
Abstract
Autophagic and proteasomal degradation constitute the major cellular proteolysis pathways. Their physiological and pathophysiological adaptation and perturbation modulates the relative abundance of apoptosis-transducing proteins and thereby can positively or negatively adjust cell death susceptibility. In addition to balancing protein expression amounts, components of the autophagic and proteasomal degradation machineries directly interact with and co-regulate apoptosis signal transduction. The influence of autophagic and proteasomal activity on apoptosis susceptibility is now rapidly gaining more attention as a significant modulator of cell death signalling in the context of human health and disease. Here we present a concise and critical overview of the latest knowledge on the molecular interplay between apoptosis signalling, autophagy and proteasomal protein degradation. We highlight that these three pathways constitute an intricate signalling triangle that can govern and modulate cell fate decisions between death and survival. Owing to rapid research progress in recent years, it is now possible to provide detailed insight into the mechanisms of pathway crosstalk, common signalling nodes and the role of multi-functional proteins in co-regulating both protein degradation and cell death.
Similar content being viewed by others


Facts
-
Apoptosis susceptibility is modulated by autophagic and proteasomal activity.
-
Pathway interplay converges at core signalling nodes which modulate Bcl-2 family signalling and initiator caspase-8 activation.
-
Pathway crosstalk is bi-directional, entails positive and negative feedback loops and previously unknown apoptosis initiation platforms.
Open Questions
-
Will we be able to progress from a qualitative to a quantitative and kinetic understanding of the signalling interplay?
-
What are the components of the novel apoptosis initiation platforms and can we exploit these as drug targets?
-
Can perturbations in these pathway interplays be associated with human pathophysiologies?
A Signalling Triangle of Apoptosis, Autophagy and Proteasomal Protein Degradation
The core molecular details of apoptosis signal transduction and its role as a programmed cell death modality have been described in great detail. Apoptosis eliminates cells primarily through the activation of proteases of the caspase family, whereby initiator caspases proteolytically activate executioner caspases. These in turn disintegrate cellular content and evoke features characteristic for apoptotic cell death, such as phosphatidylserine exposure, chromatin condensation, nuclear fragmentation, cellular shrinkage and membrane blebbing.1, 2 In recent years, it has become apparent that apoptosis signalling exhibits substantial molecular crosstalk with pathways controlling the degradation of short- and long-lived proteins, namely the ubiquitin-proteasome system (UPS) as well as macroautophagy (hereafter referred to as autophagy).3, 4 Besides the modulation of apoptosis by the UPS and by autophagic signalling, also direct inter-dependencies and co-regulations exist between these two degradation pathways. Together, these result in an intricate signalling triangle that governs and balances cell survival and death signalling (Figure 1). In the following, we review the current knowledge on the central signalling features and molecular interactions associated with these interplays. This entails an overview of well-established interactions and co-modulations that can be associated with the three main axes of the signalling triangle (autophagy–apoptosis, UPS–apoptosis and autophagy–UPS) and that primarily involve the crosstalk with apoptosis signalling at the mitochondrial interface between apoptosis initiation and execution. This is followed by an assessment of recent evidence which indicates that the apoptosis–autophagy–UPS triangle also encompasses a currently underappreciated and incompletely described signalling module that regulates the formation of upstream signalling platforms by which autophagic and UPS activities are balanced versus apoptosis initiation through the activation of initiator caspase-8.

