
Abstract
Glioblastoma multiforme is a devastating disease of the central nervous system and, at present, no effective therapeutic interventions have been identified. Celastrol, a natural occurring triterpene, exhibits potent anti-tumor activity against gliomas in xenograft mouse models. In this study, we describe the cell death mechanism employed by celastrol and identify secondary targets for effective combination therapy against glioblastoma cell survival. In contrast to the previously proposed reactive oxygen species (ROS)-dependent mechanism, cell death in human glioblastoma cells is shown here to be mediated by alternate signal transduction pathways involving, but not fully dependent on, poly(ADP-ribose) polymerase-1 and caspase-3. Our studies indicate that celastrol promotes proteotoxic stress, supported by two feedback mechanisms: (i) impairment of protein quality control as revealed by accumulation of polyubiquitinated aggregates and the canonical autophagy substrate, p62, and (ii) the induction of heat-shock proteins, HSP72 and HSP90. The Michael adduct of celastrol and N-acetylcysteine, 6-N-acetylcysteinyldihydrocelastrol, had no effect on p62, nor on HSP72 expression, confirming a thiol-dependent mechanism. Restriction of protein folding stress with cycloheximide was protective, while combination with autophagy inhibitors did not sensitize cells to celastrol-mediated cytotoxicity. Collectively, these findings imply that celastrol targets proteostasis by disrupting sulfyhydryl homeostasis, independently of ROS, in human glioblastoma cells. This study further emphasizes that targeting proteotoxic stress responses by inhibiting HSP90 with 17-N-Allylamino-17-demethoxygeldanamycin sensitizes human glioblastoma to celastrol treatment, thereby serving as a novel synergism to overcome drug resistance.
Similar content being viewed by others


Main
Glioblastoma multiforme (GBM) is the most prevalent and most aggressive malignant intracranial tumor in humans.1, 2 Despite therapeutic advances, GBM remains a fatal disease with an average survival time of 14.6 months following diagnosis.3, 4 Although the cause of primary glioblastomas is not well known, genetic abnormalities affecting growth factor receptor signaling are commonly observed,2 as is an intense resistance to apoptosis.5 At present, mainstay therapy consists of surgical resection and concomitant chemotherapy and radiotherapy with temozolomide (TMZ), a non-specific DNA alkylating agent.6, 7 Current clinical research focuses primarily on inhibiting epidermal growth factor receptor, protein kinase C and phosphoinositide 3-kinase (P13K)–protein kinase B (PKB/AKT) signaling pathways. Thus far, however, molecular targeting of GBM in this regard has no demonstratable success.8
Celastrol, a naturally occurring quinone methide triterpene derived from the Thunder of God Vine, is a pleiotropic compound showing anti-tumor,9, 10, 11, 12 anti-inflammatory,13, 14, 15, 16 anti-hypertensive,17 and anti-diabetic activity18 in numerous cellular and in vivo models. Of particular relevance to GBM treatment, celastrol inhibits the growth of human glioma xenografts in mice19, 20 and was selected from a screen of over 2000 natural products for its potential to synergistically enhance the anti-cancer response to TMZ.21
A plethora of molecular mechanisms has been described for celastrol-mediated cell death in cancer cells. The prevailing hypotheses propose activation of apoptosis via death receptor upregulation22, 23 and/or inhibition of NFκB-mediated pro-survival signaling.12, 24, 25, 26, 27 Most recently, a pathway linking mitochondrial electron transport chain inhibition, leading to reactive oxygen species (ROS) generation and subsequent c-Jun N-terminal kinase (JNK) activation, has been proposed as a unifying mechanism in human non-small cell lung carcinoma28 and mouse melanoma cell lines.11 It is conceivable that the mechanism(s) of this pleiotropic drug encompass convergent pathways in a context- and cell-dependent manner.
The goals of the present study were to gain insight into the mechanism of cell death mediated by celastrol in glioblastoma cells and, ultimately, to identify a secondary target for potential drug combinations to treat GBM. The results support the concept that celastrol-induced cell death of glioblastoma is largely ROS independent, but reveal that it is mainly dependent on both the accumulation of polyubiquitinated protein aggregates and destructive autophagy, and a marked contribution of heat-shock proteins (HSPs) to cell death resistance. We propose that a synergistic cell death effect is best achieved by combining HSP90 inhibition with celastrol, and that combination therapy targeting HSPs may serve as an effective treatment for human glioblastoma.
Results
Celastrol triggers human glioblastoma cell death independent of ROS-JNK signaling
Celastrol has been proposed as a novel pharmacological intervention to inhibit the growth and invasiveness of many different cancers, particularly human gliomas.10, 19, 20 We investigated the molecular mechanism of celastrol-induced cell death in a well-characterized human glioblastoma cell line, U251N, derived from a malignant glioblastoma tumor explanted from a 70-year-old Caucasian male patient.29, 30, 31
Celastrol is thought to orchestrate a form of cell death that is dependent on the formation of ROS and the subsequent activation of ROS-responsive stress-activated protein kinases (SAPKs), such as JNK, to mediate a pro-apoptotic response10 (Figure 1a). In the present study, we show a significant loss of cell viability with>1 μM celastrol after 24 h of treatment in U251N (Figure 1b and Supplementary Figure S1), which was particularly marked under nutrient-rich conditions (LD50: 4.07±0.29 μM (Serum−) versus 3.16±0.09 μM (Serum+); P=2.67 × 10−12) (Figure 1c). These findings are in stark contrast to those obtained in cancerous cells (e.g., PC12) under conditions of excessive ROS production following serum deprivation.32 To determine the extent to which cellular redox status influenced celastrol-mediated cytotoxicity, we manipulated the endogenous levels of glutathione (GSH), the primary cellular antioxidant. Pretreatment of human glioblastoma with 50 μM L-buthionine sulfoximine (L-BSO), an inhibitor of γ-glutamylcysteine synthetase which catalyzes the rate-limiting step in GSH synthesis, did not significantly alter the response to celastrol (LD50: 3.65±0.24 μM without L-BSO versus 3.25±0.27 μM with LBS; P=1.06; Figure 1d), although GSH synthesis was sufficiently attenuated (Figure 1e). Further corroborating these results, inhibition of celastrol-mediated JNK activation with SP600125, as measured by immunoblotting for phospho-JNK (Supplementary Figure S2), had no effect on celastrol’s dose–response in glioblastoma (LD50: 2.5±0.2 μM without SP versus 2.2±0.58 μM with 10 μM SP and 1.89±0.71 μM with 20 μM SP; P=0.0834; Figure 1f). These results suggest an alternative mechanism of cell death mediated by the triterpenoid in glioblastoma cells.

