
Abstract
Obesity is a worldwide epidemic, with the number of overweight and obese individuals climbing from just over 500 million in 2008 to 1.9 billion in 2014. Type 2 diabetes (T2D), cardiovascular disease and non-alcoholic fatty liver disease have long been associated with the obese state, whereas cancer is quickly emerging as another pathological consequence of this disease. Globally, at least 2.8 million people die each year from being overweight or obese. It is estimated that by 2020 being overweight or obese will surpass the health burden of tobacco consumption. Increase in the body mass index (BMI) in overweight (BMI>25 kg/m2) and obese (BMI>30 kg/m2) individuals is a result of adipose tissue (AT) expansion, which can lead to fat comprising >50% of the body weight in the morbidly obese. Extensive research over the last several years has painted a very complex picture of AT biology. One clear link between AT expansion and etiology of diseases like T2D and cancer is the development of insulin resistance (IR) and hyperinsulinemia. This review focuses on defining the link between obesity, IR and cancer.
Similar content being viewed by others

Facts
-
Hyperinsulinemia, along with the other obesity-related factors, is linked to the development of several types of cancers.
-
Insulin, signaling through insulin receptor A, has direct oncogenic effects on cancer cells.
-
Insulin-lowering drugs, such as metformin, may prove to be useful in lowering insulin levels and insulin resistance, decreasing body weight and improving cancer outcomes in patients with obesity and type 2 diabetes.
Open Questions
-
How are the PI3K-AKT and the Ras-MAPK pathways regulated by INSR-A in normal epithelial cells and in cancer?
-
How should obesity and T2D be treated in order to minimize the risk of cancer development, specifically keeping in mind the potential oncogenic effect of hyperinsulinemia?
-
What are the effective drugs targeting the molecular pathways that link obesity and T2D to cancer?
Obesity and Cancer
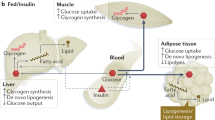
Insulin is the master regulator of energy storage and whole-body metabolism (Figure 1). It is produced and secreted by pancreatic β cells in response to a surge in blood glucose levels. Insulin stimulates glucose uptake by adipose tissue (AT) and muscle, whereas suppressing the release of glucose from the liver. It also stimulates the liver and the muscle to store excess glucose in the form of glycogen. In addition to regulating glucose homeostasis, insulin also induces fat storage. In adipocytes, it inhibits lipolysis while inducing lipogenesis and fatty acid uptake from the blood stream. Insulin thus ensures sufficient storage of energy that can be mobilized during fasting, when insulin levels are low.

The role of insulin in the control of whole-body metabolism
Perpetual caloric excess in individuals with obesity disrupts the intricate balance between energy storage and mobilization, leading to desensitization of tissues to the actions of insulin and the development of IR. The attenuation of the response of insulin target tissues to the physiological actions of insulin leads to a compensatory increase in pancreatic insulin production in an attempt to reestablish glucose homeostasis and thus overt hyperglycemia. This overproduction of insulin by the pancreatic β cells and a concomitant increase in serum insulin levels is a condition called hyperinsulinemia.
Strong clinical and epidemiological evidence links hyperinsulinemia, along with the other obesity-related factors, to the development of several types of cancers, including those of the breast, endometrium, colon, liver, esophagus, kidney and pancreas.1, 2, 3, 4, 5, 6, 7 International Agency for Research on Cancer estimated that obesity is a cause of 11% of colon, 9% of postmenopausal breast, 39% of endometrial, 25% of kidney and 37% of esophageal cancer cases.7 A prospective study of 900 000 adults in the United States reported that obesity accounts for 14% of deaths from cancer in men and 20% in women, directly linking excess body weight to cancer mortality.3 This study also highlighted a dose–response relationship between obesity and cancer, finding an increase in cancer risk with increasing body mass index (BMI). Finally, an analysis of the global burden of cancer has identified that 3.6% of all new cancers are attributable to BMI.8 Of particular interest, weight loss following a lifestyle change or bariatric surgery reduces cancer risk,9, 10, 11 whereas in the Swedish Obesity Subjects study, women who lost >30% of their body weight had a marked 41% reduction in cancer risk.11 Although some recent research suggests that the hereditary and environmental factors contribute less to the cell transformation than the cell’s division history,12 it is likely that the interplay of the cumulative changes associated with cell’s proliferative past, in the context of the genetic underpinning and the environmental factors such as obesity, underlies the development of most cancers. The beneficial effects of weight loss raise the possibility that the adverse impact of obesity on cancer can be reversed and suggests that obesity-directed therapies may impact cancer treatment and survival.
The Insulin Signaling Pathway
The diverse cellular actions of insulin are initiated by its binding to the insulin receptor (INSR) on the surface of target cells (Figure 2). The INSR signaling pathway mediates both the metabolic and the mitogenic effects of insulin and its deregulation is central to the development of insulin resistance (IR). INSR is a heterotetramer composed of two extracellular insulin-binding α subunits (130 kDa), covalently linked to two transmembrane β subunits (95 kDa) with intrinsic tyrosine kinase activity.13 There are two isoforms of INSR, INSR-A and INSR-B, differing by the presence (INSR-B) or the absence (INSR-A) of exon 11, which encodes a 12 amino-acid stretch at the C-terminal end of the α subunit (Figure 3).14, 15, 16 The lack of exon 11 in INSR-A allows this receptor to bind not only insulin but also insulin-like growth factor II (IGFII) and pro-insulin with high affinity.17, 18 Indeed, INSR-A has a 1.7-fold higher affinity for insulin than INSR-B and is internalized and recycled faster than INSR-B.19, 20, 21

The insulin signaling pathway

The link between hyperinsulinemia and cancer development
The expression of the two isoforms is regulated developmentally and in a tissue-specific manner. The INSR-A isoform is predominantly expressed in the fetal tissues where it regulates embryonic growth. Remarkably, INSR-A is also the predominant isoform overexpressed by many cancer cells.22, 23, 24 In human embryos, lack of insulin, as in the cases of pancreatic agenesis, lack of β cells or pancreatic islets, or transient neonatal diabetes, results in severe growth retardation.25, 26, 27 INSR deficiency during embryonic development results in comparable growth retardation, as seen in the infants with the Donohue syndrome.28 INSR-A also mediates the mitogenic signaling in the regulation of pancreatic β-cell proliferation, and is able to protect the myeloid 32D cells from apoptosis upon IL-3 removal more effectively than INSR-B.23, 29 INSR-B is predominantly expressed in the differentiated adult tissues, particularly the liver, the fat and the muscle, where it regulates the metabolic effects of insulin. An abnormally high INSR-A:INSR-B ratio in muscle cells of patients with myotonic dystrophy appears to be responsible for the IR seen in these patients, further highlighting the functional differences between the two isoforms of the receptor.30 In mice, INSR deletion leads to 10–20% growth retardation and metabolic abnormalities that develop after birth.31