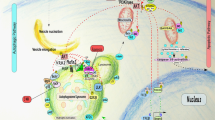
A signalling triangle of autophagy, the UPS and apoptosis modulates cell death susceptibility and balances cell death versus survival decisions
Autophagic Signalling and its Interactions with Apoptosis Signal Transduction
Autophagy typically constitutes a cytoprotective response that counteracts stresses such as nutrient deprivation or protein aggregation and, in the case of mitophagy, serves to remove old or damaged mitochondria that may otherwise cause harm by the excessive production of reactive oxygen species. Autophagosomes, double membrane vesicles that enclose material destined for degradation, form during autophagy and subsequently fuse with lysosomes. The content of the resulting autophagolysosomes is then hydrolysed to provide an intracellular source of nutrients.5 In cytoprotective scenarios, autophagy may suppress, avert or delay cell death. However, if autophagy is excessively induced and if autophagic flux remains elevated over prolonged times, autophagy can ultimately likewise result in cell death. Such conditions may also prime cells for apoptosis, as demonstrated, for example, by enhanced apoptosis sensitivity at conditions of endoplasmatic reticulum stress.6 The context-dependent role of autophagy in promoting or antagonising cell death susceptibility therefore needs attention when interpreting the functional role of autophagic features that may manifest in cell death scenarios.7, 8
A well-described molecular interplay between autophagy and apoptosis manifests at the mitochondrial interface between apoptosis initiation and execution. This interplay links anti-apoptotic Bcl-2 family members, which prevent apoptotic mitochondrial engagement, into the regulation of autophagic flux through interactions with Beclin-1. Beclin-1 is one of several protein components required for autophagy initiation and drives the early steps of autophagosome formation through its interaction with Vps34, a phosphoinositide 3-kinase.9 Vps34 in turn recruits additional autophagy-related (Atg) proteins such as Atg3, 4, 5, 7, 10, 12 and 16, and their interplay coordinates autophagosomal membrane nucleation and expansion.5 Beclin-1 interacts with anti-apoptotic members of the Bcl-2 protein family such as Bcl-2, Bxl-xL and Mcl-1 through its BH3 domain motif, and these interactions suppress Beclin-1-dependent autophagy initiation10, 11, 12, 13, 14 (Figure 2a). Anti-apoptotic Bcl-2 proteins thereby obtain significant regulatory influence over cellular autophagic flux. In particular, Mcl-1 is rapidly degraded in response to nutrient starvation, resulting in Beclin-1 liberation and efficient autophagy induction.14 Importantly, it was shown that Beclin-1 fails to reciprocally reduce the anti-apoptotic potential of Bcl-2, demonstrating that the interaction with anti-apoptotic Bcl-2 proteins uni-directionally limits autophagy initiation.15 The suppression of autophagy can be alleviated by the BH3-only proteins Bad and BNIP3. Bad and BNIP3 competitively bind to anti-apoptotic Bcl-2 members promoting apoptosis through the activation and oligomerisation of Bax and Bak and can reinstate Beclin-1-driven autophagy (Figure 2a).13, 16, 17 Similar to Bad and BNIP3, also synthetic BH3 mimetics such as ABT-737 and HA14-1 induce autophagy by Beclin-1 liberation in addition to triggering apoptotic responses.18 Interestingly, the BH3-only protein Bim, which needs to be phosphorylated by JNK to become pro-apoptotic, can directly interact with Beclin-1 and LC8 in its apoptosis-inactive form.19 Bim thereby suppresses autophagy by retaining Beclin-1 at the dynein motor complex. Upon nutrient starvation Bim is phosphorylated, Beclin-1 is liberated and promotes autophagy19 (Figure 2a). Apoptotic signalling can therefore be accompanied by autophagy induction. Of note, nutrient depletion can also suppress apoptosis induction. Increases in mTOR activity upon starvation result in the phosphorylation and inactivation of Bad.20 Therefore, the nutrient sensing machinery upstream of autophagosome formation can actively drive or suppress apoptosis induction through Bim and Bad. As Bcl-2 family members can modulate bioenergetic signalling independent of their apoptotic functions,21 the interplay and regulation between autophagic and apoptotic responses may be more complex than is currently understood.

Crosstalk and interactions in the autophagy–apoptosis and UPS–apoptosis interplays. (a) The major components regulating the interplay of autophagy and apoptosis at the level of Beclin-1 and Bcl-2 family members are shown. Unphosphorylated Bim forms a complex with Beclin-1 and the adaptor protein LC8. Free Beclin-1 can be antagonised by anti-apoptotic Bcl-2 family proteins. The latter can be inhibited by phosphorylated Bim and other BH3-only proteins such as Bad and Bnip3, resulting in liberation of Beclin-1 and induction of autophagy. Phosphorylated Bim can activate Bax and Bak, resulting in caspase activation and apoptosis execution. Beclin-1 can be cleaved by effector caspases and is thereby converted to a pro-apoptotic protein. The interaction of positive and negative regulatory processes therefore determines the induction of autophagy and/or apoptosis. (b) The main processes and players involved in the control and modulation of apoptosis by proteasomal activity are displayed. (i) The steady state expression amounts of both pro- and anti-apoptotic proteins are co-regulated by proteasomal activity. Pro-apoptotic proteins that are strongly regulated include Bax and the BH3-only proteins Bim, Noxa and Bik, anti-apoptotic proteins include Mcl-1, cFLIP and cIAPs. (ii) Inhibition of proteasomal activity disturbs the steady state. As a consequence, high turn-over proteins accumulate. In addition, proteasome inhibition can actively induce the expression of pro-apoptotic proteins. Proteasome inhibition can induce apoptosis and is accompanied by compensatory induction of autophagy
Although Beclin-1 does not limit the anti-apoptotic potency of Bcl-2 family proteins, it can strengthen cell death responses subsequent to apoptosis initiation. Caspases can cleavage Beclin-1 and thereby convert it from a pro-autophagic to a pro-apoptotic protein. The COOH-terminal cleavage fragment of Beclin-1 integrates into the outer mitochondrial membrane and promotes cytochrome c release and apoptosis execution22 (Figure 2a). A similar proteolysis-driven conversion of function has been described for Atg5, an E3 ubiquitin ligase component of the LC3 lipidation complex that is required for autophagosome formation. During apoptosis scenarios that are associated with calpain activation, calpains cleave Atg5 and the cleavage product antagonises anti-apoptotic Bcl-2 family proteins.23 Together, this highlights a substantial interplay between autophagic and apoptotic signalling through a number of well-characterised molecular interactions. These interactions can modulate the thresholds for autophagy and apoptosis induction.