Celastrol mediates a ROS and JNK-independent mechanism of cell death in human glioblastoma cells. (a) In a number of different cell types, it has been proposed that celastrol generates ROS and subsequently activates stress-dependent pathways (e.g., JNK) and inhibits pro-survival signaling (e.g., decreased AKT/PKB expression). (b) U251N cells were treated with celastrol at the indicated concentrations (0.3–10 μM) and labeled with Hoechst (nuclear stain). Scale bar in top left panel (200 μm) is representative for all panels shown. (c) U251N cells were treated with celastrol at the indicated concentrations (0.3–10 μM) in serum-containing and serum-deprived media for 24 h. Cell viability was assessed with the MTT assay. LD50: 4.07±0.29 μM (Serum−) versus 3.16±0.09 μM (Serum+); P=2.67 × 10−12 (see Statistical analysis in Materials and Methods section). (d), U251N cells were treated with serum-deprived media with or without 50 μM L-BSO, an inhibitor of gamma-glutamylcysteine synthetase, for 3 h, after which celastrol was added at various concentrations (0.3–10 μM) for another 24 h. Cell viability was assessed as in panel (c). L-BSO treatment did not have a significant effect (LD50: 3.65±0.24 μM without L-BSO versus 3.25±0.27 μM with L-BSO; P=1.06). (e) Intracellular GSH content was assessed as described in the Materials and Methods section, following 24 h treatment with serum-containing or serum-deprived media in the presence and absence of 50 μM L-BSO and 3 μM celastrol. L-BSO led to a significant reduction in intracellular GSH relative to untreated cells. No significant change in GSH content was noted for celastrol alone. (f) U251N cells were treated with serum-deprived media with or without SP600125, an inhibitor of JNK, at the indicated concentrations for 3 h, after which celastrol was added at various concentrations (0.1–10 μM) for another 24 h. Cell viability was assessed as in panels (c) and (d). No significant effect was seen with SP600125 (LD50: 2.5±0.2 μM without SP versus 2.2±0.58 μM with 10 μM SP and 1.89±0.71 μM with 20 μM SP; P=0.0834). Average values and S.D. are reported for triplicate measurements (N=3). Results are representative of at least three independent experiments. ***P<0.001
To further our investigation, we looked at the pro-apoptotic caspase-3. Poly(ADP-ribose) polymerase-1 (PARP-1) cleavage (Figure 2a) in response to celastrol confirmed caspases-3 activation, albeit to a reduced extent relative to the prototypical ATP-competitive kinase inhibitor staurosporine (STS). Direct measurement of caspase-3 activity using an optogenetic reporter assay showed no increase in enzymatic activity in cells exposed to celastrol but significant increase after STS exposure. These data support celastrol-induced cell death in a caspase-3-independent manner. (Supplementary Figure S3A). The specific small peptide inhibitor Ac-DEVD-CMK diminished celastrol-induced cell death by<5% (3 μM), independent of peptide concentration (LD50: 1.12±0.19 μM without DEVD versus 1.45±0.28 μM with 20 μM DEVD and 1.35±0.14 μM with 50 μM DEVD; P=0.000113; Figure 2b). Finally, membrane permeabilization associated with necrotic forms of cell death was not observed with celastrol (Supplementary Figure S3B). These findings suggest that the ROS-JNK-caspase-3 pathway is not required for celastrol-induced glioblastoma cell death. We therefore considered the evidence from previous studies supporting the proposed ROS-dependent mechanism of action; primarily, the capacity of thiol antioxidants to abolish the activity of a quinone methide triterpenoid, like celastrol.

Caspase-3 is dispensable for celastrol-mediated cell death in human glioblastoma cells. (a) U251N cells were incubated with either serum-containing (Serum+) or serum-deprived (Serum−) media or treated with 1 μM STS for 12 h or 3 μM celastrol for 24 h in serum-deprived media. Total cell lysates were collected and immunoblotted for PARP-1 cleavage, characteristic of caspase-3 activation. Actin is used as a control for protein loading. (b) U251N cells were treated with serum-deprived media with or without the caspase-3 selective inhibitor, Ac-DEVD-CMK, at the indicated concentrations for 3 h, after which celastrol was added at various concentrations (0.1–10 μM) for another 24 h. Cell viability was assessed by MTT assay. Caspase-3 inhibition led to a small but significant effect in shifting the dose–response to celastrol relative to treatment with celastrol alone (LD50: 1.12±0.19 μM with DEVD versus 1.45±0.28 μM with 20 μM DEVD and 1.35±0.14 μM with 50 μM DEVD; P=0.000113). Average values and S.Ds. are reported for triplicate measurements (N=3). Results are representative of at least three independent experiments
Excess free thiols, but not antioxidants, delay human glioblastoma cell death induced by celastrol
Thiol-based antioxidants, such as N-acetylcysteine (NAC), block celastrol’s activity in vitro and in vivo,11, 22, 33 leading some to conclude that its mechanism of action is ROS dependent. However, NAC, and free thiols in general, readily modify the reactive quinone methide moiety by nucleophilic Michael addition.34 We sought to differentiate between NAC’s antioxidant capacity and its reactive thiol by pretreating cells for 24 h with NAC and removing NAC-containing media before the addition of celastrol, markedly reducing the probability of their direct interaction (Figure 3a). Although pretreatment with 2 mM NAC did not prevent cell loss induced by celastrol (LD50: 3.38±0.11 μM without NAC versus 3.55±0.19 μM with NAC pretreatment; P=0.0856), co-treatment with NAC led to a remarkable delay in this regard (LD50: 12.6±161 μM; P=1.6 × 10−23), suggesting that thiol reactivity is pivotal to celastrol’s mechanism of action in glioblastoma. Notably, the NAC pretreatment paradigm diminished cell loss induced by paraquat, a potent herbicide known to mediate cell death via a ROS-dependent mechanism (Figure 3b).35 In addition, the non-thiol-based antioxidant 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) was incapable of delaying the response, meanwhile co-treatment with free thiols in the form of dithiothreitol (DTT) mimicked the effect of NAC co-treatment (LD50: 5.65±0.08 μM without DTT versus 12.6±14.1 μM with DTT; P=5.13 × 10−8; Figure 3c). These findings indicate that NAC interacts directly with celastrol, serving as a source of free thiols and not an antioxidant under these conditions.

Celastrol-mediated glioblastoma cell death is dependent on thiol reactivity and is abrogated by co-treatment or preincubation of the drug with free thiols. (a) U251N cells were pretreated with either 2 mM NAC (NAC PreTreat) or serum-deprived media (Serum− and NAC Co-Treat) for 24 h. Media was replenished with celastrol at various concentrations (0.3–10 μM) with (NAC Co-Treat) or without (Serum−, NAC PreTreat) 2 mM NAC for a subsequent 24 h. Cell viability was assessed with the MTT assay. Significant differences in cell viability are noted when NAC is co-treated with celastrol relative to cells treated with celastrol alone (LD50: 12.6±161 μM; P=1.6E-23) but not when NAC is removed following pretreatment and before the addition of celastrol (LD50: 3.38±0.11 μM without NAC versus 3.55±0.19 μM with NAC pretreatment; P=0.0856). (b) U251N cells were pretreated with either 2 mM NAC or serum-deprived media for 24 h. Media was replenished with paraquat at various concentrations (0–1 mM) for a subsequent 24 h. Cell viability was assessed with the MTT assay. Significant rescue of cell viability was noted with NAC pretreatment compared with no pretreatment for the two highest doses of paraquat. (c) U251N cells were treated with 1 mM Trolox, 50 μM DTT or serum-deprived media in combination with celastrol at various concentrations (0.1–10 μM) for 24 h. Cell viability was assessed as in panels (b) and (c). No significant effect was seen with trolox (P=0.0102), whereas the presence of DTT significantly delayed the response to celastrol (LD50: 5.65±0.08 μM without DTT versus 12.6±14.1 μM with DTT; P=5.13 × 10−8) (see Statistical analysis section in Materials and Methods). Average values and S.Ds. are reported for triplicate measurements (N=3). Results are representative of at least three independent experiments. **P<0.01. (d) Chemical structure for celastrol–NAC adduct (6-N-Acetylcysteinyldihydrocelastrol) formed upon Michael addition of NAC to C6 electrophilic center of the celastrol quinone methide moeity
Our in vitro studies clearly show that direct interaction between NAC and celastrol is necessary to attenuate its biological activity, suggesting that an inactive end product is produced. Because of the α,β-unsaturated carbonyl of the quinone methide, position C-6 is highly electrophilic and thus susceptible to conjugate addition by NAC to form a Michael adduct, likely 6-N-Acetylcysteinyldihydrocelastrol (Figure 3d), based on Klaić et al.’s findings.34 To validate this assumption, celastrol (10 μM) was incubated with excess NAC (2 mM) in methanol for 1 h and was subsequently analyzed by electrospray ionization mass spectrometry (ESI-MS) (see Materials and Methods). ESI-MS revealed a peak at 636.2964, corresponding to the molecular weight of the Michael addition product [C34H47O7NS+Na+]+, and thereby confirming formation of the adduct (Supplementary Figure S4). This product was also observed when the same experiment was performed in deionized water or in phosphate buffer (data not shown). Collectively, these findings point to a mode of cell death that is dependent on celastrol’s thiol reactivity and not on the induction of ROS in human glioblastoma, prompting our investigation to reveal alternative mechanisms of cell death.