INSR can form hybrid receptors from INSR-A or B but also with a related tyrosine kinase, insulin-like growth factor 1 receptor (IGF1R), thus resulting in a complex, tissue-specific regulation of the metabolic and the mitogenic signaling pathways. Upon insulin binding, INSR undergoes a conformational change and autophosphorylates several residues in the C-terminal tail of the β subunit, leading to recruitment and further phosphorylation of a number of effector proteins. Insulin receptor substrates (IRSs), which bind INSR and are directly phosphorylated by it on tyrosine residues, present sites for binding of adaptor molecules containing Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domains, which further propagate the signals.32, 33 One such effector is the regulatory subunit (p85) of phosphoinositide 3-kinase (PI3K), which, when bound to tyrosine phosphorylated IRS, relieves inhibition of the catalytic PI3K subunit (p110) leading to its activation.34 PI3K then phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to produce phosphatidylinositol 3,4,5-triphosphate (PIP3), a cell membrane-associated lipid second messenger that attracts proteins containing pleckstrin homology (PH) domains, including protein kinase B/AKT (PKB/AKT) and the 3-phosphoinositide-dependent protein kinase 1 (PDK1).35 PIP3 recruits AKT and PDK1 to the plasma membrane where PDK1 phosphorylates AKT on threonine 308 (T308), leading to its partial activation.36 Mammalian target of rapamycin complex 2 (mTORC2) then phosphorylates AKT on serine 473 (S473), fully activating the protein.37
AKT signaling through numerous downstream targets governs the metabolic effects of insulin. For example, the AKT substrate of 160 kDa (AS160) regulates the glucose transporter 4 (GLUT4) translocation to the plasma membrane, a process that initiates cellular glucose uptake in response to insulin.38 Phosphorylation of the phosphofructokinase 2 by AKT induces glycolysis, enabling cells to metabolize glucose into usable energy in the form of adenosine triphosphate (ATP).39 AKT also phosphorylates and inhibits the glycogen synthase kinase 3 (GSK3), a negative regulator of glycogen synthesis and lipogenesis.40, 41 In the liver, PDK1-dependent disassembly of the cAMP-response-element-binding protein (CREB) complex and phosphorylation of the forkhead box O (FOXO) by AKT suppress gluconeogenesis.42, 43, 44 Finally, AKT phosphorylates and downregulates the GTPase activator protein (GAP) activity of tuberous sclerosis complex 2 (TSC2) toward Ras homolog enriched in brain (Rheb), a G protein that regulates the activity of mammalian target of rapamycin complex 1 (mTORC1).45, 46, 47, 48 This allows for a Rheb-mediated activation of mTORC1, which in turn phosphorylates the sterol regulatory element-binding protein 1c (SREBP1c), a transcription factor that induces the transcription of genes involved in lipogenesis and represses those involved in lipolysis.49, 50
The PI3K-AKT pathway also mediates some of the mitogenic effects of insulin. mTORC1 regulates cell growth not only through SREBP1-mediated lipid synthesis but also by controlling mRNA translation, via direct phosphorylation of the S6 kinase (p70S6K) and the 4E binding proteins (4EBPs).51, 52 In addition, AKT-mediated phosphorylation and deactivation of the transcription factor FOXO results in its nuclear export and proteasomal degradation, thus releasing cells from FOXO-mediated cell cycle arrest.53, 54, 55 Deactivation of FOXO, along with another target of Akt activity, the BCL2-associated agonist of cell death (BAD), coordinately represses the cellular apoptotic program.56 Finally, AKT-mediated inhibition of GSK3 results in nuclear accumulation of cyclin D1, which regulates G1/S phase transition.57
The mitogenic effects of insulin are mainly mediated by the recruitment of SH2 domain-containing adaptor (SHC) to INSR and activation of the rat sarcoma-mitogen-activated protein kinase/ERK (Ras-MAPK/ERK) signaling pathway.58 SHC interacts with the activated INSR via its PTB domain and recruits growth factor receptor bound 2 – son of sevenless (GRB2-SOS) complexes to the cell membrane where SOS acts as a guanine nucleotide exchange factor (GEF) for Ras, converting GDP-bound Ras into the active GTP-bound form.59, 60, 61, 62 Ras may also be activated through the interaction of the GRB2-SOS complex with IRSs.62 Ras then recruits and activates Raf, which phosphorylates and activates MEK, which in turn phosphorylates and activates MAPK/ERK.63, 64 Activated MAPK translocates to the nucleus where it phosphorylates several transcription factors that regulate genes involved in cell growth, proliferation, differentiation and survival.65 Activated Ras can also feed into the PI3K-AKT signaling pathway through its direct interaction with PI3K p110, independently of p85.66 Multiple negative feedback loops and crosstalk between the PI3K-AKT and RAS-MAPK pathways orchestrate the dynamic and intricate tissue-specific effects of insulin, governing both cell metabolism and proliferation.
How the two isoforms of INSR differentially activate the mitogenic versus the metabolic signaling pathways in cells is under active investigation, especially as many cancer cells overexpress INSR-A. In the pancreatic β cells, insulin upregulates the expression of its own gene through INSR-A and the IRS2-PI3K class Ia-mTORC1-p70S6K signaling pathway,67, 68 whereas the upregulation of the glucokinase (βGK) gene requires the INSR-B signaling through the PI3K class II C2α-PDK1-PKB/AKT signaling pathway.69 Work in MEFs engineered to only express either INSR-A or INSR-B (R- cells), revealed that long-acting insulin analogs (administered to many patients with diabetes) preferentially stimulated cell proliferation and led to higher ERK:AKT phosphorylation ratios through INSR-A.70 Differential activation of the signaling pathways by the two INSR isoforms can, at least in part, be attributed to their distinct localization within the plasma membrane and discrete internalization and recycling dynamics.21, 71
Insulin Signaling and Cancer
INSR is often overexpressed in tumor cells, particularly that of the breast.72, 73, 74, 75, 76 Increased INSR expression in breast tumors is associated with poor survival in patients.74 INSR is also overexpressed and highly phosphorylated in mammary tumors from diabetic mice.77 Preferential expression of the INSR-A isoform has been demonstrated in cancers of the breast, lung, colon, ovaries, endometrium, thyroid and muscle.18, 73, 78, 79, 80, 81, 82 In vitro, INSR-A has been shown to be essential for the growth and survival of many cancer cell lines, including those of the breast.82, 83, 84, 85 Overexpression of INSR-A in NIH 3T3 fibroblasts or in immortalized human breast epithelial cells, induced a ligand-dependent transformed phenotype, which could be reversed with anti-INSR antibodies.86, 87 INSR is also essential for virus-induced transformation of vascular endothelial cells.88 Finally, the knockdown of INSR in MDA-MB-435 breast cancer cell line results in smaller xenograft tumors and fewer pulmonary metastases.89
A pathway initiated by the IGFIR receptor operates in parallel to the INSR, sharing several downstream components. This pathway can be activated by three ligands: insulin, IGFI and IGFII (Figure 3). Hyperinsulinemia has been shown to increase hepatic production and bioavailability of IGFI, in part by inhibiting hepatic production of IGF binding proteins 1 and 2 (IGFBP1 and IGFBP2), which sequester IGFs in the serum.90, 91 This excess IGFI may hyperactivate IGF1R and INSR/IGFIR and their proliferative and anti-apoptotic programs in both premalignant and malignant tissues. Strong evidence for a direct and independent impact of hyperinsulinemia on cancer development came out of studies in the MKR mice.77 These animals express a dominant negative form of IGF1R in the muscle, thus disrupting signaling through the IGF1R receptors and INSR/IGF1R hybrid receptors. This results in severe muscle IR and systemic hyperinsulinemia, without obesity, hyperglycemia or hyperlipidemia, thus allowing an assessment of the impact of high insulin levels on cancer, without the confounding effects of other obesity-related factors. When implanted with xenografts, MKR mice formed larger mammary tumors, with increased lung metastases compared with their non-hyperinsulinemic wild-type counterparts. Chronic treatment of MKR mice with a fast-acting insulin analog (AspB10) results in even bigger tumors and a further increase in INSR but not IGF1R phosphorylation. In this model, tumors display greater INSR but not IGF1R phosphorylation.92, 93 Accordingly, knockdown of INSR, but not IGF1R, in mammary carcinoma Mvt-1 cell line led to the considerably smaller xenografted tumors, both in wild-type and hypersinsulinemic mice.94 Similarly, in breast cancer patients, increased INSR but not IGFIR expression and higher phosphorylation of INSR/IGFIR hybrid receptors correlate with poor survival.74