The UPS and Interactions with Apoptosis Signal Transduction
Besides autophagy, the UPS is a second evolutionarily conserved degradation pathway which likewise is interlinked with apoptosis signal transduction by co-modulating relationships.4 Proteasomes are multi-protein complexes responsible for the selective degradation of mostly cytoplasmic proteins, in particular misfolded and high-turnover proteins.24, 25 Proteasomes possess multiple proteolytically active sites that degrade proteins into peptides with a length of 3–25 amino acids. Proteins are marked for proteasomal degradation when covalently labelled with K48-linked poly-ubiquitin chains by substrate-specific E3 ubiquitin ligases.25 However, non-canonical ubiquitin-based post-translational modifications that target proteins for proteasomal degradation have been described as well.26 The steady-state expression amounts of various pro- and anti-apoptotic proteins, such as high-turnover BH3-only proteins Bik, Bim and Noxa and also Bax, Mcl-1, cFLIP species or cIAPs are strongly influenced by changes in proteasomal activity27, 28, 29, 30, 31, 32 (Figure 2b). Changes in the activity and in the selectivity of the ubiquitin-labelling and proteolytic machinery of the UPS therefore provide the possibility to swiftly increase or decrease apoptosis responsiveness. For example, the accumulation of cFLIP species can delay or prevent caspase-8 activation during extrinsic apoptosis,32, 33 Mcl-1 is degraded upon interleukin-3 withdrawal and increases cell death susceptibility,34 and BH3-only protein Bim-EL accumulates upon proteasome inhibition to trigger the intrinsic apoptosis pathway.35 During apoptosis, executioner caspases cleave and inactivate the proteasome,36 thereby stabilising pro-apoptotic proteins such as cytosolic Smac, BH3-only proteins and active caspases. In addition, the prolonged inhibition of the proteasome in itself can induce apoptosis by active transcriptional responses such as p53-dependent and -independent upregulations of BH3-only proteins and by the parallel suppression of NFκB-dependent survival signalling through IκBα stabilisation37, 38 (Figure 2b). Proteasome inhibitors also synergistically enhance the susceptibility to various intrinsic and extrinsic apoptosis stimuli in co-treatment scenarios.39, 40 The interplay between the UPS and apoptosis is therefore complex and, depending on the signalling context, UPS activity can enhance or reduce apoptosis susceptibility.
Interplay of Autophagic and Proteasomal Protein Degradation Pathways
Besides the interplay of autophagy and the UPS with apoptosis signal transduction, significant co-modulation and crosstalk exists also between autophagic and UPS signalling at the level of substrate labelling and recognition. Ubiquitin has a central role in this interplay due to its dual functionality in channelling substrates either through proteasomal or autophagic degradation. Although K48 ubiquitin chains target labelled proteins for proteasomal degradation, K63-linked ubiquitin chains direct proteins towards autophagy-dependent lysosomal degradation.41 A dual functionality that carries relevance for both proteasomal and autophagic degradation pathways has also been observed at the level of E3 ubiquitin ligases, with the E3 ligase CHIP being capable of generating both K48- and K63-linked ubiquitin chains.42 Although this already indicates that autophagic and proteasomal flux can be cross-modulated, the ubiquitin binding protein and autophagy receptor p62/SQSTM1 exerts further control on this interplay. p62/SQSTM1 effectively binds to K63-ubiquitin chains to shuttle labelled proteins towards autophagic degradation. However, p62/SQSTM1 also binds K48 ubiquitin chains, albeit with lower affinity.43 Although it was reported that p62/SQSTM1 can assist in channelling substrates through the proteasomal degradation pathway,44 it also delays or reduces proteasomal degradation of short-lived proteins, including cell death regulator p53, when accumulating excessively as a consequence of autophagy inhibition.45, 46 It was suggested that the inhibition of proteasomal flux might arise from two factors. First, p62/SQSTM1-associated protein aggregates may become too bulky for proteasomal degradation and, second, high amounts of p62/SQSTM1 may outcompete other ubiquitin binding proteins that promote efficient proteasomal protein degradation.47 Overall, the influence of p62/SQSTM1 at the interface of autophagy and proteasomal degradation therefore appears substantial. In contrast, an inhibition of autophagy is not observed when inhibiting the UPS. Rather, proteasome inhibition results in increased steady-state levels of p62 and elevated autophagic flux,48, 49 indicating that cells may actively respond to compensate for impaired UPS function.