Celastrol promotes accumulation of polyubiquitinated protein aggregates and leads to destructive autophagy
Celastrol is proposed to be an inhibitor of proteasomal function, resulting in the accumulation of polyubiquitinated substrates.36, 37 We therefore tested this hypothesis in human glioblastoma cells. Celastrol caused a dose-dependent increase in ubiquitin-positive protein aggregates, in a similar manner to the prototypical proteasome inhibitor MG-132, (Figure 4a). Polyubiquitinated cargo is destined for one of two selective protein quality-control pathways: proteasomal degradation or autophagy. Recent reports suggest that celastrol can initiate autophagy.38, 39 We first sought to measure the change in levels of the lipidated form of LC3B (LC3B-II), a marker of autophagy initiation, by western blotting. Notably, within 3 h of celastrol (3 μM) treatment, levels of the lipidated form of LC3B (LC3B-II), a marker of autophagy initiation, became elevated (Figure 4b and Supplementary Figure S5). In parallel with the observed LC3B-II increase, there was a significant accumulation of p62, an autophagy substrate and ubiquitin receptor, which peaked within 12 h (Figure 4c and Supplementary Figure S5). This effect was sensitive to the presence of free thiols (Supplementary Figure S6). In conjunction, p62 aggregate formation can be seen upon immunolabeling in situ (Figure 4d). Importantly, rapamycin, a well studied mTORC1 inhibiter and autophagy inducer, did not affect LC3B processing or p62 homeostasis (Figures 4b and c and Supplementary Figure S5), even in the presence of a sustained increase in lysosome content (Supplementary Figure S7). Observations with celastrol are characteristic of a blockade in autophagic flux and are comparable to the effects exerted by chloroquine (CQ), a lysosomotropic agent and inhibitor of endosomal acidification (Figure 4c, right).

Celastrol blocks protein degradation and promotes accumulation of poly-ubiquinated substrates in human glioblastoma cells. (a) U251N cells were treated with serum-containing and serum-deprived media in the presence and absence of 500 nM 17-AAG (Hsp90 inhibitor), 10 μM MG-132 (proteasome inhibitor) and various concentrations of celastrol for 24 h. Total cell lysates were collected and immunoblotted for ubiquitin. (b and c) Cells were treated with serum-containing and serum-deprived media in the presence and absence of 10 μM MG-132 and 3 μM celastrol for 3–24 h. Total cell lysates were collected and immunoblotted for LC3B (a) and p62 (b). Actin is used as a control for protein loading. (d) In situ immunolabeling of p62 in U251N cells reveals accumulation of aggresomes, as indicated by white arrowheads. Cel, Celastrol 3 μM for 8 h; Rap, Rapamycin 200 nM for 24 h. Scale bar is representative for all four panels. (e) U251N cells were treated with increasing concentrations of celastrol for 24 h and analyzed for lysosomal content as described in the Materials and Methods section. Lysosome number was normalized to untreated cells (100%). Significant differences relative to untreated cells are denoted. (f) U251N cells labeled with LysoTracker Red DND-99 are depicted with cytoplasmic membrane outlines highlighted to clarify cell delineations. Scale bar in the first panel (50 μM) is representative for all the four panels shown. (g) U251N cells were treated with serum-deprived media with or without 50 μM CQ (inhibitor of lysosomal acidification) or 5 mM 3MA (inhibitor of the class III P13K, Vps34) for 1 h after which celastrol was added at various concentrations (0.5–10 μM) for another 24 h. Cell viability was assessed using the MTT assay. No significant differences were observed with CQ or 3MA relative to celastrol alone (LD50: 3.66±0.31 μM with serum deprivation versus 3.50±0.30 μM with CQ and 3.69±0.24 μM with 3MA; P=2.728). (h) U251N cells were treated with serum-deprived media with or without 1 μg/ml CHX, an inhibitor of protein translation, for 1 h, after which celastrol was added at various concentrations (0.3–10 μM) for another 24 h. Cell viability was assessed using the MTT assay. A significant difference in the response to celastrol was noted with CHX-treated cells relative to treatment with celastrol alone (LD50: 5.01±0.24 μM without CHX versus 14.8±88.3 μM with CHX; P=4.38 × 10−4) (see Statistical analysis section in Materials and Methods). Average values and S.Ds. are reported for triplicate measurements (N=3). Results are representative of at least three independent experiments. **P<0.01, ***P<0.001
In line with this notion of impeding autophagy completion, triterpenoids, such as the betulinic acid derivative, B10, reportedly destabilize lysosomes and subsequently disrupt autophagic flux-triggering cathepsin release in glioblastoma cells.40 To test this hypothesis for celastrol, we stained cells with the weakly basic amine dye, LysoTracker Red, which preferentially accumulates in acidic lysosomal compartments. In contrast to CQ, celastrol treatment increased lysosomal content at minimally toxic doses (<3 μM) (Figures 4e and f and Supplementary Figure S7). Not surprisingly, lysosomal integrity diminished with increasing toxicity (3–10 μM), suggesting that celastrol’s effects indirectly overburden lysosomal degradative pathways, thereby interfering with autophagy-mediated degradation. Additionally, pretreatment with leupeptin, an inhibitor of the lysosomal-specific cathepsin B, did not block celastrol’s effects (Supplementary Figure S8), arguing against a lysosomal destabilizing mechanism of necrotic cell-death. Blocking autophagic flux upstream with 5 mM 3-methyladenine (3MA), an inhibitor of class III phosphatidylinositol kinase or downstream with 50 μM CQ does not further sensitize cells to celastrol-induced toxicity (LD50: 3.66±0.31 μM with serum deprivation versus 3.50±0.30 μM with CQ and 3.69±0.24 μM with 3MA; P=2.728; Figure 4g), confirming that autophagy is already compromised by celastrol. It should be noted that both of these autophagic modulators are cytotoxic to glioblastoma within the concentration range used (Supplementary Figures S9 and S10). Furthermore, cycloheximide (CHX), which arrests de novo protein synthesis and thereby reduces the burden on homeostatic protein-folding mechanisms,41 significantly delayed the cell death response to celastrol (LD50: 5.01±0.24 μM without CHX versus 14.8±88.3 μM with CHX; P=4.38 × 10−4; Figure 4h). These observations in U251N suggest a mechanism whereby celastrol promotes proteoteoxic stress by interfering with quality-control pathways, such as autophagy.
To generalize our findings to the treatment of GBM, we also tested two other human glioblastoma and astrocytoma cell lines, U87 and U343, respectively. Accumulation of polyubiquitinated substrates and p62 upon celastrol treatment in both cell types (Figures 5a and b and Supplementary Figure S11) was observed, confirming our conclusions from experiments in U251N. CHX, meanwhile, blocked celastrol’s response in U343 (Figure 5c; LD50: 5.30±0.28 μM without CHX versus 7.32±0.1 μM with CHX; P=8.36 × 10−6) but not in U87 (Figure 5d; LD50: 5.94±0.09 μM without CHX versus 75.44±0.22 μM with CHX; P=0.18). To broaden the implication of these results, we also examined the response to CHX in three GBM cancer stem cells (GSCs) (see Materials and Methods for more details): GSC1, GSC2, and 48 EF. In all the three GSC lines, CHX was protective against celastrol-mediated toxicity (Figure 5e), confirming our previous findings from U251N and U343. These results support a mechanism implicating a destructive form of autophagy, prompting our investigation into novel mechanisms of resistance against celastrol-induced glioblastoma cell death.