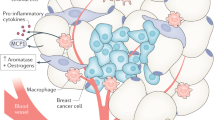
In addition to the possible direct oncogenic effects of insulin on the proliferative and anti-apoptotic signaling in cancer cells, insulin is also implicated in various aspects of the maintenance of whole-body homeostasis, including the action of sex hormones, which may contribute to carcinogenesis. For over 100 years, sex hormones have been known to affect cancer development and progression.95 Hyperinsulinemia inhibits hepatic production and secretion of the sex hormone-binding globulin (SHBG; Figure 3).96, 97 A reduction in serum SHBG results in increased bioavailability of estradiol (in men and in women) and testosterone (in women only).98, 99 High insulin levels can also increase ovarian and adrenal androgen production.100 Moreover, adipocytes express a number of sex steroid stabilizing enzymes that convert androgenic precursors into estrogens, thus AT can act as a sink or a source of lipid-soluble sex hormones.101 Remarkably, AT is the main site of estrogen production in men and postmenopausal women.102, 103 Considering that >70% of all breast cancers express the estrogen receptor (ER) and that activation of this receptor initiates a proliferative and anti-apoptotic transcription program, in such tumors, deregulated insulin signaling and its effects on AT may have considerable impact on prognosis and outcome.99, 104
Hyperinsulinemia Drugs as Cancer Therapeutics
Metformin is the most commonly used drug for treatment of type 2 diabetes (T2D). It is cheap, widely available and well tolerated. Growing preclinical, clinical and epidemiologic evidence suggests that metformin may also prove to be a valuable drug for cancer therapy. Patients with diabetes that are treated with metformin have a lower incidence and mortality of breast, pancreatic, hepatocellular and colorectal cancer.105, 106, 107, 108, 109, 110 Metformin may impact tumor cells via direct and indirect mechanisms, both involving the activation of 5′-AMP-activated protein kinase (AMPK), the major cellular sensor of energy stress. Metformin is transported into cells via the organic cation transporter 1 (OCT1), where it accumulates in the mitochondria and inhibits the complex 1 of the mitochondrial respiratory chain.111, 112 Inhibition of mitochondrial ATP synthesis by metformin leads to a rise in intracellular AMP, which binds AMPK and facilitates its phosphorylation by the liver kinase B1 (LKB1) on threonine 172, thus activating the protein.113, 114 In the liver, AMPK-mediated suppression of gluconeogenesis results in a decrease in the levels of fasting blood glucose, leading to a reduction in circulating insulin levels and resensitization of insulin target tissues to the action of insulin.115, 116
A decrease in circulating insulin levels may also result in the downregulation of INSR signaling pathways in cancers expressing INSR and attenuation of the proliferative and anti-apoptotic signals117, 118 Supporting such a possibility, non-diabetic breast cancer patients given metformin for 6 months displayed an average 22% reduction in insulin levels.119 Moreover, a 2-week administration of metformin in between diagnosis and surgery led to a reduction in circulating insulin levels, a decrease in INSR expression and downregulation of AKT and MAPK signaling pathways in their tumors.117
Metformin may also have direct effects on tumor cells, involving AMPK-mediated stabilization of TSC2 and concomitant inhibition of mTORC1. This results in the downregulation of p70S6K and 4EBP1 activities and inhibition of protein synthesis and cell proliferation.120, 121, 122, 123 Limited evidence also suggests that there may be other, AMPK independent, direct effects of metformin on cancer cells.124 In vitro data on metformin action should be interpreted with caution though, as the concentrations of the drug used in such studies far exceed tolerable doses achievable in humans.125
To further probe the utility of metformin in cancer therapy, the NCIC Clinical Trials Group, as part of the North American Breast Cancer Group, is leading an ongoing phase III, randomized and placebo-controlled clinical trial (MA.32).115 The effect of metformin on disease-free survival and other outcomes is monitored in 3649 non-diabetic women with early-stage breast cancer receiving metformin versus placebo for 5 years. Analyses of key metabolic parameters after 6 months of treatment showed that women on metformin have a reduction in body weight (−3.0%), glucose (−3.8%), insulin (−11.1%), IR (homeostasis model assessment; −17.1%) and leptin (−20.2%). Furthermore, there was a significant reduction in the levels of highly sensitive C-reactive protein (−6.7%), a marker of chronic inflammation. Further analyses in this cohort are aimed at identifying the link(s) between metformin’s metabolic and anti-inflammatory impact on cancer-free survival.
Sources of IR in Obesity
A series of confounding factors and a complex interplay between many tissues contributes to the development of IR and hyperinsulinemia in obesity. The mechanisms have not been fully elucidated, but the major factors center on the changes in AT biology, disruption of the normal endocrine function of adipocytes, as well as deregulated lipolysis and overproduction of free fatty acids (FFAs; Figure 4).

The sources of IR in obesity
AT is the body’s main energy reserve. There are two major types of AT, white AT (WAT) and brown AT (BAT). BAT is primarily responsible for heat production in infants, although small BAT depots have recently also been identified in adults.126 The majority of body fat is stored in WAT, where adipocytes store energy in the form of triglycerides (TAGs). When there is energetic demand, TAGs are broken down into glycerol and FFAs through the process of lipolysis. Adipocytes then release glycerol and FFAs into the blood stream for transporting to target tissues, mainly the liver and the muscle. Oxidative metabolism in these tissues breaks down the FFAs to produce ATP, whereas gluconeogenesis in the liver converts glycerol into glucose.
Fat storage is not the only function of AT, as adipocytes are also secretory cells. They produce a number of hormones and cytokines, now termed adipokines, which influence AT biology, as well as cellular metabolism and function in the brain, liver, muscle, vasculature, reproductive organs and β cells of the pancreas.127 This function of adipocytes came to light when leptin was discovered as the gene mutated in obese ob/ob mice.128 Leptin is a peptide hormone, produced primarily by the adipocytes, which opposes many actions of insulin. It acts in the hypothalamus to repress appetite, and in the liver and AT to stimulate lipolysis and inhibit lipogenesis through activation of AMPK.129, 130 When administered to ob/ob mice or human patients with a mutation in the leptin gene, leptin lowers blood glucose levels and resensitizes cells to insulin.131, 132 Obesity in humans often leads to high leptin levels and leptin resistance.133, 134 Adiponectin is another hormone product of adipocytes.135 In the muscle and liver, adiponectin binding to its receptor activates two signaling pathways, involving LKB1 and the Ca2+/calmodulin-dependent protein kinase kinase (CaMKK), which converge on AMPK activation, which then induces glucose uptake and lipolysis, inhibits gluconeogenesis and promotes FFA oxidation.136, 137 Adiponectin also sensitizes cells to the action of insulin by inducing transcription of IRS and GLUT4, as well as translocation of GLUT4 to the membrane.138, 139, 140, 141 Paradoxically, adiponectin levels are low in patients with obesity and/or T2D, which likely contributes to the development of IR.142, 143 A perturbed balance in leptin and adiponectin levels is a hallmark of obesity and IR.