The Interplay of Autophagic and Proteasomal Activities Controls Lethal and Non-lethal Caspase-8 Activation
Although the interplay of apoptosis with autophagy and the UPS at the mitochondrial interface between apoptosis initiation and execution has been described in great detail, several recent studies have demonstrated that so far poorly characterised signalling platforms may form upstream or in parallel to this interplay. These platforms promote the activation of initiator caspase-848, 50, 51 (Figure 3). Although the apical activation of caspase-8 may have implications for apoptosis initiation signalling, these platforms could also be involved in regulating non-lethal caspase-8 activities that could be relevant for cellular proliferation and differentiation.52

Proposed components and composition of a caspase-8 activation platform that forms as a signalling node in the interplay of apoptosis, autophagy and the UPS. On the basis of the accumulated experimental evidence of a number of recent studies, the molecular composition and formation mechanism for a novel caspase-8 activation platform is proposed. The formation of the activation platform requires the interplay of a number of Atg proteins and LC3 as well as core proteins that are relevant also during extrinsic apoptosis induction (Fadd, cFLIP, caspase-8). Lipidated LC3II, together with Atg5-12, Atg16, Fadd and p62 appear to contribute to caspase-8 activation and may form a multi-molecular activation platform on autophagosomal membranes. Details on these interactions and their implications for caspase-8 activation and apoptosis induction are provided in the text
Caspase-8 is predominantly known as the primary initiator caspase of the extrinsic apoptosis pathway that is triggered by death ligands binding to their respective cell surface receptors. The activation of caspase-8 in the context of proteasome inhibition and autophagic signalling instead occurs independently of death ligands on an alternative intracellular activation platform.48, 51 Caspase-8 appears to contribute substantially towards the potency of proteasome inhibitors in eliminating human cancer cells, in particular those that express high amounts of anti-apoptotic Bcl-2 family proteins.48, 50 Caspase-8 activation upon proteasome inhibition requires FADD, an adaptor protein that is also required for procaspase-8 aggregation and activation during extrinsic apoptosis, and also depends on the induction of autophagy.48 Inhibition of autophagy by the PI3K-inhibitor 3-methyl adenine or inhibition of autophagosome formation by Atg5 deletion significantly reduce caspase-8 activation and apoptotic responses upon proteasome inhibition.48, 51 In this context, it is interesting to note that FADD was previously shown to bind to Atg5 in vitro and in vivo in a study that investigated cell death regulation in response to interferon γ.53 In addition to FADD and Atg5, microtubule-associated protein 1 light chain 3 (LC3) and p62/SQSTM1 further promote caspase-8 activation upon proteasome inhibition.50 p62/SQSTM1 accumulates upon proteasome inhibition,48 and p62/SQSTM1 has previously also been shown to promote the efficient activation of poly-ubiquitylated caspase-8 during canonical extrinsic apoptosis.54 A recent study further supports the role of p62/SQSTM1 in promoting caspase-8-dependent apoptosis in scenarios of autophagy induction in co-treatment scenarios with proteasome inhibitors.55 p62/SQSTM1 may therefore be a molecular link that not only has a role in the interplay between UPS and autophagy, but also co-regulates caspase-8 activation both during extrinsic caspase-8-dependent apoptosis and during intrinsic scenarios of apical caspase-8 activation.