Celastrol promotes proteotoxic stress in a panel of human glioblastoma cell lines and GSCs. (a) U343 and (b) U87 cells were treated with serum-containing and serum-deprived media in the presence and absence of 50 μM CQ (inhibitor of lysosomal acidification), 500 nM 17-AAG (Hsp90 inhibitor), and 3 μM celastrol for 24 h. Total cell lysates were collected and immunoblotted for proteasomal substrates (ubiquitin) and autophagy substrates (p62). Actin is used as a control for protein loading. (c) U343 and (d) U87 cells were treated with serum-deprived media with or without 1 μg/ml CHX, an inhibitor of protein translation, for 1 h, after which celastrol was added at various concentrations (0.3–10 μM) for another 24 h. Cell viability was assessed using the MTT assay. A significant difference in the response to celastrol was noted with CHX-treated U343 cells relative to treatment with celastrol alone (LD50: 5.30±0.28 μM without CHX versus 7.32±0.1 μM with CHX; P=8.36 × 10−6) (see Statistical analysis section in Materials and Methods). Average values and S.Ds. are reported for triplicate measurements (N=3). (e) GSCs were treated with serum-deprived media with or without 1 μg/ml CHX 1 h, after which celastrol (3 μM) was added for another 24 h. Cell viability was assessed using the MTT assay and reported as the percentage of control (untreated cells for celastrol−CHX and CHX-treated cells for celastrol+CHX). A significant difference in the response to celastrol was noted with CHX-treated GSCs as noted in the annotations. At right, the response to 1 μg/ml CHX alone is observed to reduce cell viability and proliferation by∼30–40%. Average values and S.Ds. are reported for triplicate measurements (N=3)
HSP function protects glioblastoma from celastrol-induced cell death
Celastrol activates the HSP response in various cell types in a heat-shock factor 1 (HSF-1)-dependent manner,36, 42 for which the mechanism involved is thought to depend on thiol reactivity.43 In human glioblastoma cells, celastrol augments HSP72 protein levels in a time-dependent manner by a mechanism that is sensitive to protein synthesis blockade with CHX (Figure 6a and Supplementary Figure S12) and preincubation with free thiols, NAC and DTT (Supplementary Figure S13). These findings confirm that preservation of celastrol’s thiol-reactive moiety (i.e., quinone methide) is necessary for the upregulation of HSP72 expression.

Celastrol upregulates the expression of heat-shock response genes, which delay cell death in human glioblastoma cells. (a) U251N cells were treated with serum-containing or serum-deprived media in the presence and absence of 1 μg/ml CHX, an inhibitor of protein translation, for 1 h, after which cells were treated with celastrol for 3–24 h. CQ (50 μM) for 24 h controls for protein accumulation following inhibition of autophagy. Total cell lysates were collected and immunoblotted for Hsp72. Actin is used as a control for protein loading. (b) U251N cells were treated with serum-containing and serum-deprived media in the presence and absence of 500 nM 17-AAG (Hsp90 inhibitor) or 10 μM MG-132 (proteasome inhibitor) for 24 h and 3 μM celastrol for 3–24 h. Total cell lysates were collected and immunoblotted for Hsp90. Actin is used as a control for protein loading. (c) U251N cells were treated with serum-deprived media with or without 500 nM 17-AAG for 1 h, after which celastrol was added at various concentrations (0.3–10 μM) for another 24 h. Cell viability was assessed using the MTT assay. 17AAG significantly shifted the dose–response to celastrol treatment relative to treatment with celastrol alone (LD50: 1.03±0.12 μM without 17-AAG versus 0.69±0.11 μM with 17-AAG; P=1.23 × 10−6). Average values and S.Ds. are reported for triplicate measurements (N=3). Results are representative of at least three independent experiments
Celastrol also induced HSP90, in a similar manner to MG-132, suggesting that proteasomal inhibition is sufficient to activate the HSP response (Figure 6b and Supplementary Figure S14). This was in contrast to studies in a prostate cancer cell line, where HSP90 protein expression remained stable with celastrol treatment for up to 24 h.27 The function of the 90-kDa HSP is to stabilize various oncogenic kinases and works in concert with proteasomal degradation to regulate protein stability.44, 45 We hypothesized that a broad-spectrum selective HSP90 inhibitor, like 17-N-Allylamino-17-demethoxygeldanamycin (17-AAG), could act in concert with celastrol to kill human glioblastoma. Although 17-AAG alone is minimally toxic to human glioblastoma (Supplementary Figure S15), combining with celastrol potentiates cell death mediated by the triterpenoid (LD50: 1.03±0.12 μM without 17-AAG versus 0.69±0.11 μM with 17-AAG; P=1.23 × 10−6) (Figure 6c). This set of experiments highlights a novel synergistic mechanism between celastrol and the inhibition of HSP function to mediate a more potent cytotoxic response in glioblastoma cells.
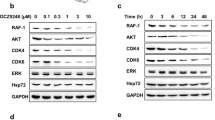
Celastrol was reported to induce chaperone proteins primarily by inhibiting HSP90, subsequently releasing and activating the bound transcription factor, HSF-1.36, 42, 46 Activated HSF-1 in trimeric form undergoes nuclear localization and transactivates gene expression at loci downstream of heat-shock responsive elements (HSEs), such as HSPA1A, leading to the expression of the HSP72 protein. The inhibition of HSP90 by celastrol has been confirmed by others using purified preparations of HSP90 and its co-chaperone Cdc37 to show disruption of the interaction.47, 48 Using 17-AAG, a canonical inhibitor of HSP90,49, 50 we directly compare the effects of HSP90 inhibition to celastrol, and show a remarkable difference. Primarily, 17-AAG (500 nM) blocks HSP90, as revealed by the decrease in the stability of protein kinase clients such as Akt (Supplementary Figure S16), without robust toxicity at this concentration (Supplementary Figure S15). In agreement with these findings, at celastrol’s LD50 (3 μM), Akt expression is stable, which also suggests no inhibition of HSP90 (Supplementary Figure S16). Unlike 17-AAG, celastrol directly affects proteostasis by increasing polyubiquitinated aggregates (Figures 4a, 5a and b) and p62 accumulation (Figures 4c, 5a and b). Finally, the novel synergism revealed by combining these two compounds (Figure 6c) confirms that they do not target the same pathway but rather converging pathways implicated in proteotoxic stress. As illustrated in Figure 7, these are protein degradation and protein folding. Therefore, these data suggest an alternative mode of action for celastrol-mediated cell death, apart from HSP90 inhibition, and firmly corroborate evidence by others that 17-AAG effectively sensitizes glioma cells to chemotherapeutic drugs.51, 52

Celastrol causes proteotoxicity via a thiol-sensitive mechanism in human glioblastoma. Poly-ubiquitinated protein aggregates along with the autophagy substrate and stress sensor, p62, accumulate as a result of defective protein degradation quality-control mechanisms, namely autophagy and proteasomal degradation. The disruption in proteostasis is further substantiated by the activation of cytoprotective heat-shock responses (HSP72/90)
Discussion
In the present study, we show that celastrol drives proteotoxicity in glioblastoma cells. This was revealed both by the accumulation of polyubiquitinated protein aggregates and by the induction of cytoprotective molecular chaperones (HSPs). Autophagic flux was further impaired as revealed by loss of lysosomal integrity and the buildup of p62, a canonical autophagy receptor for protein aggregates. Importantly, cell death occurred independently of apoptosis, indicating the importance of alternate cell death pathways. Furthermore, we identify HSPs as a key resistance mechanism to cell death mediated by celastrol, thereby highlighting a secondary target in GBM and a novel drug synergism between celastrol and the geldanamycin analog, 17-AAG.
Recent studies have suggested that celastrol mediates cell death via the induction of ROS and stress-activated kinases.11, 22, 33 However, these studies reported marginal ROS generation by celastrol and used the thiol-containing antioxidant NAC to illustrate dependence on this mechanism, in some cases using concentrations as high as 20 mM.33 Evidence provided herein and by others34 demonstrates unequivocally that NAC mediates a nucleophilic attack on the electrophilic center of celastrol’s quinone methide moiety (Figure 3d and Supplementary Figure S4), thereby acting as a source of free thiols and not an antioxidant. To this purpose, celastrol was also reported to deplete cysteine-containing peptides, including the reduced form of the primary cellular antioxidant GSH, and induce subsequent radiosensitization in human lung cancer cell lines.53 Conversely, in human glioblastoma cells, we did not detect a change in GSH levels (normalized to cell number) in response to celastrol (Figure 1e). Moreover, depletion of GSH with the well-characterized inhibitor of γ-glutamylcysteine synthetase, L-BSO, did not sensitize cells to celastrol (Figure 1d). Together, these observations indicate that celastrol acts through a mechanism reliant on the preservation of its reactivity with thiols.