Adipocytes also produce pro-inflammatory cytokines, such as the tumor necrosis factor alpha (TNFα), monocyte chemoattractant protein 1 (MCP-1), interleukins 1β and 6 (IL1β and IL6) and others.144, 145, 146 TNFα, like leptin, stimulates lipolysis and inhibits lipogenesis in AT.147 Enlarged adipocytes from the obese rodents and humans overexpress TNFα, which is thought to have a major role in the development of IR.148, 149 In the muscle, TNFα leads to activation of the Jun N-terminal kinase 1 (JNK1) and mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) pathway, which inhibits GLUT4 translocation to the membrane and induces inhibitory serine (S270, S307 and S636/639) phosphorylation of IRSs, thereby inhibiting INSR signaling.150, 151, 152, 153, 154, 155 In adipocytes, TNFα inhibits IRS and GLUT4 expression.156 Highlighting the physiological relevance of these relationships, knockout of TNFα or MCP-1 in high-fat diet-fed mice, or knockdown of MAP4K4 in the isolated muscles from patients with diabetes, ameliorates IR.149, 150, 157 TNFα also impedes expression and protein stability of the nuclear hormone receptor peroxisome proliferator-activated receptor gamma (PPARγ).158, 159 This transcription factor regulates expression of genes involved in lipogenesis and lipid sequestration in adipocytes and thus a reduction in its expression leads to overproduction of FFAs. PPARγ-activating thiazolidinediones or the activation of PPARγ by deletion of NCoR, corepressors of transcription, can block sensing of cytokines and improve insulin sensitivity in adipocytes.160, 161
MCP-1, which is also overexpressed in AT during obesity, acts as a chemoattractant for macrophages and other immune cells that infiltrate AT, leading to the development of inflammation, another hallmark of obesity and IR.146, 162, 163 AT of individuals with obesity consists of up to 50% of macrophages, whereas they only make up 5–10% of AT cells in lean subjects.164 Macrophages locally secret TNFα, a process that requires signaling by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and JNK-MAP4K4-AP1 (activator protein 1) pathways.165, 166, 167 Indicative of the importance of TNFα in AT biology, knockout of JNK1 in macrophages partially protects from high-fat diet-induced IR in mice.167 Consistent with such a notion, anti-inflammatory drugs like the salsalate, an NF-kB inhibitor, have shown some efficacy in improving IR in patients with obesity and T2D.168, 169, 170
Rapid expansion of AT mass and enlarged adipocyte size in obesity impact oxygen delivery and can create hypoxic stress, with further effects on the development of IR. In adipocytes in culture, hypoxia leads to reduced INSR and IRS1 tyrosine phosphorylation, directly affecting their ability to sense insulin.171 Hypoxia has also been found to affect the glucose transport machinery via the reduction in AS160 phosphorylation and eventually lowered GLUT4 expression.171 Finally, low O2 pressure in adipocytes stabilizes the O2-sensitive transcription factor hypoxia-inducible factor 1 alpha (HIF1α), the master regulator of cellular response to hypoxia.172 HIF1α, in concert with HIF1β, induces expression of TNFα, IL6 and leptin, whereas repressing expression of adiponectin, further driving IR development.173, 174
Another major hallmark of AT expansion is an increase in circulating FFA levels, because of elevated lipolysis, and their uptake by the muscle, liver and pancreatic cells. Excessive FFA accumulation in these cells results in lipotoxicity and development of IR.175 There are several hypotheses as to how excess FFAs lead to IR. One suggests that an increase in diacyglycerol levels in the muscle, likely due to a backlog in the FFA reesterification pathway, leads to activation of protein kinase C theta, beta2 and delta (PKCθ,β2,δ).176, 177, 178, 179 These novel PKCs phosphorylate INSR and IRSs on inhibitory serine residues thereby reducing their activity. Another hypothesis suggests that FFAs induce the pro-inflammatory response through activation of NF-kB signaling and production of pro-inflammatory cytokines like TNFα, MCP-1, IL6 and IL1β.180 The development of IR in response to FFAs is thus a complex process, involving deactivation of INSR signaling pathway components and induction of inflammation and cellular stress.
Insufficiency in AT, as seen in the cases of lipodystrophy, can also lead to IR. As AT is a storage depot for fat, physiologically low AT levels lead to elevated circulating concentrations of TAG and FFAs and development of IR.181, 182 Healthy AT is also required for proper secretion and the physiological balance of adipokines like adopinectin and leptin, which sensitize cells to insulin.183, 184 Leptin replacement therapy is now an approved treatment for patients with lipodystrophy, and significantly attenuates IR in those patients.185 Functional AT in proportion to body size is thus essential for normal insulin sensitivity and whole-body metabolism.
Outlook and Treatment Options
Special considerations have to be made for the prevention, detection and treatment of cancers in patients with obesity and T2D. In 2007, the World Cancer Research Fund/American Institute for Cancer Research Second Expert Report provided the guidelines for cancer prevention, with the main recommendation being ‘maintaining a healthy weight throughout life’.6 Thus, patients should be encouraged to lose weight and undergo a lifestyle change, incorporating exercise and healthy diet into their daily lives.
There are several challenges in cancer detection in the overweight/obese patients. Women with obesity and T2D are less likely to use the preventative services for cancer screening.186, 187 Among over 700 000 Canadian women, those with diabetes were 32% less likely to receive routine mammography screening.186 This highlights a need for better patient education and organization of primary healthcare to ensure early cancer detection in this high-risk group. Obesity is also linked to a reduction in tumor biomarkers, like the carcinoembryonic antigen in colorectal cancer.188 The ability to detect cancer in individuals with obesity is further complicated by the restrictions in the weight and the diameter of most imaging modalities, as well as poorer quality of the collected images.189 A considerable challenge in the treatment of cancer in these patients is the ability to achieve proper chemotherapy dosing. In a study of over 9000 women with breast cancer, reduced chemotherapy dosing was found in up to 40% of patients with obesity.190 Furthermore, patients with T2D and obesity may present for cancer treatment with pre-existing renal, cardiovascular or neurological complications, conditions that can be further exacerbated by the chemotherapeutic agents. The use of glucocorticoids should be carefully considered, as they are known to cause IR, decrease insulin production and secretion, as well as increase gluconeogenesis and glycogenolysis.191 Inflammation has long been associated with neoplastic transformation and the inflammatory cytokines are known to activate oncogenic signaling in cells. Thus, targeting the obesity-associated inflammation may reduce the development of IR while decreasing the oncogenic input from the cytokines in the tumor microenvironment, together inhibiting cancer development.192
The ability to treat cancers in patients with obesity and T2D will hinge on the development of effective drugs targeting the molecular pathways that link obesity and T2D to cancer. For example, targeting INSR-A or the PI3K-AKT and/or RAS-MAPK pathways that are downstream of INSR-A represents such an option. Moreover, supplementing the standard of care treatments with metformin or other insulin-lowering drugs may proof to be useful in lowering insulin levels and IR, decreasing body weight and improving cancer outcomes in patients with obesity and T2D.