Interestingly, it was reported that cellular FLICE inhibitory protein (cFLIP), an inactive caspase-8 homologue, can bind to Atg3, an E2-like conjugating enzyme for LC3. cFLIP thereby prevents LC3I-II conversion and limits autophagy induction and autophagic flux.56 cFLIP is known to accumulate upon proteasome inhibition, and we have previously demonstrated that even though proteasome inhibition induces autophagy, LC3-I is inefficiently converted to LC3-II.48 This indicates that cFLIP may limit increases in autophagic flux following proteasome inhibition. It can be speculated that because of the structural similarities to cFLIP also caspase-8 may interact with Atg3, but experimental proof so far is missing. Nevertheless, similar to the loss of Atg5, also Atg3 deficiency reduces caspase-8 activity, indicating that it has a role in the activation process and probably is an important part of the activation platform. As Atg3 can be cleaved by caspases, probably including caspase-8,57 Atg3 cleavage may further reduce overall autophagic flux and could contribute to establishing an additional layer of control at the interface of autophagy and apoptosis induction. Of note, it has been reported that during canonical extrinsic apoptosis induction active caspase-8 is eliminated by autophagy.58 Limiting autophagic flux in parallel to or directly by caspase-8 activation could therefore also be a prerequisite to maintain caspase-8 activities for prolonged times.
An autophagy-dependent activation of caspase-8 was also reported in response to sphingosine kinase inhibition.51 However, it is currently unclear whether sphingosine kinase inhibition generally triggers autophagy and whether apoptosis is the dominant resulting cell death modality. The observed responses appear to depend substantially on the chosen experimental system.59, 60 Another report demonstrated that glucose starvation can trigger caspase-8 activation.61 Although the underlying molecular mechanism has not yet been identified, glucose deprivation is known to potently induce autophagic responses. A similar signalling scenario therefore may be implicated in this condition. Interestingly, interaction complexes between FADD and Atg5-12 that drive caspase-8 activation have also been reported in proliferating T cells.62 Here, caspase-8 activity was shown to limit autophagic flux and thereby to contribute to cell survival. T cell death in absence of caspase-8 activity could be prevented by inhibition of RIP kinase 1 by necrostatin, indicating a role for necroptosis in shaping T-cell populations. Although the interplay of necroptosis and apoptosis and their dependence on caspase-8 and RIP kinase signalling has been further elucidated in the meantime,63 it needs to be noted that RIP-mediated necroptosis appears not to contribute to cancer cell death in response to proteasome inhibition.48 However, cancer cells frequently appear to lack expression of the crucial RIPK1 substrate and downstream necroptosis mediator RIPK3.64 RIPK1 was also reported to be part of non-canonical caspase-8 activation complexes, termed ripoptosomes, that form in response to genotoxic stress or during innate immune responses, in particular when IAPs are depleted.65, 66 RIPK1 activity is required for the formation of these complexes, and proteasome inhibition further enhances caspase-8 activation on ripoptosomes.65, 66 However, ripoptosomes contain only a fraction of the total cellular caspase-8 pool,65, 66 and the potency of proteasome inhibitors in inducing apical caspase-8 processing appears to be significantly higher than what can be achieved with genotoxic drugs such as 5-fluorouracil or cisplatin.48 It is therefore currently unclear whether proteasome inhibition enhances casapase-8 activation upon genotoxic stress by a more efficient formation of ripoptosomes or rather by inducing an independent autophagy-associated response of caspase-8 activation as described above.
Conclusions and Perspectives
We here described and discussed the most central aspects of the interplay of apoptosis signalling, autophagy and proteasomal protein degradation. This interplay is complex and provides a multi-factorial control over the modulation of apoptosis susceptibility and cell fate decisions between death and survival. Of note, the currently published data indicate that this modulation appears to manifest predominantly through interactions with the apoptosis initiation network. With the exception of the multifunctional caspase inhibitor x-linked inhibitor of apoptosis protein that has now been described to possess an autophagy suppressor function,67 components of apoptosis execution signalling so far have not been implicated as central regulators of this interplay. However, as effector caspases cleave and thereby inactivate or functionally transform critical components of the autophagy and UPS signalling systems,22, 36, 68 downstream feedbacks from drivers of apoptosis execution may significantly contribute towards ensuring strict and efficient cell death decisions by attenuating proteasomal and autophagic fluxes.