Sulfhydryl reactivity is sufficient to regulate cytoprotective responses, such as antioxidant and heat-shock responses.54 Zhang et al.54 tested the hypothesis that electrophiles, like celastrol, trigger a common cysteine-reactive mechanism to induce cytoprotective signaling mechanisms: HSF-1/heat-shock response element (HSE) and the Kelch-like-ECH-associated protein 1 (KEAP-1)/NRF2/antioxidant response element pathways. Serving as a sensor in the ‘electrophile counterattack response,’ key cysteine residues of KEAP-1 chemically react with electrophiles, barring its inhibitory effect on NRF2.55 Zhang et al.54 showed that various electrophiles, independent of overall chemical structure, readily induced both NRF2 and HSF-1-dependent genes. Like NRF2, HSF-1 is also activated by a wide range of electrophiles, including celastrol; this activation, however, does not require the expression of KEAP-1, suggesting that distinct electrophile sensors exist for both NRF2 and HSF-1 pathways. In fact, the yeast HSP70 chaperone Ssa1 serves as an electrophile sensor for HSF-1,43 similar to the role of KEAP-1 in NRF2-mediated antioxidant gene induction. In the yeast homolog of HSP70, Ssa1, mutations to cysteine residues (C264 and C303) were individually sufficient to render HSF-1 unresponsive both to celastrol and 4-hydroxynonenal (4-HNE), a model organic electrophile. Similarly, conserved cysteine residues (C267 and C306) in HSP72, but not HSP70, were necessary for redox sensing in response to oxidation by methylene blue.56 Nonetheless, in contrast to 4-HNE,43 celastrol was shown by us to disrupt proteostasis in human glioblastoma, in a manner which was dependent on sulfhydryl reactivity, and was associated with the induction of HSPs. These findings suggest that celastrol may function by a mechanism that is distinct from other cysteine-reactive compounds, which directly target conserved redox-sensing mechanisms.
To provide a clear picture of celastrol’s effects in human glioblastoma cells, one that unifies observations by others with the present findings, the link between autophagy, proteostasis and endoplasmic reticulum (ER) stress must be considered. Most notably, Zhu et al.57 showed previously that celastrol activates the ER-stress response in human hepatocellular carcinoma cells via a mechanism that is dependent on preserved eIF2α-ATF4-Noxa signaling. In line with these results, celastrol induces cytoplasmic vacuolation in several human cancer cell lines in conjunction with elevated membrane-bound LC3 (LC3-II).39 This cell phenotype is characteristic of an ER-stress response termed cytoplasmic vacuolation death or paraptosis, which is dependent on LC3.39, 58, 59 The delay of celastrol-induced cell death with CHX treatment in glioblastoma cells (Figures 4g, 5d and e) is directly in line with findings by Wang et al.39 that inhibition of protein translation attenuates cytoplasmic vacuolation. It is therefore not surprising that serum deprivation protected against celastrol-induced cell death (Figure 1c); nutrient-rich conditions favor de novo protein synthesis, placing an increased burden on mechanisms countering protein misfolding and aggregation. Indications of excess polyubiquitinated protein aggregates (Figures 4a, 5a and b), induction of LC3 maturation (Figure 4b), and accumulation of p62 (Figures 4c, 5a and b) in human glioblastoma cells further validate this mechanism of cell death and explain why celastrol is more effective than a selective proteasome inhibitor (i.e., MG132) in killing cancer cells.
Serving a critical role in selective degradation of ubiquitinated substrates via autophagy,60, 61 p62 (also called SQSTM1) is a receptor for Ub and LC3, potentially acting as a sensor of protein stress at the crossroads of cell survival and cell death.62, 63, 64, 65 The inhibition of autophagy leading to the accumulation of autophagy substrates and receptors may lie upstream of proteasomal dysfunction and programmed cell death in certain cases.66 Under these conditions, p62 is believed to act by sequestering and delaying the delivery of substrates destined for proteasomal degradation, an effect which can be rescued by p62 knockdown and aggravated by overexpression.66 It is hence conceivable that p62 acts at a critical junction between tumor-promoting autophagic processing and the buildup of aggregated substrates leading to cell death by paraptosis; celastrol apparently tips the balance in favor of the latter. Future studies may reveal alternate modes of glioblastoma cell death,67 by employing chemotherapeutic drug combinations to simultaneously and/or sequentially silence chaperones and disrupt autophagy.
The present results suggest a mechanism whereby celastrol triggers the accumulation of polyubiquitinated protein aggregates by disrupting sulfhydryl homeostasis and exerting widespread proteotoxicity in glioblastoma cells, as summarized in Figure 6. Combining celastrol with drugs that place additional stress on homeostatic mechanisms (i.e., inhibition of HSPs) could exacerbate the response, whereas suppressing protein misfolding stress could protect cells from celastrol-mediated toxicity. We show that celastrol-mediated cell death in glioblastoma cells occurs independently of ROS and also highlight what we regard as the misuse of thiol-containing antioxidants (e.g., NAC) as pharmacological tools to study the mechanism of action of celastrol and other electrophilic drugs. Questions still remain as to whether or not celastrol directly inhibits autophagy signaling upstream of lysosomal fusion, thereby promoting protein aggregate deposition and proteotoxic stress. This proposal does not exclude the possibility that protein aggregates accumulate in response to proteasomal inhibition and other sulfhydryl-dependent mechanisms that subsequently place an excess burden on homeostatic pathways. Both selective autophagy and proteasomal degradation are dependent on thiol-mediated protein conjugation reactions, which are proposed new targets for celastrol’s mode of action.
Materials and Methods
Solutions, media, and reagents
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), NAC, L-BSO, Hoechst 33342, methyl viologen dichloride hydrate (paraquat dichloride), (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (trolox), 17-AAG, MG-132, CHX, rapamycin from Streptomyces hygroscopicus (powder form), and GSH assay kit were purchased from Sigma-Aldrich (Oakville, ON, Canada). Celastrol was purchased from Cayman Chemical (Ann Arbor, MI, USA). CQ diphosphate was purchased from TOCRIS Biosciences through Cedarlane Laboratories (Burlington, ON, Canada). Caspase-3 Inhibitor III (Ac-DEVD-CMK), JNK Inhibitor II (SP600125), and anti-Actin antibody, clone C4 were purchased from Millipore (Burlington, MA, USA). Anti-PARP-1 antibody (H-250) and anti-SAPK/JNK antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-ubiquitin and anti-Hsp70/72 antibodies were purchased from Enzo Life Sciences through Cedarlane Laboratories (Burlington, ON, USA). Anti-LC3B antibody and anti-Phospho-SAPK/JNK (Thr183/Tyr185) were purchased from Cell Signaling Technologies through Cedarlane Laboratories (Burlington, ON, Canada). Anti-p62-SQSTM1 antibody was purchased from MBL International (Des Plaines, IL, USA). Anti-Hsp90 antibody was purchased BD Transduction Laboratories through BD Biosciences (Mississauga, ON, Canada). Cell culture media, penicillin–streptomycin, heat-inactivated fetal bovine serum (FBS), DTT, and LysoTracker Red DND-99 were purchased from Invitrogen (Burlington, ON, Canada).
Cell culture and treatments
U251N human glioblastoma cells and U343 human astrocytoma cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Life Technologies Inc., Burlington, ON, Canada) supplemented with 10% (v/v) FBS (Gibco) and 1% (v/v) penicillin–streptomycin (Gibco). U87 cells were cultured in Minimum Essential Medium (Gibco) supplemented with 10% (v/v) FBS (Gibco) and 1% (v/v) penicillin–streptomycin (Gibco). Cells were seeded 24 h before treatment/media change according to the appropriate density for the indicated assay (described in detail below). They were maintained at 37 °C with 5% CO2 and>95% relative humidity. Medium was changed to serum free for all treatments and lasted for 0–48 h, depending on treatment paradigm (details provided below for specific methods).