Abbreviations
- T2D:
-
type 2 diabetes
- BMI:
-
body mass index
- AT:
-
adipose tissue
- IR:
-
insulin resistance
- INSR:
-
insulin receptor
- INSR-B:
-
insulin receptor B isoform
- INSR-A:
-
insulin receptor A isoform
- IGFII:
-
insulin-like growth factor II
- IGF1R:
-
insulin-like growth factor receptor 1
- IRSs:
-
insulin receptor substrates
- SH2:
-
Src homology 2
- PTB:
-
phosphotyrosine-binding
- p85:
-
p85 regulatory subunit of PI3K
- PI3K:
-
phosphoinositide 3-kinase
- p110:
-
p110 catalytic subunit of PI3K
- PIP2:
-
phosphatidylinositol 4,5-bisphosphate
- PIP3:
-
phosphatidylinositol 3,4,5-triphosphate
- PH:
-
pleckstrin homology
- PKB/AKT:
-
protein kinase B
- PDK1:
-
3-phosphoinositide-dependent protein kinase 1
- mTORC2:
-
mammalian target of rapamycin complex 2
- AS160:
-
AKT substrate of 160 kDa
- GLUT4:
-
glucose transporter 4
- GSK3:
-
glycogen synthase kinase 3
- CREB:
-
cAMP-response-element-binding
- FOXO:
-
forkhead box O
- GAP:
-
GTPase activator protein
- TSC2:
-
tuberous sclerosis complex 2
- Rheb:
-
Ras homolog enriched in brain
- mTOCR1:
-
mammalian target of rapamycin complex 1
- SREBP1c:
-
sterol regulatory element-binding protein 1c
- p70S6K:
-
S6 kinase
- 4EBPs:
-
4E binding protein
- BAD:
-
BCL2-associated agonist of cell death
- SHC:
-
Src homology 2 domain-containing
- Ras:
-
rat sarcoma
- MAPK/ERK:
-
mitogen-activated protein kinase
- GRB2:
-
growth factor receptor bound 2
- SOS:
-
son of sevenless
- GEF:
-
guanine nucleotide exchange factor
- GK:
-
glucokinase
- IGFBP:
-
IGF binding protein
- AspB10:
-
fast-acting insulin analog
- SHBG:
-
sex hormone-binding globulin
- ER:
-
estrogen receptor
- AMPK:
-
5′-AMP-activated protein kinase
- OCT1:
-
organic cation transporter 1
- LKB1:
-
liver kinase B1
- FFAs:
-
free fatty acids
- WAT:
-
white adipose tissue
- BAT:
-
brown adipose tissue
- TAGs:
-
triglycerides
- ATP:
-
adenosine triphosphate
- CaMKK:
-
Ca2+/calmodulin-dependent protein kinase kinase
- MCP-1:
-
monocyte chemoattractant protein 1
- IL:
-
interleukin
- JNK1:
-
Jun N-terminal kinase
- MAP4K4:
-
mitogen-activated protein kinase kinase kinase kinase 4
- PPARγ:
-
proliferator-activated receptor gamma
- NF-kB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- AP1:
-
activator protein 1
- HIF1α:
-
hypoxia-inducible factor 1 alpha
- PKCθ:
-
protein kinase C theta
- PKCβ2:
-
PKC beta 2
- PKCδ:
-
PKC delta.
References
World Health Organization. Obesity and Overweight: Fact Sheet N 311 2015.
Stewart ST, Cutler DM, Rosen AB . Forecasting the effects of obesity and smoking on U.S. life expectancy. N Engl J Med 2009; 361: 2252–2260.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ . Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003; 348: 1625–1638.
Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol 2002; 20: 42–51.
Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M . Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 2008; 371: 569–578.
World Cancer Research Fund, American Institute for Cancer Research Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective 2007.
International Agency for Research on Cancer IARC Handbooks of Cancer Prevention: Weight Control and Physical Activity 2002.
Arnold M, Pandeya N, Byrnes G, Renehan AG, Stevens GA, Ezzati M et al. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol 2015; 16: 36–46.
Byers T, Sedjo RL . Does intentional weight loss reduce cancer risk? Diabetes Obes Metab 2011; 13: 1063–1072.
Eliassen AH, Colditz GA, Rosner B, Willett WC, Hankinson SE . Adult weight change and risk of postmenopausal breast cancer. JAMA 2006; 296: 193–201.
Sjostrom L, Gummesson A, Sjostrom CD, Narbro K, Peltonen M, Wedel H et al. Effects of bariatric surgery on cancer incidence in obese patients in Sweden (Swedish Obese Subjects Study): a prospective, controlled intervention trial. Lancet Oncol 2009; 10: 653–662.
Tomasetti C, Vogelstein B . Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015; 347: 78–81.
Kasuga M, Hedo JA, Yamada KM, Kahn CR . The structure of insulin receptor and its subunits. Evidence for multiple nonreduced forms and a 210,000 possible proreceptor. J Biol Chem 1982; 257: 10392–10399.
Ebina Y, Edery M, Ellis L, Standring D, Beaudoin J, Roth RA et al. Expression of a functional human insulin receptor from a cloned cDNA in Chinese hamster ovary cells. Proc Natl Acad Sci USA 1985; 82: 8014–8018.
Seino S, Seino M, Nishi S, Bell GI . Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci USA 1989; 86: 114–118.
Ullrich A, Bell JR, Chen EY, Herrera R, Petruzzelli LM, Dull TJ et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985; 313: 756–761.
Malaguarnera R, Sacco A, Voci C, Pandini G, Vigneri R, Belfiore A . Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway. Endocrinology 2012; 153: 2152–2163.
Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene 1999; 18: 2471–2479.
Mosthaf L, Grako K, Dull TJ, Coussens L, Ullrich A, McClain DA . Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J 1990; 9: 2409–2413.
Vogt B, Carrascosa JM, Ermel B, Ullrich A, Haring HU . The two isotypes of the human insulin receptor (HIR-A and HIR-B) follow different internalization kinetics. Biochem Biophys Res Commun 1991; 177: 1013–1018.
Yamaguchi Y, Flier JS, Yokota A, Benecke H, Backer JM, Moller DE . Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology 1991; 129: 2058–2066.
Pandini G, Medico E, Conte E, Sciacca L, Vigneri R, Belfiore A . Differential gene expression induced by insulin and insulin-like growth factor-II through the insulin receptor isoform A. J Biol Chem 2003; 278: 42178–42189.
Sciacca L, Prisco M, Wu A, Belfiore A, Vigneri R, Baserga R . Signaling differences from the A and B isoforms of the insulin receptor (IR) in 32D cells in the presence or absence of IR substrate-1. Endocrinology 2003; 144: 2650–2658.
Wang Y, Hua S, Tian W, Zhang L, Zhao J, Zhang H et al. Mitogenic and anti-apoptotic effects of insulin in endometrial cancer are phosphatidylinositol 3-kinase/Akt dependent. Gynecol Oncol 2012; 125: 734–741.
Dodge JA, Laurence KM . Congenital absence of islets of Langerhans. Arch Dis Child 1977; 52: 411–413.
Fosel S . Transient and permanent neonatal diabetes. Eur J Pediatr 1995; 154: 944–948.
Hill DE . Effect of insulin on fetal growth. Semin Perinatol 1978; 2: 319–328.
Taylor SI, Cama A, Accili D, Barbetti F, Quon MJ, de la Luz Sierra M et al. Mutations in the insulin receptor gene. Endocr Rev 1992; 13: 566–595.
Escribano O, Guillen C, Nevado C, Gomez-Hernandez A, Kahn CR, Benito M . Beta-cell hyperplasia induced by hepatic insulin resistance: role of a liver-pancreas endocrine axis through insulin receptor A isoform. Diabetes 2009; 58: 820–828.
