as new generation sequencing with functional experiments through iPS cells will elucidate key pathways in the initiation and development of many genetic disorders including neurodegenerative diseases.
Despite many advances in MS field have been achieved in the last few years, a number of open questions regarding disease initiation, disease subtypes and response to therapy still need to be answered. The generation of MS phenotype in a dish represents the biggest challenge to overcome all the limitations associated with the autoimmune-mediated demyelinating animal models used so far. iPS cells derived from MS patients34 will make accessible neural populations that so far have been poorly analyzed owing to inaccessibility of the brain tissues. Analyzing neural populations with the specific patient’s genetic background will undoubtedly shed light on disease-pathogenetic mechanisms promoting the generation of new effective treatments for this debilitating disease.
Change history
14 April 2016
This article has been corrected since Online Publication and an erratum has also been published
Abbreviations
- iPS cells:
-
induced pluripotent stem cells
- MS:
-
multiple sclerosis
- CNS:
-
central nervous system
- ES cells:
-
embryonic stem cells
- EBs:
-
embryoid bodies
- DMRs:
-
differentially methylated regions
- IL:
-
interleukin
- TYK2:
-
tyrosine kinase 2
- ACCN1 :
-
amiloride-sensitive cation channel 1, neuronal
- KIF1B :
-
kinesin family member 1B
- miRNAs:
-
microRNAs
- SMA:
-
spinal muscular athrophy
References
Nylander A, Hafler DA . Multiple sclerosis. J Clin Invest 2012; 122: 1180–1188.
Ebers GC . Environmental factors and multiple sclerosis. Lancet Neurol 2008; 7: 268–277.
Donati D, Jacobson S . Virus and multiple sclerosis. In: Brogden KA, Guthmiller JM (eds). Polymicrobial Diseases. ASM Press: Washington, DC, USA, 2002; Chapter 6, pp 1–49.
Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS et al. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain 2009; 132: 3318–3328.
Goldberg P . Multiple sclerosis: vitamin D and calcium as environmental determinants of prevalence. Int J Environ Studies 1974; 6: 121–129.
Handel AE, Giovannoni G, Ebers GC, Ramagopalan SV . Environmental factors and their timing in adult-onset multiple sclerosis. Nat Rev Neurol 2010; 6: 156–166.
Patti F . Cognitive impairment in multiple sclerosis. Mult Scler 2009; 15: 2–8.
Wekerle H . Lessons from multiple sclerosis: models, concepts, observations. Ann Rheum Dis 2008; 67: iii56–iii60.
Amabile G, Meissner A . Induced pluripotent stem cells: current progress and potential for regenerative medicine. Trends Mol Med 2009; 15: 59–68.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872.
Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 2008; 26: 101–106.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318: 1917–1920.
Feng B, Jiang J, Kraus P, Ng JH, Heng JC, Chan YS et al. Reprogramming of fibroblasts into induced pluripotent stem cells with orphan nuclear receptor Esrrb. Nat Cell Biol 2009; 11: 197–203.
Wernig M, Meissner A, Cassady JP, Jaenisch R . c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2008; 2: 10–12.
Zhou W, Freed CR . Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009; 27: 2667–2674.
Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M . Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 2009; 85: 348–362.
Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009; 324: 797–801.
Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010; 7: 618–630.
Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K . Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 2009; 458: 771–775.
Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 2008; 26: 1269–1275.
Ichida JK, Tcw J, Williams LA, Carter AC, Shi Y, Moura MT et al. Notch inhibition allows oncogene-independent generation of iPS cells. Nat Chem Biol 2014; 10: 632–639.
Taura D, Sone M, Homma K, Oyamada N, Takahashi K, Tamura N et al. Induction and isolation of vascular cells from human induced pluripotent stem cells—brief report. Arterioscler Thromb Vasc Biol 2009; 29: 1100–1103.
Narazaki G, Uosaki H, Teranishi M, Okita K, Kim B, Matsuoka S et al. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation 2008; 118: 498–506.
Amabile G, Welner RS, Nombela-Arrieta C, D’Alise AM, Di Ruscio A, Ebralidze AK et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 2013; 121: 1255–1264.
Tateishi K, He J, Taranova O, Liang G, D’Alessio AC, Zhang Y . Generation of insulin-secreting islet-like clusters from human skin fibroblasts. J Biol Chem 2008; 283: 31601–31607.
Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 2010; 51: 297–305.
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008; 321: 1218–1221.
Wernig M, Zhao JP, Pruszak J, Hedlund E, Fu D, Soldner F et al. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson's disease. Proc Natl Acad Sci USA 2008; 105: 5856–5861.
Suzuki N, Yamazaki S, Yamaguchi T, Okabe M, Masaki H, Takaki S et al. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther 2013; 21: 1424–1431.
Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 2008; 26: 1276–1284.
Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011; 471: 68–73.
Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010; 467: 285–290.
Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A et al. Disease-specific induced pluripotent stem cells. Cell 2008; 134: 877–886.
Song B, Sun G, Herszfeld D, Sylvain A, Campanale NV, Hirst CE et al. Neural differentiation of patient specific iPS cells as a novel approach to study the pathophysiology of multiple sclerosis. Stem Cell Res 2012; 8: 259–273.
Oksenberg JR, Baranzini SE . Multiple sclerosis genetics—is the glass half full, or half empty? Nature Rev Neurol 2010; 6: 429–437.
International Multiple Sclerosis Genetics C, Sawcer S, Hellenthal G, Pirinen M, Spencer CC et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011; 476: 214–219.