Isolation and maintenance of GSC
GSC1 and GSC2 were isolated directly from patients undergoing surgical resection according to a Montreal Neurological Institute and Hospital research ethics board-approved protocol. 48EF stem cells were kindly provided by Dr. Samuel Weiss (University of Calgary). GSC1, GSC2, and 48EF stem cells were isolated as previously performed68 and expanded in neurosphere cultures. When spheres appear large enough for passaging (<300 μm in diameter), they were collected in tube and spun at 1200 r.p.m. for 3 min. To dissociate the spheres, Accumax (Millipore, Burlington, MA, USA) was added to the cell pellet and incubated for 5 min at 37 °C, they were then washed with phosphate-buffered saline (PBS), centrifuged and resuspended in complete Neurocult-NS-A proliferation medium and seeded at a concentration of 200 000 cells/flask.
Labeling nuclei with fluorescent dye Hoechst 33342 for cell viability determinations
U251N cells were seeded at a density of 10 000 per well into 96-well cell culture plates and left to adhere for 24 h in a final volume of 0.2 ml serum-supplemented DMEM media (10% FBS, 1% Pen-Strep). Media was replaced with serum-deprived DMEM and treated as indicated. Following treatment, cells were washed once with PBS and treated with fixative (4% formaldehyde) for 10 min at 37 °C. Fixative was subsequently aspirated, and cells were washed three times with PBS and stained with 10 μM Hoechst 33342 (Sigma-Aldrich) diluted in PBS for 10 min. Cells were washed once and imaged using Operetta High Content Screening System (Perkin Elmer, Woodbridge, ON, Canada) and analyzed using Harmony High Content Imaging and Analysis Software (Perkin Elmer). Data output was in text file format and graphed using the Microsoft Excel (Microsoft Canada Inc., Mississauga, ON, Canada) and Igor Pro software (Wavemetrics, Tigard, OR, USA).
Intracellular GSH content analysis
U251N cells were seeded at a density of 300 000 per well into six-well cell culture plates and left to adhere for 24 h in a final volume of 2 ml serum-supplemented DMEM media (10% FBS, 1% Pen-Strep). Media was replaced with serum-deprived DMEM and treated as indicated. Following treatment, cells were trypsinized, washed and centrifuged at 700 × g. Pellets were resuspended in 5% 5-sulfosalicylic acid solution, followed by three cycles of freeze–thaw, alternating between 5 min at −80 °C and 5 min at +37 °C. Samples were centrifuged at 10 000 × g, and supernatants were analyzed for GSH content using the GSH Assay Kit (Sigma-Aldrich; Cat. C S0260), following the manufacturer’s protocol. Data were tabulated and graphed using the Microsoft Excel and Igor Pro software.
MTT assay for mitochondrial metabolic activity as a measure of cell viability
U251N, U87, and U343 cells were seeded at a density of 50 000 per well into 24-well cell culture plates and left to adhere for 24 h in a final volume of 0.5 ml serum-supplemented DMEM media (10% FBS, 1% Pen-Strep). Media was replaced with serum-deprived DMEM and treated as indicated. Following treatment, media was replenished with serum-deprived media containing MTT (0.5 mg/ml). Cells were incubated at 37 °C with 5% CO2 and>95% relative humidity for 30 min, after which media was removed. DMSO was added to lyse the cells and dissolve the formazan produced. Triplicates from each well were collected into a 96-well plate (Sarstedt, Montreal, QC, Canada), and the absorbance at 595 nm of each well was measured using a Benchmark microplate reader (Bio-Rad, Mississauga, ON, Canada). Results from MTT were comparable to those obtained with cell counting (Figure 1c, Serum− versus Supplementary Figure S1). Data are expressed as the percentage of change from untreated controls and were tabulated and graphed using the Microsoft Excel and Igor Pro software.
GSCs cells were plated at a density of 20 000 cells/well in a 96-well plated overnight in stem cell culture media. 17AAG, CHX, and celastrol treatment were added at their final concentrations (as indicated) in a final volume of 100 μl. After 24 h incubation at 37 °C, 20 μl of MTT (2.5 mg/ml; in PBS) was added and further incubated for 3 h at 37 °C. The media containing GSCs was transferred to an eppendorf tube and spun down to pellet the cells. Media was then aspirated, and the insoluble formazan product was solubilized in 100 μl DMSO for 1 h at 37 °C. The absorbance at 595 nM was analyzed on an Epoch (Biotek Instruments Inc., Winooski, VT, USA) plate reader. Data expressed as the percentage of change from untreated controls were tabulated and graphed using the Microsoft Excel and Igor Pro software.
Confirming 6-N-Acetylcysteinyldihydrocelastrol formation (NAC addition to celastrol)
A methanol solution of celastrol (10 μM) and NAC (2 mM) was allowed to react at 37 °C for 1 h and was directly injected into an ESI-MS (Exactive Orbitrap, ThermoFisher Scientific (Ottawa, ON, Canada), Department of Chemistry/McGill University). The high-resolution and high-accuracy ESI-MS results suggest the presence of 6-Nz-acetylcysteinyldihydrocelastrol, the Michael addition product of NAC and celastrol (see Supplementary Figure S1) ([C34H47O7NS+Na+]+ requires: 636.2965, Found: 636.2964). The NAC–celastrol adduct was also observed when the same experiment was performed in deionized water or in phosphate buffer. In the latter case, extraction with ethyl acetate had to be performed before the MS analysis.
Western blotting
Cells were seeded at a density of 300 000 per well into six-well cell culture plates and left to adhere for 24 h in a final volume of 2 ml serum-supplemented DMEM media (10% FBS, 1% Pen-Strep). Media was replaced with serum-deprived DMEM and treated as indicated. Following treatment, cells were trypsinized, pelleted, resuspended in lysis buffer, sonicated briefly, centrifuged, and supernatants collected. Of the total cell extracts, 15–25 μg of protein (measured by bicinchoninic acid assay) was loaded onto a 12% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Membranes were washed with 0.1% Tween-20+TBS and blocked with 5% milk+0.5% Tween-20+TBS for 1 h at room temperature (RT) and incubated with primary antibodies diluted 1 : 1000–2000 in blocking solution overnight at 4 °C. After five washes with 0.1% Tween-20+TBS, the membranes were incubated with the HRP-conjugated anti-rabbit/mouse (Bio-Rad) diluted 1 : 3000 in 5% milk+0.5% Tween-20+TBS for 1 h at RT. After five washes with 0.1% Tween-20+TBS, HRP substrate (Luminata, Millipore, Billerica, MA, USA) was added and incubated for 5 min, following which the membranes were exposed to film (Harvard, Holliston, MA, USA). Blotting with mouse anti-actin (dilution 1 : 1000, Millipore, Burlington, MA, USA) was used as a housekeeping protein to control for global protein expression levels. Densitometry was performed using ImageJ (NIH, Bethesda, MD, USA), and data were tabulated and graphed using the Microsoft Excel and Igor Pro software.
Immunocytochemistry for p62
Cells were seeded at a density of 15 000 onto 12 mm Bo-Si glass coverslides placed at the base of tissue culture plate wells and left to adhere for 24 h in a final volume of 0.5 ml serum-supplemented DMEM media (10% FBS, 1% Pen-Strep). Media was replaced with serum-deprived DMEM and treated as indicated. Following treatment, cells were fixed in 4% paraformaldehyde for 10 min at RT, followed by washing with PBS. Permeabilization was done using 0.1% triton-X-100 at RT for 5 min, followed by washing again with PBS. Nonspecific binding sites were blocked for 60 min with 10% goat serum diluted in PBS. The samples were then incubated overnight at 4 °C with the primary antibodies (rabbit anti-p62-SQSTM1 pAb). Following the incubation, samples were washed three times in PBS and incubated for 60 min at RT with the secondary antibodies (anti-rabbit AlexaFluor 488, 1 : 800, Invitrogen). Following three washes in PBS, the samples were counterstained with Hoechst 33342 (10 uM, 10 min; Invitrogen), rinsed once with distilled water, and mounted onto glass slides using Aqua-Poly/Mount (Polysciences; Warrington, PA, USA). Samples were placed at 4 °C before imaging. Images were captured on a Confocor LSM 510 META mounted onto an Axiovert 200 M inverted microscope (Carl Zeiss Canada Ltd., Toronto, ON, Canada).