Savkur RS, Philips AV, Cooper TA . Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 2001; 29: 40–47.
Louvi A, Accili D, Efstratiadis A . Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev Biol 1997; 189: 33–48.
Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991; 352: 73–77.
Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG Jr., Glasheen E et al. Role of IRS-2 in insulin and cytokine signalling. Nature 1995; 377: 173–177.
Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM . Regulation of the p85/p110 phosphatidylinositol 3'-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol 1998; 18: 1379–1387.
Hawkins PT, Jackson TR, Stephens LR . Platelet-derived growth factor stimulates synthesis of PtdIns(3,4,5)P3 by activating a PtdIns(4,5)P2 3-OH kinase. Nature 1992; 358: 157–159.
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997; 7: 261–269.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM . Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307: 1098–1101.
Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 2003; 278: 14599–14602.
Deprez J, Vertommen D, Alessi DR, Hue L, Rider MH . Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signaling cascades. J Biol Chem 1997; 272: 17269–17275.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA . Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995; 378: 785–789.
Embi N, Rylatt DB, Cohen P . Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 1980; 107: 519–527.
He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009; 137: 635–646.
Nakae J, Park BC, Accili D . Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem 1999; 274: 15982–15985.
Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003; 423: 550–555.
Inoki K, Li Y, Zhu T, Wu J, Guan KL . TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4: 648–657.
Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J . Rheb binds and regulates the mTOR kinase. Curr Biol 2005; 15: 702–713.
Manning BD, Cantley LC . Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 2003; 28: 573–576.
Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D et al. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem 1994; 269: 16333–16339.
Kim JB, Spiegelman BM . ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 1996; 10: 1096–1107.
Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 2008; 8: 224–236.
Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ et al. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 1997; 277: 99–101.
Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE . Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 1992; 257: 973–977.
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96: 857–868.
Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP . FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem 2003; 278: 35959–35967.
Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ et al. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol 2002; 22: 2025–2036.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91: 231–241.
Diehl JA, Cheng M, Roussel MF, Sherr CJ . Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12: 3499–3511.
Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G et al. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992; 70: 93–104.
Pronk GJ, McGlade J, Pelicci G, Pawson T, Bos JL . Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem 1993; 268: 5748–5753.
Sasaoka T, Draznin B, Leitner JW, Langlois WJ, Olefsky JM . Shc is the predominant signaling molecule coupling insulin receptors to activation of guanine nucleotide releasing factor and p21ras-GTP formation. J Biol Chem 1994; 269: 10734–10738.
Sasaoka T, Rose DW, Jhun BH, Saltiel AR, Draznin B, Olefsky JM . Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem 1994; 269: 13689–13694.
Skolnik EY, Batzer A, Li N, Lee CH, Lowenstein E, Mohammadi M et al. The function of GRB2 in linking the insulin receptor to Ras signaling pathways. Science 1993; 260: 1953–1955.
Moodie SA, Willumsen BM, Weber MJ, Wolfman A . Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993; 260: 1658–1661.
Warne PH, Viciana PR, Downward J . Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993; 364: 352–355.
Dhillon AS, Hagan S, Rath O, Kolch W . MAP kinase signalling pathways in cancer. Oncogene 2007; 26: 3279–3290.
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994; 370: 527–532.
Leibiger B, Leibiger IB, Moede T, Kemper S, Kulkarni RN, Kahn CR et al. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol Cell 2001; 7: 559–570.
Leibiger IB, Leibiger B, Moede T, Berggren PO . Exocytosis of insulin promotes insulin gene transcription via the insulin receptor/PI-3 kinase/p70 s6 kinase and CaM kinase pathways. Mol Cell 1998; 1: 933–938.
Uhles S, Moede T, Leibiger B, Berggren PO, Leibiger IB . Selective gene activation by spatial segregation of insulin receptor B signaling. FASEB J 2007; 21: 1609–1621.
Sciacca L, Cassarino MF, Genua M, Pandini G, Le Moli R, Squatrito S et al. Insulin analogues differently activate insulin receptor isoforms and post-receptor signalling. Diabetologia 2010; 53: 1743–1753.
Uhles S, Moede T, Leibiger B, Berggren PO, Leibiger IB . Isoform-specific insulin receptor signaling involves different plasma membrane domains. J Cell Biol 2003; 163: 1327–1337.
Cox ME, Gleave ME, Zakikhani M, Bell RH, Piura E, Vickers E et al. Insulin receptor expression by human prostate cancers. Prostate 2009; 69: 33–40.
Kalli KR, Falowo OI, Bale LK, Zschunke MA, Roche PC, Conover CA . Functional insulin receptors on human epithelial ovarian carcinoma cells: implications for IGF-II mitogenic signaling. Endocrinology 2002; 143: 3259–3267.
Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res 2008; 68: 10238–10246.
Mulligan AM, O'Malley FP, Ennis M, Fantus IG, Goodwin PJ . Insulin receptor is an independent predictor of a favorable outcome in early stage breast cancer. Breast Cancer Res Treat 2007; 106: 39–47.
Papa V, Pezzino V, Costantino A, Belfiore A, Giuffrida D, Frittitta L et al. Elevated insulin receptor content in human breast cancer. J Clin Invest 1990; 86: 1503–1510.
Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res 2010; 70: 741–751.
Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol 1999; 19: 3278–3288.
Moller DE, Yokota A, Caro JF, Flier JS . Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol 1989; 3: 1263–1269.
Sciacca L, Mineo R, Pandini G, Murabito A, Vigneri R, Belfiore A . In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene 2002; 21: 8240–8250.
Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. J Clin Endocrinol Metabol 2002; 87: 245–254.
Wang CF, Zhang G, Zhao LJ, Qi WJ, Li XP, Wang JL et al. Overexpression of the insulin receptor isoform A promotes endometrial carcinoma cell growth. PloS One 2013; 8: e69001.
Frisch CM, Zimmermann K, Zillessen P, Pfeifer A, Racke K, Mayer P . Non-small cell lung cancer cell survival crucially depends on functional insulin receptors. Endocrine-Related Cancer 2015; 22: 609–621.
Milazzo G, Giorgino F, Damante G, Sung C, Stampfer MR, Vigneri R et al. Insulin receptor expression and function in human breast cancer cell lines. Cancer Res 1992; 52: 3924–3930.
Osborne CK, Monaco ME, Lippman ME, Kahn CR . Correlation among insulin binding, degradation, and biological activity in human breast cancer cells in long-term tissue culture. Cancer Res 1978; 38: 94–102.
Frittitta L, Vigneri R, Stampfer MR, Goldfine ID . Insulin receptor overexpression in 184B5 human mammary epithelial cells induces a ligand-dependent transformed phenotype. J Cell Biochem 1995; 57: 666–669.
Giorgino F, Belfiore A, Milazzo G, Costantino A, Maddux B, Whittaker J et al. Overexpression of insulin receptors in fibroblast and ovary cells induces a ligand-mediated transformed phenotype. Mol Endocrinol 1991; 5: 452–459.
Rose PP, Carroll JM, Carroll PA, DeFilippis VR, Lagunoff M, Moses AV et al. The insulin receptor is essential for virus-induced tumorigenesis of Kaposi's sarcoma. Oncogene 2007; 26: 1995–2005.
Zhang H, Fagan DH, Zeng X, Freeman KT, Sachdev D, Yee D . Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene 2010; 29: 2517–2527.