Although a number of recent studies provide evidence which points out that caspase-8 is a central player that links autophagy-UPS signalling to apoptosis signal transduction, further fundamental studies will be required to elucidate and understand the control and regulation of this interplay at the molecular level. For example, even though some key components required for caspase-8 activation in these scenarios are known, information on their stoichiometry within the caspase-8 activation platforms is not available. Quantitative biochemical and structural biological studies were successfully conducted in recent years to determine the relative protein abundances and quaternary structures of caspase-8 activation platforms during canonical extrinsic apoptosis initiation.69, 70, 71 Adapted accordingly, further insight into the molecular mechanisms of alternative modes of caspase-8 activation, and their control by autophagic and proteasomal flux can be expected in the future. Besides the molecular composition of such caspase-8 activation platforms, also the regulation of post-translational modifications will likely constitute an additional layer of control. Experimental evidence indicates that posttranslational control over extrinsic, DISC-dependent caspase-8 activation encompasses glycosylation, nitrosylation, ubiquitylation, and phosphorylation, and that these modifications can be physiologically and pathophysiologically relevant.72, 73 For example, it is well established that caspase-8 can be phosphorylated on conserved tyrosine residues by several kinases, including Src kinases, to prevent or delay its activation during extrinsic apoptosis.74, 75 Interestingly, Src kinase can be eliminated by autophagy.76 It therefore appears to be warranted to further investigate whether the autophagic elimination of Src kinase activity is required to allow an efficient activation of caspase-8 activation on autophagosomal membranes. In general, investigations taking into account posttranslational modifications therefore could provide further important insight into the modulation of caspase-8 activation and the interplay of apoptosis initiation with proteasomal and autophagic degradation pathways. As caspase-8 cleaves and activates the Bcl-2 family protein Bid, the modulation of apoptosis initiation through the formation of caspase-8 on non-canonical activation platforms (Figure 3) will also need to be investigated in the context of the co-regulation of protein degradation pathways and apoptosis initiation at the level of Bcl-2 family proteins (Figure 2).
As the work discussed here has shown, the consequences of perturbed autophagic or proteasomal signalling on apoptosis susceptibility can be ambiguous (Figure 4). Care therefore needs to be taken when investigating and interpreting experimental data on causes and effects in these scenarios. In particular, generalised statements on whether apoptosis susceptibility will be enhanced or decreased by modulating the main cellular proteolytic pathways may not be warranted. Translational and clinical studies investigating the role of autophagic signalling in cancer have already highlighted that whether autophagy promotes or inhibits cancer development and progression must be assessed individually for each scenario under investigation.77 For example, autophagy is required for tumour growth in pancreatic cancers,78 whereas in early-stage cutaneous melanoma higher levels of autophagy correlate with prolonged progression-free survival times.79 Besides these examples, deregulations in autophagic signalling and in the UPS have widely been associated with human pathophysiologies, including neurodegenerative diseases, cardiovascular diseases, and infectious diseases.80, 81, 82 In the light of these findings, further research on the interplay of autophagy and the UPS with cell death signalling pathways will likely be highly relevant to understand how the respective disease phenotypes are established. A detailed understanding of these interplays may therefore allow us to associate quantitative information on pathway crosstalks and activities with pathophysiological consequences. Ultimately, this may serve to devise novel targeted interventions that therapeutically may be broadly applicable.

Perturbation–response relationships in the apoptosis–autophagy–UPS triangle. An overview of the perturbations and their consequences in the apoptosis–autophagy–UPS triangle demonstrates that signalling responses can be either definite or ambiguous. Downward red arrows represent inhibitions or reductions, upward green arrows indicate activation or upregulations. Care therefore needs to be taken when investigating and interpreting experimental data on cause and effect relationships, and when generalising such findings
Abbreviations
- Atg:
-
autophagy-related
- cFLIP:
-
cellular FLICE inhibitory protein
- LC3:
-
microtubule-associated protein 1 light chain 3
- UPS:
-
ubiquitin-proteasome system
References
Taylor RC, Cullen SP, Martin SJ . Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008; 9: 231–241.
Hellwig CT, Passante E, Rehm M . The molecular machinery regulating apoptosis signal transduction and its implication in human physiology and pathophysiologies. Curr Mol Med 2011; 11: 31–47.
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N . Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 2013; 1833: 3448–3459.
Bader M, Steller H . Regulation of cell death by the ubiquitin-proteasome system. Curr Opin Cell Biol 2009; 21: 878–884.
Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P . Autophagy: for better or for worse. Cell Res 2012; 22: 43–61.
Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007; 282: 4702–4710.
Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009; 16: 1093–1107.
Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol 2008; 445: 29–76.
Kang R, Zeh HJ, Lotze MT, Tang D . The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18: 571–580.
Oberstein A, Jeffrey PD, Shi Y . Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 2007; 282: 13123–13132.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122: 927–939.
Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S et al. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007; 3: 561–568.
Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. Embo J 2007; 26: 2527–2539.
Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS et al. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. Embo J 2011; 30: 395–407.
Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM . Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009; 28: 2128–2141.
Sinha S, Levine B . The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene 2008; 27 (Suppl 1): S137–S148.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283: 10892–10903.
Malik SA, Orhon I, Morselli E, Criollo A, Shen S, Marino G et al. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 2011; 30: 3918–3929.
Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell 2012; 47: 359–370.
Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ . p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA 2001; 98: 9666–9670.
Kilbride SM, Prehn JH . Central roles of apoptotic proteins in mitochondrial function. Oncogene 2013; 32: 2703–2711.
Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 2010; 1: e18.
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8: 1124–1132.
Adams J . The proteasome: a suitable antineoplastic target. Nat Rev Cancer 2004; 4: 349–360.
Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ . Catalytic mechanism and assembly of the proteasome. Chem Rev 2009; 109: 1509–1536.
Kravtsova-Ivantsiv Y, Ciechanover A . Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci 2012; 125 (Pt 3): 539–548.
Fennell DA, Chacko A, Mutti L . BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene 2007; 27: 1189–1197.
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006; 124: 601–613.
Blankenship JW, Varfolomeev E, Goncharov T, Fedorova AV, Kirkpatrick DS, Izrael-Tomasevic A et al. Ubiquitin binding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 and c-IAP2(1). Biochem J 2009; 417: 149–160.
Breitschopf K, Zeiher AM, Dimmeler S . Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J Biol Chem 2000; 275: 21648–21652.
Li B, Dou QP . Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci USA 2000; 97: 3850–3855.
Laussmann MA, Passante E, Hellwig CT, Tomiczek B, Flanagan L, Prehn JH et al. Proteasome inhibition can impair Caspase-8 activation upon submaximal stimulation of apoptotic tumor necrosis factor-related apoptosis inducing ligand (TRAIL) signaling. J Biol Chem 2012; 287: 14402–14411.
Fricker N, Beaudouin J, Richter P, Eils R, Krammer PH, Lavrik IN . Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J Cell Biol 2010; 190: 377–389.
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR . Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21: 749–760.
Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G et al. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene 2003; 22: 6785–6793.
Sun XM, Butterworth M, MacFarlane M, Dubiel W, Ciechanover A, Cohen GM . Caspase activation inhibits proteasome function during apoptosis. Mol Cell 2004; 14: 81–93.
Brancolini C . Inhibitors of the ubiquitin-proteasome system and the cell death machinery: how many pathways are activated? Curr Mol Pharmacol 2008; 1: 24–37.
Cusack JC Jr, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J et al. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Cancer Res 2001; 61: 3535–3540.
Frankland-Searby S, Bhaumik SR . The 26S proteasome complex: an attractive target for cancer therapy. Biochim Biophys Acta 2012; 1825: 64–76.
de Wilt LH, Kroon J, Jansen G, de Jong S, Peters GJ, Kruyt FA . Bortezomib and TRAIL: a perfect match for apoptotic elimination of tumour cells? Crit Rev Oncol/Hematol 2013; 85: 363–372.
Kirkin V, McEwan DG, Novak I, Dikic I . A role for ubiquitin in selective autophagy. Mol Cell 2009; 34: 259–269.
Zhang M, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou C et al. Chaperoned ubiquitylation—crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell 2005; 20: 525–538.
Long J, Gallagher TR, Cavey JR, Sheppard PW, Ralston SH, Layfield R et al. Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J Biol Chem 2008; 283: 5427–5440.
Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW . Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004; 24: 8055–8068.
Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC . Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33: 517–527.
Qiao L, Zhang J . Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett 2009; 456: 15–19.
Korolchuk VI, Menzies FM, Rubinsztein DC . Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010; 584: 1393–1398.
Laussmann MA, Passante E, Dussmann H, Rauen JA, Wurstle ML, Delgado ME et al. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ 2011; 18: 1584–1597.
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447: 859–863.
Pan JA, Ullman E, Dou Z, Zong WX . Inhibition of protein degradation induces apoptosis through a microtubule-associated protein 1 light chain 3-mediated activation of caspase-8 at intracellular membranes. Mol Cell Biol 2011; 31: 3158–3170.
Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem 2012; 287: 12455–12468.
Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P . Caspases in cell survival, proliferation and differentiation. Cell Death Differ 2007; 14: 44–55.
Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280: 20722–20729.
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR et al. Cullin3-based polyubiquitination and p62-dependent aggregation of Caspase-8 mediate extrinsic apoptosis signaling. Cell 2009; 137: 721–735.