Lysosomal content analysis
Cells were seeded at a density of 15 000 per well into 96-well cell culture plates and left to adhere for 24 h in a final volume of 0.2 ml serum-supplemented DMEM media (10% FBS, 1% Pen-Strep). Media was replaced with serum-deprived DMEM and treated as indicated. Following treatment, media was removed and replenished with 500 nM LysoTracker Red DND99 (Molecular Probes, Life Technologies Inc., Burlington, ON, Canada) diluted in fresh media, and cells were incubated at 37 °C for 3 min. Cells were subsequently washed once with PBS and treated with fixative (4% formaldehyde) for 10 min at 37 °C. Fixative was aspirated, and cells were washed three times with PBS and stained with 10 μM Hoechst 33342 (Sigma-Aldrich) diluted in PBS for 10 min. Cells were washed once and imaged using the Operetta High Content Screening System (Perkin Elmer) and analyzed using Harmony High Content Imaging and Analysis Software (Perkin Elmer). Relative numbers of lysosomes were quantified as the number of lysotracker-positive-stained spots above the intensity threshold set by 1 h treatment with 50 μM CQ. Data output was in text file format and graphed using the Microsoft Excel and Igor Pro software.
Statistical analysis
All values presented in the Results section and figures were obtained from minimum triplicate analysis (real n is mentioned in each figure legend as it pertains to individual experiments) and from at least three independent experiments. All data were expressed as mean±S.D. or S.E.M. When a significant effect was obtained with one-way ANOVA, Dunnett’s test was used to compare all values to the relative control (comparisons are clearly indicated in the figure legends and Results section. Alternatively, Student’s t-test was used to analyze significant differences between the means of 2–4 groups, and Bonferroni’s correction was applied. For labeling in figures, the following premise was applied ubiquitously: *P<0.05, **P<0.01, ***P<0.001. To measure effects between celastrol dose–response groups, two-way ANOVA was applied, and the P-value for the measured between-groups effect is reported with Bonferroni’s correction applied (due to number of times (>10), this type of comparison was applied in the overall study). For this type of comparison only, an alpha level of 0.01 was set when determining if the null hypothesis was true or false.
Abbreviations
- 17-AAG:
-
17-N-Allylamino-17-demethoxygeldanamycin
- DTT:
-
dithiothreitol
- ER:
-
endoplasmic reticulum
- ESI-MS:
-
electrospray ionization mass spectrometer
- GBM:
-
glioblastoma multiforme
- GSH:
-
glutathione
- HSE:
-
heat-shock response element
- HSF-1:
-
heat-shock factor 1
- HSP:
-
heat-shock protein
- KEAP-1:
-
Kelch-like-ECH-associated protein 1
- L-BSO:
-
L-buthionine sulfoximine
- NAC:
-
N-acetylcysteine
- PARP-1:
-
poly(ADP-ribose) polymerase-1
- ROS:
-
reactive oxygen species
- SAPK:
-
stress-activated protein kinase
References
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114: 97–109.
Wen PY, Kesari S . Malignant gliomas in adults. N Engl J Med 2008; 359: 492–507.
Lima FR, Kahn SA, Soletti RC, Biasoli D, Alves T, da Fonseca AC et al. Glioblastoma: therapeutic challenges, what lies ahead. Biochim Biophys Acta 2012; 1826: 338–349.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–996.
Wang Y, Jiang T . Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett 2013; 331: 139–146.
Easaw JC, Mason WP, Perry J, Laperriere N, Eisenstat DD, Del Maestro R et al. Canadian recommendations for the treatment of recurrent or progressive glioblastoma multiforme. Curr Oncol 2011; 18: e126–e136.
Norden AD, Lesser GJ, Drappatz J, Ligon KL, Hammond SN, Lee EQ et al. Phase 2 study of dose-intense temozolomide in recurrent glioblastoma. Neuro Oncol 2013; 15: 930–935.
Bai RY, Staedtke V, Riggins GJ . Molecular targeting of glioblastoma: drug discovery and therapies. Trends Mol Med 2011; 17: 301–312.
Rajendran P, Li F, Shanmugam MK, Kannaiyan R, Goh JN, Wong KF et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev Res (Phila) 2012; 5: 631–643.
Kannaiyan R, Manu KA, Chen L, Li F, Rajendran P, Subramaniam A et al. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 2011; 16: 1028–1041.
Lee JH, Won YS, Park KH, Lee MK, Tachibana H, Yamada K et al. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis 2012; 17: 1275–1286.
Tozawa K, Sagawa M, Kfki M . Quinone methide tripterine, celastrol, induces apoptosis in human myeloma cells via NF-kappaB pathway. Int J Oncol 2011; 39: 1117–1122.
Jung HW, Chung YS, Kim YS, Park YK . Celastrol inhibits production of nitric oxide and proinflammatory cytokines through MAPK signal transduction and NF-kappaB in LPS-stimulated BV-2 microglial cells. Exp Mol Med 2007; 39: 715–721.
Kannaiyan R, Shanmugam MK, Sethi G . Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett 2010; 303: 9–20.
Kim DH, Shin EK, Kim YH, Lee BW, Jun JG, Park JH et al. Suppression of inflammatory responses by celastrol, a quinone methide triterpenoid isolated from Celastrus regelii. Eur J Clin Invest 2009; 39: 819–827.
Paimela T, Hyttinen JM, Viiri J, Ryhanen T, Karjalainen RO, Salminen A et al. Celastrol regulates innate immunity response via NF-kappaB and Hsp70 in human retinal pigment epithelial cells. Pharmacol Res 2011; 64: 501–508.
Kagan VE, Konduru NV, Feng W, Allen BL, Conroy J, Volkov Y et al. Carbon nanotubes degraded by neutrophil myeloperoxidase induce less pulmonary inflammation. Nat Nanotechnol 2010; 5: 354–359.
Kim JE, Lee MH, Nam DH, Song HK, Kang YS, Lee JE et al. Celastrol, an NF-kappaB inhibitor, improves insulin resistance and attenuates renal injury in db/db Mice. PLoS One 2013; 8: e62068.
Ge P, Ji X, Ding Y, Wang X, Fu S, Meng F et al. Celastrol causes apoptosis and cell cycle arrest in rat glioma cells. Neurol Res 2010; 32: 94–100.
Huang Y, Zhou Y, Fan Y, Zhou D . Celastrol inhibits the growth of human glioma xenografts in nude mice through suppressing VEGFR expression. Cancer Lett 2008; 264: 101–106.
Chen M, Rose AE, Doudican N, Osman I, Orlow SJ . Celastrol synergistically enhances temozolomide cytotoxicity in melanoma cells. Mol Cancer Res 2009; 7: 1946–1953.
Sung B, Park B, Yadav VR, Aggarwal BB . Celastrol, a triterpene, enhances TRAIL-induced apoptosis through the down-regulation of cell survival proteins and up-regulation of death receptors. J Biol Chem 2010; 285: 11498–11507.
Zhu H, Liu XW, Ding WJ, Xu DQ, Zhao YC, Lu W et al. Up-regulation of death receptor 4 and 5 by celastrol enhances the anti-cancer activity of TRAIL/Apo-2L. Cancer Lett 2010; 297: 155–164.
Kim Y, Kang H, Jang SW, Ko J . Celastrol inhibits breast cancer cell invasion via suppression of NF-kB-mediated matrix metalloproteinase-9 expression. Cell Physiol Biochem 2011; 28: 175–184.
Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol 2006; 72: 1311–1321.
Sethi G, Ahn KS, Pandey MK, Aggarwal BB . Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood 2007; 109: 2727–2735.
Shao L, Zhou Z, Cai Y, Castro P, Dakhov O, Shi P et al. Celastrol suppresses tumor cell growth through targeting an AR-ERG-NF-kappaB pathway in TMPRSS2/ERG fusion gene expressing prostate cancer. PLoS One 2013; 8: e58391.
Chen G, Zhang X, Zhao M, Wang Y, Cheng X, Wang D et al. Celastrol targets mitochondrial respiratory chain complex I to induce reactive oxygen species-dependent cytotoxicity in tumor cells. BMC Cancer 2011; 11: 170.
Ponten J, Macintyre EH . Long term culture of normal and neoplastic human glia. Acta Pathol Microbiol Scand 1968; 74: 465–486.