Lincoln MR, Montpetit A, Cader MZ, Saarela J, Dyment DA, Tiislar M et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet 2005; 37: 1108–1112.
Fogdell-Hahn A, Ligers A, Gronning M, Hillert J, Olerup O . Multiple sclerosis: a modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens 2000; 55: 140–148.
Fugger L, Friese MA, Bell JI . From genes to function: the next challenge to understanding multiple sclerosis. Nat Rev Immunol 2009; 9: 408–417.
International Multiple Sclerosis Genetics C Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007; 357: 851–862.
Ban M, Goris A, Lorentzen AR, Baker A, Mihalova T, Ingram G et al. Replication analysis identifies TYK2 as a multiple sclerosis susceptibility factor. Eur J Hum Genet 2009; 17: 1309–1313.
De Jager PL, Baecher-Allan C, Maier LM, Arthur AT, Ottoboni L, Barcellos L et al. The role of the CD58 locus in multiple sclerosis. Proc Natl Acad Sci USA 2009; 106: 5264–5269.
Bernardinelli L, Murgia SB, Bitti PP, Foco L, Ferrai R, Musu L et al. Association between the ACCN1 gene and multiple sclerosis in Central East Sardinia. PloS One 2007; 2: e480.
Aulchenko YS, Hoppenbrouwers IA, Ramagopalan SV, Broer L, Jafari N, Hillert J et al. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat Genet 2008; 40: 1402–1403.
Denham M, Dottori M . Neural differentiation of induced pluripotent stem cells. Methods Mol Biol 2011; 793: 99–110.
Koch MW, Metz LM, Kovalchuk O . Epigenetic changes in patients with multiple sclerosis. Nat Rev Neurol 2013; 9: 35–43.
Jenuwein T, Allis CD . Translating the histone code. Science 2001; 293: 1074–1080.
Smith ZD, Meissner A . DNA methylation: roles in mammalian development. Nat Rev Genet 2013; 14: 204–220.
Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013; 503: 371–376.
Guttman M, Rinn JL . Modular regulatory principles of large non-coding RNAs. Nature 2012; 482: 339–346.
Huynh JL, Casaccia P . Epigenetic mechanisms in multiple sclerosis: implications for pathogenesis and treatment. Lancet Neurol 2013; 12: 195–206.
Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 2010; 464: 1351–1356.
Liggett T, Melnikov A, Tilwalli S, Yi Q, Chen H, Replogle C et al. Methylation patterns of cell-free plasma DNA in relapsing-remitting multiple sclerosis. J Neurol Sci 2010; 290: 16–21.
Mastronardi FG, Wood DD, Mei J, Raijmakers R, Tseveleki V, Dosch HM et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci 2006; 26: 11387–11396.
Sharma P, Azebi S, England P, Christensen T, Moller-Larsen A, Petersen T et al. Citrullination of histone H3 interferes with HP1-mediated transcriptional repression. PLoS Genet 2012; 8: e1002934.
Jr Ode F, Moore CS, Kennedy TE, Antel JP, Bar-Or A, Dhaunchak AS . MicroRNA dysregulation in multiple sclerosis. Front Genet 2012; 3: 311.
Cox MB, Cairns MJ, Gandhi KS, Carroll AP, Moscovis S, Stewart GJ et al. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PloS One 2010; 5: e12132.
Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P et al. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008; 454: 49–55.
Stricker SH, Feber A, Engstrom PG, Caren H, Kurian KM, Takashima Y et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev 2013; 27: 654–669.
Amabile G, Di Ruscio A, Muller F, Welner RS, Yang H, Ebralidze AK et al. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat Commun 2015; 6: 7091.
Haycock JW . 3D cell culture: a review of current approaches and techniques. Methods Mol Biol 2011; 695: 1–15.
Hutchinson L, Kirk R . High drug attrition rates—where are we going wrong? Nat Rev Clin Oncol 2011; 8: 189–190.
Aggarwal BB, Danda D, Gupta S, Gehlot P . Models for prevention and treatment of cancer: problems vs promises. Biochem Pharmacol 2009; 78: 1083–1094.
Ebert AD, Yu J, Rose FF Jr., Mattis VB, Lorson CL, Thomson JA et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009; 457: 277–280.
Sawcer S, Franklin RJ, Ban M . Multiple sclerosis genetics. Lancet Neurol 2014; 7: 700–709.
Hafler DA, De Jager PL . Applying a new generation of genetic maps to understand human inflammatory disease. Nat Rev Immunol 2005; 1: 83–91.
Acknowledgements
We thank all colleagues and friends of Biogen for input and scientific discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Di Ruscio, A., Patti, F., Welner, R. et al. Multiple sclerosis: getting personal with induced pluripotent stem cells. Cell Death Dis 6, e1806 (2015). https://doi.org/10.1038/cddis.2015.179
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.179
This article is cited by
-
The Post-GWAS Era: How to Validate the Contribution of Gene Variants in Lupus
Current Rheumatology Reports (2019)
-
Neural Stem Cell-Based Regenerative Approaches for the Treatment of Multiple Sclerosis
Molecular Neurobiology (2018)
-
Viruses and Multiple Sclerosis: From Mechanisms and Pathways to Translational Research Opportunities
Molecular Neurobiology (2017)
-
Differential regulated microRNA by wild type and mutant p53 in induced pluripotent stem cells
Cell Death & Disease (2016)