Huang S, Okamoto K, Yu C, Sinicrope FA . p62/sequestosome-1 upregulation promotes ABT-263-induced Caspase-8 aggregation/activation on the autophagosome. J Biol Chem 2013; 288: 33654–33666.
Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 2009; 11: 1355–1362.
Oral O, Oz-Arslan D, Itah Z, Naghavi A, Deveci R, Karacali S et al. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012; 17: 810–820.
Hou W, Han J, Lu C, Goldstein LA, Rabinowich H . Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010; 6: 891–900.
Coward J, Ambrosini G, Musi E, Truman JP, Haimovitz-Friedman A, Allegood JC et al. Safingol (L-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of PKC and the PI3-kinase pathway. Autophagy 2009; 5: 184–193.
Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R et al. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem 2006; 281: 8518–8527.
Caro-Maldonado A, Tait SW, Ramirez-Peinado S, Ricci JE, Fabregat I, Green DR et al. Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-, Bak-deficient cells. Cell Death Differ 2010; 17: 1335–1344.
Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci USA 2008; 105: 16677–16682.
Vanlangenakker N, Vanden Berghe T, Vandenabeele P . Many stimuli pull the necrotic trigger, an overview. Cell Death Differ 2012; 19: 75–86.
He S, Wang L, Miao L, Wang T, Du F, Zhao L et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009; 137: 1100–1111.
Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M et al. cIAPs block ripoptosome formation, a RIP1/Caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 2011; 43: 449–463.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 2011; 43: 432–448.
Huang X, Wu Z, Mei Y, Wu M . XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. Embo J 2013; 32: 2204–2216.
Luo S, Rubinsztein DC . Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ 2009; 17: 268–277.
Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell 2012; 47: 291–305.
Schleich K, Warnken U, Fricker N, Ozturk S, Richter P, Kammerer K et al. Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell 2012; 47: 306–319.
Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol 2010; 17: 1324–1329.
Azijli K, Weyhenmeyer B, Peters GJ, de Jong S, Kruyt FA . Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell Death Differ 2013; 20: 858–868.
Pennarun B, Meijer A, de Vries EG, Kleibeuker JH, Kruyt F, de Jong S . Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim Biophys Acta 2010; 1805: 123–140.
van Raam BJ, Salvesen GS . Proliferative versus apoptotic functions of caspase-8 Hetero or homo: the caspase-8 dimer controls cell fate. Biochim Biophys Acta 2012; 1824: 113–122.
Cursi S, Rufini A, Stagni V, Condo I, Matafora V, Bachi A et al. Src kinase phosphorylates Caspase-8 on Tyr380: a novel mechanism of apoptosis suppression. Embo J 2006; 25: 1895–1905.
Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 2012; 14: 51–60.
White E . Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12: 401–410.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25: 717–729.
Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon H-U . Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Translat Med 2013; 5: 202ra123.
Nixon RA . The role of autophagy in neurodegenerative disease. Nat Med 2013; 19: 983–997.
Choi AM, Ryter SW, Levine B . Autophagy in human health and disease. N Engl J Med 2013; 368: 651–662.
Ciechanover A . Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim Biophys Acta 2012; 1824: 3–13.
Acknowledgements
This work was supported by grants from Science Foundation Ireland (09/RFP/BIC2375), the Royal College of Surgeons in Ireland Research Committee, and the European Union (FP7 Health – APO-DECIDE; FP7 Marie Curie IAPP SYS-MEL). LD was supported by the European Community Action Scheme for the Mobility of University Students (ERASMUS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Delgado, M., Dyck, L., Laussmann, M. et al. Modulation of apoptosis sensitivity through the interplay with autophagic and proteasomal degradation pathways. Cell Death Dis 5, e1011 (2014). https://doi.org/10.1038/cddis.2013.520
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.520
Keywords
This article is cited by
-
Marizomib sensitizes primary glioma cells to apoptosis induced by a latest-generation TRAIL receptor agonist
Cell Death & Disease (2021)
-
LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases
Cell Death & Differentiation (2020)
-
The proton pump inhibitor pantoprazole disrupts protein degradation systems and sensitizes cancer cells to death under various stresses
Cell Death & Disease (2018)
-
Lup-20(29)-en-3β,28-di-yl-nitrooxy acetate affects MCF-7 proliferation through the crosstalk between apoptosis and autophagy in mitochondria
Cell Death & Disease (2018)
-
High perfluorooctanoic acid exposure induces autophagy blockage and disturbs intracellular vesicle fusion in the liver
Archives of Toxicology (2017)