Clemmons DR, Underwood LE . Nutritional regulation of IGF-I and IGF binding proteins. Ann Rev Nutr 1991; 11: 393–412.
Leung KC, Doyle N, Ballesteros M, Waters MJ, Ho KK . Insulin regulation of human hepatic growth hormone receptors: divergent effects on biosynthesis and surface translocation. J Clin Endocrinol Metabol 2000; 85: 4712–4720.
Ferguson RD, Novosyadlyy R, Fierz Y, Alikhani N, Sun H, Yakar S et al. Hyperinsulinemia enhances c-Myc-mediated mammary tumor development and advances metastatic progression to the lung in a mouse model of type 2 diabetes. Breast Cancer Res 2012; 14: R8.
Gallagher EJ, Alikhani N, Tobin-Hess A, Blank J, Buffin NJ, Zelenko Z et al. Insulin receptor phosphorylation by endogenous insulin or the insulin analog AspB10 promotes mammary tumor growth independent of the IGF-I receptor. Diabetes 2013; 62: 3553–3560.
Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, Dakwar M et al. Highly specific role of the insulin receptor in breast cancer progression. Endocrine-Related Cancer 2015; 22: 145–157.
Jensen EV, Jacobson HI, Walf AA, Frye CA . Estrogen action: a historic perspective on the implications of considering alternative approaches. Physiol Behav 2010; 99: 151–162.
Plymate SR, Matej LA, Jones RE, Friedl KE . Inhibition of sex hormone-binding globulin production in the human hepatoma (Hep G2) cell line by insulin and prolactin. J Clin Endocrinol Metabol 1988; 67: 460–464.
Weaver JU, Holly JM, Kopelman PG, Noonan K, Giadom CG, White N et al. Decreased sex hormone binding globulin (SHBG) and insulin-like growth factor binding protein (IGFBP-1) in extreme obesity. Clin Endocrinol 1990; 33: 415–422.
Evans DJ, Hoffmann RG, Kalkhoff RK, Kissebah AH . Relationship of androgenic activity to body fat topography, fat cell morphology, and metabolic aberrations in premenopausal women. J Clin Endocrinol Metabol 1983; 57: 304–310.
Key TJ, Appleby PN, Reeves GK, Roddam A, Dorgan JF, Longcope C et al. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J Natl Cancer Inst 2003; 95: 1218–1226.
Moghetti P, Castello R, Negri C, Tosi F, Spiazzi GG, Brun E et al. Insulin infusion amplifies 17 alpha-hydroxycorticosteroid intermediates response to adrenocorticotropin in hyperandrogenic women: apparent relative impairment of 17,20-lyase activity. J Clin Endocrin Metab 1996; 81: 881–886.
Belanger C, Luu-The V, Dupont P, Tchernof A . Adipose tissue intracrinology: potential importance of local androgen/estrogen metabolism in the regulation of adiposity. Horm Metab Res 2002; 34: 737–745.
Grodin JM, Siiteri PK, MacDonald PC . Source of estrogen production in postmenopausal women. J Clin Endocrinol Metab 1973; 36: 207–214.
Schindler AE, Ebert A, Friedrich E . Conversion of androstenedione to estrone by human tissue. J Clin Endocrinol Metab 1972; 35: 627–630.
Henderson BE, Ross RK, Pike MC, Casagrande JT . Endogenous hormones as a major factor in human cancer. Cancer Res 1982; 42: 3232–3239.
Bosco JL, Antonsen S, Sorensen HT, Pedersen L, Lash TL . Metformin and incident breast cancer among diabetic women: a population-based case-control study in Denmark. Cancer Epidemiol Biomarkers Prev 2011; 20: 101–111.
Choi Y, Kim TY, Oh DY, Lee KH, Han SW, Im SA et al. The impact of diabetes mellitus and metformin treatment on survival of patients with advanced pancreatic cancer undergoing chemotherapy. Cancer Res Treat 2015 [e-pub ahead of print].
Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res 2010; 3: 1451–1461.
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD . Metformin and reduced risk of cancer in diabetic patients. BMJ 2005; 330: 1304–1305.
Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA et al. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat 2011; 126: 215–220.
Vissers PA, Cardwell CR, van de Poll-Franse LV, Young IS, Pouwer F, Murray LJ . The association between glucose-lowering drug use and mortality among breast cancer patients with type 2 diabetes. Breast Cancer Res Treat 2015; 150: 427–437.
El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X . Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000; 275: 223–228.
Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest 2007; 117: 1422–1431.
Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 1996; 271: 27879–27887.
Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005; 310: 1642–1646.
Goodwin PJ, Parulekar WR, Gelmon KA, Shepherd LE, Ligibel JA, Hershman DL et al. Effect of metformin vs placebo on and metabolic factors in NCIC CTG MA.32. J Natl Cancer Inst 2015; 107: djv006.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108: 1167–1174.
Dowling RJ, Niraula S, Chang MC, Done SJ, Ennis M, McCready DR et al. Changes in insulin receptor signaling underlie neoadjuvant metformin administration in breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res 2015; 17: 32.
Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA . Metformin prevents tobacco carcinogen—induced lung tumorigenesis. Cancer Prev Res 2010; 3: 1066–1076.
Goodwin PJ, Pritchard KI, Ennis M, Clemons M, Graham M, Fantus IG . Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer 2008; 8: 501–505.
Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N . Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 2007; 67: 10804–10812.
Mohammed A, Janakiram NB, Brewer M, Ritchie RL, Marya A, Lightfoot S et al. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl Oncol 2013; 6: 649–659.
Zakikhani M, Blouin MJ, Piura E, Pollak MN . Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat 2010; 123: 271–279.
Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M . Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 2006; 66: 10269–10273.
Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 2010; 11: 390–401.
Stambolic V, Woodgett JR, Fantus IG, Pritchard KI, Goodwin PJ . Utility of metformin in breast cancer treatment, is neoangiogenesis a risk factor? Breast Cancer Res Treat 2009; 114: 387–389.
Cohade C, Osman M, Pannu HK, Wahl RL . Uptake in supraclavicular area fat ("USA-Fat"): description on 18F-FDG PET/CT. J Nucl Med 2003; 44: 170–176.
Bluher M, Mantzoros CS . From leptin to other adipokines in health and disease: facts and expectations at the beginning of the 21st century. Metabolism 2015; 64: 131–145.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM . Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432.
Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P . Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science 1995; 269: 546–549.
Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002; 415: 339–343.
Andreev VP, Paz-Filho G, Wong ML, Licinio J . Deconvolution of insulin secretion, insulin hepatic extraction post-hepatic delivery rates and sensitivity during 24- h standardized meals: time course of glucose homeostasis in leptin replacement treatment. Horm Metab Res 2009; 41: 142–151.
Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995; 269: 543–546.
Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1995; 1: 1155–1161.
Myers MG Jr., Leibel RL, Seeley RJ, Schwartz MW . Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metabol 2010; 21: 643–651.
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF . A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 1995; 270: 26746–26749.
Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L . Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest 2001; 108: 1875–1881.
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002; 8: 1288–1295.
Awazawa M, Ueki K, Inabe K, Yamauchi T, Kubota N, Kaneko K et al. Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway. Cell Metab 2011; 13: 401–412.
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE . The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 2001; 7: 947–953.
Fu Y, Luo N, Klein RL, Garvey WT . Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J Lipid Res 2005; 46: 1369–1379.
Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 2001; 7: 941–946.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999; 257: 79–83.
Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 2000; 20: 1595–1599.