Westermark B, Ponten J, Hugosson R . Determinants for the establishment of permanent tissue culture lines from human gliomas. Acta Pathol Microbiol Scand A 1973; 81: 791–805.
Ponten J, Westermark B . Properties of human malignant glioma cells in vitro. Med Biol 1978; 56: 184–193.
Khatchadourian A, Krumova K, Boridy S, Ngo AT, Maysinger D, Cosa G . Molecular imaging of lipid peroxyl radicals in living cells with a BODIPY-alpha-tocopherol adduct. Biochemistry 2009; 48: 5658–5668.
Seo WY, Goh AR, Ju SM, Song HY, Kwon DJ, Jun JG et al. Celastrol induces expression of heme oxygenase-1 through ROS/Nrf2/ARE signaling in the HaCaT cells. Biochem Biophys Res Commun 2011; 407: 535–540.
Klaic L, Trippier PC, Mishra RK, Morimoto RI, Silverman RB . Remarkable stereospecific conjugate additions to the Hsp90 inhibitor celastrol. J Am Chem Soc 2011; 133: 19634–19637.
Castello PR, Drechsel DA, Patel M . Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem 2007; 282: 14186–14193.
Walcott SE, Heikkila JJ . Celastrol can inhibit proteasome activity and upregulate the expression of heat shock protein genes, hsp30 and hsp70, in Xenopus laevis A6 cells. Comp Biochem Physiol A Mol Integr Physiol 2010; 156: 285–293.
Yang H, Chen D, Cui QC, Yuan X, Dou QP . Celastrol, a triterpene extracted from the Chinese ‘Thunder of God Vine,’ is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 2006; 66: 4758–4765.
Deng YN, Shi J, Liu J, Qu QM . Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy. Neurochem Int 2013; 63: 1–9.
Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH et al. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J Cell Physiol 2012; 227: 2196–2206.
Gonzalez P, Mader I, Tchoghandjian A, Enzenmuller S, Cristofanon S, Basit F et al. Impairment of lysosomal integrity by B10, a glycosylated derivative of betulinic acid, leads to lysosomal cell death and converts autophagy into a detrimental process. Cell Death Differ 2012; 19: 1337–1346.
Conn CS, Qian SB . Nutrient signaling in protein homeostasis: an increase in quantity at the expense of quality. Sci Signal 2013; 6: ra24.
Chow AM, Brown IR . Induction of heat shock proteins in differentiated human and rodent neurons by celastrol. Cell Stress Chaperones 2007; Autumn 12: 237–244.
Wang Y, Gibney PA, West JD, Morano KA . The yeast Hsp70 Ssa1 is a sensor for activation of the heat shock response by thiol-reactive compounds. Mol Biol Cell 2012; 23: 3290–3298.
Arora S, Yang JM, Hait WN . Identification of the ubiquitin-proteasome pathway in the regulation of the stability of eukaryotic elongation factor-2 kinase. Cancer Res 2005; 65: 3806–3810.
Whitesell L, Lindquist SL . HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005; 5: 761–772.
Ding QH, Cheng Y, Chen WP, Zhong HM, Wang XH . Celastrol, an inhibitor of heat shock protein 90beta potently suppresses the expression of matrix metalloproteinases, inducible nitric oxide synthase and cyclooxygenase-2 in primary human osteoarthritic chondrocytes. Eur J Pharmacol 2013; 708: 1–7.
Sreeramulu S, Gande SL, Gobel M, Schwalbe H . Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl 2009; 48: 5853–5855.
Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D . Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem 2009; 284: 35381–35389.
Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E et al. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem 1997; 272: 23843–23850.
Schnur RC, Corman ML, Gallaschun RJ, Cooper BA, Dee MF, Doty JL et al. erbB-2 oncogene inhibition by geldanamycin derivatives: synthesis, mechanism of action, and structure-activity relationships. J Med Chem 1995; 38: 3813–3820.
Cruickshanks N, Shervington L, Patel R, Munje C, Thakkar D, Shervington A . Can hsp90alpha-targeted siRNA combined with TMZ be a future therapy for glioma? Cancer Invest 2010; 28: 608–614.
Jane EP, Pollack IF . The heat shock protein antagonist 17-AAG potentiates the activity of enzastaurin against malignant human glioma cells. Cancer Lett 2008; 268: 46–55.
Seo HR, Seo WD, Pyun BJ, Lee BW, Jin YB, Park KH et al. Radiosensitization by celastrol is mediated by modification of antioxidant thiol molecules. Chem Biol Interact 2011; 193: 34–42.
Zhang Y, Ahn YH, Benjamin IJ, Honda T, Hicks RJ, Calabrese V et al. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem Biol 2011; 18: 1355–1361.
Motohashi H, Yamamoto M . Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 2004; 10: 549–557.
Miyata Y, Rauch JN, Jinwal UK, Thompson AD, Srinivasan S, Dickey CA et al. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem Biol 2012; 19: 1391–1399.
Zhu H, Yang W, He LJ, Ding WJ, Zheng L, Liao SD et al. Upregulating Noxa by ER stress, celastrol exerts synergistic anti-cancer activity in combination with ABT-737 in human hepatocellular carcinoma cells. PLoS One 2012; 7: e52333.
Kar R, Singha PK, Venkatachalam MA, Saikumar P . A novel role for MAP1 LC3 in nonautophagic cytoplasmic vacuolation death of cancer cells. Oncogene 2009; 28: 2556–2568.
Singha PK, Pandeswara S, Venkatachalam MA, Saikumar P . Manumycin A inhibits triple-negative breast cancer growth through LC3-mediated cytoplasmic vacuolation death. Cell Death Dis 2013; 4: e457.
Shaid S, Brandts CH, Serve H, Dikic I . Ubiquitination and selective autophagy. Cell Death Differ 2013; 20: 21–30.
Komatsu M, Kageyama S, Ichimura Y . p62/SQSTM1/A170: physiology and pathology. Pharmacol Res 2012; 66: 457–462.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137: 1062–1075.
Moscat J, Diaz-Meco MT . p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137: 1001–1004.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282: 24131–24145.
Su H, Wang X . p62 Stages an interplay between the ubiquitin-proteasome system and autophagy in the heart of defense against proteotoxic stress. Trends Cardiovasc Med 2011; 21: 224–228.
Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC . Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33: 517–527.
Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G . Cell death assays for drug discovery. Nat Rev Drug Discov 2011; 10: 221–237.
Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL et al. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells 2009; 27: 1722–1733.
Acknowledgements
The authors acknowledge: (1) Mr. Wolfgang Ernst Reintsch for assistance with Operetta High Content Screening System in the Department of Pharmacology and Therapeutics Green Chemistry and Green Chemicals Research Facility. Purchase of equipment in this facility was made possible with funding from the Canadian Foundation for Innovation (CFI) and the Ministère du Développement économique, innovation et exportation Québec (MDEIE), (2) Dr. Annie Castonguay for measurements and analysis with electrospray ionization mass spectrometer (ESI-MS), and (3) Dr. Mirela Matokanovic for insightful discussions. This work was supported by funding from the Canadian Institutes of Health Research (MOP-142626, MOP-119425). SB is supported by a Vanier Canada Graduate Scholarship through the Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Finazzi-Agrò
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Boridy, S., Le, P., Petrecca, K. et al. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis 5, e1216 (2014). https://doi.org/10.1038/cddis.2014.182
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.182
Keywords
This article is cited by
-
Exploring the combined anti-cancer effects of sodium butyrate and celastrol in glioblastoma cell lines: a novel therapeutic approach
Medical Oncology (2024)
-
Celastrol exerts a neuroprotective effect by directly binding to HMGB1 protein in cerebral ischemia–reperfusion
Journal of Neuroinflammation (2021)
-
Gedunin isolated from the mangrove plant Xylocarpus granatum exerts its anti-proliferative activity in ovarian cancer cells through G2/M-phase arrest and oxidative stress-mediated intrinsic apoptosis
Apoptosis (2020)
-
Celastrol enhances TRAIL-induced apoptosis in human glioblastoma via the death receptor pathway
Cancer Chemotherapy and Pharmacology (2019)
-
Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule
Cell Death Discovery (2018)