Hotamisligil GS, Shargill NS, Spiegelman BM . Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993; 259: 87–91.
Juge-Aubry CE, Somm E, Giusti V, Pernin A, Chicheportiche R, Verdumo C et al. Adipose tissue is a major source of interleukin-1 receptor antagonist: upregulation in obesity and inflammation. Diabetes 2003; 52: 1104–1110.
Sartipy P, Loskutoff DJ . Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA 2003; 100: 7265–7270.
Torti FM, Dieckmann B, Beutler B, Cerami A, Ringold GM . A macrophage factor inhibits adipocyte gene expression: an in vitro model of cachexia. Science 1985; 229: 867–869.
Bullo M, Garcia-Lorda P, Peinado-Onsurbe J, Hernandez M, Del Castillo D, Argiles JM et al. TNFalpha expression of subcutaneous adipose tissue in obese and morbid obese females: relationship to adipocyte LPL activity and leptin synthesis. Int J Obes Relat Metab Disord 2002; 26: 652–658.
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS . Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997; 389: 610–614.
Bouzakri K, Zierath JR . MAP4K4 gene silencing in human skeletal muscle prevents tumor necrosis factor-alpha-induced insulin resistance. J Biol Chem 2007; 282: 7783–7789.
Hotamisligil GS, Johnson RS, Distel RJ, Ellis R, Papaioannou VE, Spiegelman BM . Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science 1996; 274: 1377–1379.
Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM . IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996; 271: 665–668.
Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE et al. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc Natl Acad Sci USA 2001; 98: 4640–4645.
Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A et al. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 2001; 107: 181–189.
Zhang J, Gao Z, Yin J, Quon MJ, Ye J . S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem 2008; 283: 35375–35382.
Stephens JM, Lee J, Pilch PF . Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem 1997; 272: 971–976.
Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006; 116: 1494–1505.
Tang X, Guilherme A, Chakladar A, Powelka AM, Konda S, Virbasius JV et al. An RNA interference-based screen identifies MAP4K4/NIK as a negative regulator of PPARgamma, adipogenesis, and insulin-responsive hexose transport. Proc Natl Acad Sci USA 2006; 103: 2087–2092.
Zhang B, Berger J, Hu E, Szalkowski D, White-Carrington S, Spiegelman BM et al. Negative regulation of peroxisome proliferator-activated receptor-gamma gene expression contributes to the antiadipogenic effects of tumor necrosis factor-alpha. Mol Endocrinol 1996; 10: 1457–1466.
Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 2011; 147: 815–826.
Oakes ND, Kennedy CJ, Jenkins AB, Laybutt DR, Chisholm DJ, Kraegen EW . A new antidiabetic agent, BRL 49653, reduces lipid availability and improves insulin action and glucoregulation in the rat. Diabetes 1994; 43: 1203–1210.
Curat CA, Miranville A, Sengenes C, Diehl M, Tonus C, Busse R et al. From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes 2004; 53: 1285–1292.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003; 112: 1821–1830.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr . Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796–1808.
Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005; 11: 191–198.
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K et al. A central role for JNK in obesity and insulin resistance. Nature 2002; 420: 333–336.
Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab 2007; 6: 386–397.
Faghihimani E, Aminorroaya A, Rezvanian H, Adibi P, Ismail-Beigi F, Amini M . Reduction of insulin resistance and plasma glucose level by salsalate treatment in persons with prediabetes. Endocr Pract 2012; 18: 826–833.
Fleischman A, Shoelson SE, Bernier R, Goldfine AB . Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 2008; 31: 289–294.
Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J et al. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 2008; 1: 36–43.
Regazzetti C, Peraldi P, Gremeaux T, Najem-Lendom R, Ben-Sahra I, Cormont M et al. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes 2009; 58: 95–103.
Huang LE, Gu J, Schau M, Bunn HF . Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sc USA 1998; 95: 7987–7992.
Chen B, Lam KS, Wang Y, Wu D, Lam MC, Shen J et al. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem Biophys Res Commun 2006; 341: 549–556.
Wang B, Wood IS, Trayhurn P . Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Archiv 2007; 455: 479–492.
Unger RH . Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrin Metab 2003; 14: 398–403.
Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999; 48: 1270–1274.
Itani SI, Ruderman NB, Schmieder F, Boden G . Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002; 51: 2005–2011.
Schmitz-Peiffer C, Browne CL, Oakes ND, Watkinson A, Chisholm DJ, Kraegen EW et al. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes 1997; 46: 169–178.
Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 2002; 277: 50230–50236.
Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001; 293: 1673–1677.
Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B et al. Life without white fat: a transgenic mouse. Genes Dev 1998; 12: 3168–3181.
Sovik O, Vestergaard H, Trygstad O, Pedersen O . Studies of insulin resistance in congenital generalized lipodystrophy. Acta Paediatr 1996; 413: 29–37.
Arioglu E, Rother KI, Reitman ML, Premkumar A, Taylor SI . Lipoatrophy syndromes: when 'too little fat' is a clinical problem. Pediatr Diabetes 2000; 1: 155–168.
Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med 2002; 346: 570–578.
Diker-Cohen T, Cochran E, Gorden P, Brown RJ . Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metabol 2015; 100: 1802–1810.
Lipscombe LL, Hux JE, Booth GL . Reduced screening mammography among women with diabetes. Arch Intern Med 2005; 165: 2090–2095.
Wee CC, McCarthy EP, Davis RB, Phillips RS . Screening for cervical and breast cancer: is obesity an unrecognized barrier to preventive care? Ann Intern Med 2000; 132: 697–704.
Chang IH, Ahn SH, Han JH, Kim TH, Kim YS, Myung SC . The clinical significance in healthy men of the association between obesity related plasma hemodilution and tumor marker concentration. J Urol 2009; 181: 567–572 discussion 72-73.
Uppot RN, Sahani DV, Hahn PF, Gervais D, Mueller PR . Impact of obesity on medical imaging and image-guided intervention. Am J Roentgenol 2007; 188: 433–440.
Griggs JJ, Sorbero ME, Lyman GH . Undertreatment of obese women receiving breast cancer chemotherapy. Arch Intern Med 2005; 165: 1267–1273.
Qi D, Rodrigues B . Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism. Am J Physiolo Endocrinol Metab 2007; 292: E654–E667.
Yamauchi H, Cristofanilli M, Nakamura S, Hortobagyi GN, Ueno NT . Molecular targets for treatment of inflammatory breast cancer. Nature Rev Clinical Oncol 2009; 6: 387–394.
Acknowledgements
This work was supported by the grants from the Canadian Cancer Society and the Canadian Breast Cancer Foundation (Ontario chapter) to VS. YP is a recipient of a post-doctoral fellowship from the Canadian Breast Cancer Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Poloz, Y., Stambolic, V. Obesity and cancer, a case for insulin signaling. Cell Death Dis 6, e2037 (2015). https://doi.org/10.1038/cddis.2015.381
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.381
This article is cited by
-
Prognostic Significance of Adipose Tissue Distribution and Metabolic Activity in PET/CT in Patients with Metastatic Colorectal Cancer
Journal of Gastrointestinal Cancer (2023)
-
Dietary iron overload enhances Western diet induced hepatic inflammation and alters lipid metabolism in rats sharing similarity with human DIOS
Scientific Reports (2022)
-
Sedentary behavior and cancer–an umbrella review and meta-analysis
European Journal of Epidemiology (2022)
-
Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer
Nature Communications (2021)
-
Targeting obesity-related dysfunction in hormonally driven cancers
British Journal of Cancer (2021)
