
Abstract
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is a cytokine and a selective inducer of apoptosis in a range of tumour cells, but not in normal, untransformed cells. A large number of chemotherapeutics as well as biological agents are being tested for their potential to sensitise resistant tumour cells to TRAIL as a means to broaden the range of tumours treatable with TRAIL. However, because of the incomplete understanding of the mechanism(s) underlying TRAIL resistance in non-malignant cells, it is unpredictable whether the effect of these sensitisers will be restricted to tumour cells or they would also sensitise non-transformed cells causing unwanted toxicity. In this study, we carried out a systematic analysis of the mechanisms driving TRAIL resistance in non-transformed cells. We found that cellular FLICE-like inhibitory protein, anti-apoptotic B-cell lymphoma 2 proteins, and X-linked inhibitor of apoptosis protein were independently able to provide resistance to TRAIL. Deficiency of only one of these proteins was not sufficient to elicit TRAIL sensitivity, demonstrating that in non-transformed cells multiple pathways control TRAIL resistance and they act in a redundant manner. This is contrary to the resistance mechanisms found in tumour cell types, many of them tend to rely on a single mechanism of resistance. Supporting this notion we found that 76% of TRAIL-resistant cell lines (13 out of 17) expressed only one of the above-identified anti-apoptotic proteins at a high level (≥1.2-fold higher than the mean expression across all cell lines). Furthermore, inhibition or knockdown of the single overexpressed protein in these tumour cells was sufficient to trigger TRAIL sensitivity. Therefore, the redundancy in resistance pathways in non-transformed cells may offer a safe therapeutic window for TRAIL-based combination therapies where selective sensitisation of the tumour to TRAIL can be achieved by targeting the single non-redundant resistance pathway.
Similar content being viewed by others


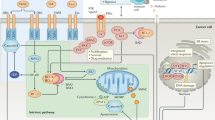
Main
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is a death ligand member of the TNF ligand superfamily.1 Similar to other death ligands, TRAIL activates the extrinsic apoptotic pathway upon binding to its cell surface receptors, death receptor (DR) 4 and DR5. Upon ligation of the receptor, pro-caspase-8 and -10 are recruited to the receptors where they become activated.2, 3 Once active, caspase-8/-10 translocates to the cytosol to activate executioner caspases or to cleave the BH3-only protein Bid and thus trigger Bcl-2-associated X protein (Bax)/Bcl-2 homologous antagonist/killer (Bak)-mediated cytochrome c release and consequent execution of apoptosis.4, 5
TRAIL is attracting attention as a potential anti-cancer agent because of its property of inducing apoptosis selectively in tumour cells, but not in healthy, non-transformed cells.6 Corroborating this attribute of TRAIL, pre-clinical studies and phase I clinical studies have demonstrated no systemic toxicity of TRAIL to organs and tissues, even at very high administered doses.6, 7 In vitro studies, however, revealed that approximately 60–70% of tumour cell lines are resistant to TRAIL, and thus the therapeutic potential of TRAIL may be limited to a small subset of TRAIL-sensitive tumours.8, 9
Our understanding of resistance mechanisms in tumour cells has greatly increased over the past 15 years. First, activation of DR4 and DR5 can be regulated by the decoy receptors, DcR1 and DcR2, which are able to sequester TRAIL from the DRs as well as to form inactive, heteromeric complexes with them.10, 11, 12 Activation of caspase-8 (or -10) is another target for regulation by at least three different proteins: the caspase-8 homologue cellular FLICE-like inhibitory protein (cFLIP), phosphorylated MAPK-activating death domain protein or the complex of glycogen synthase kinase-3, DDX3 and cellular inhibitor of apoptosis protein-1. All these proteins act by binding to DR4/DR5 and prevent Fas-associated protein with death domain and/or caspase-8 recruitment.13, 14, 15, 16 In addition to the inhibitors that act at the level of the receptor, anti-apoptotic B-cell lymphoma 2 (Bcl-2) proteins can block Bax/Bak activation induced by caspase-8-processed tBid and thus block the activation of the mitochondrial amplification loop.17, 18 Finally, in a number of tumour cells TRAIL-mediated apoptosis has been shown to be blocked by the caspase inhibitor, X-linked inhibitor of apoptosis protein (XIAP) that can directly bind to caspase-9 and -3 and block their activation or activity.19
Drugs that reduce the expression of such anti-apoptotic proteins, either by inducing cellular stress or inhibiting the oncogene that drives their expression can restore sensitivity of tumour cells to TRAIL.20 Although healthy, non-transformed cells are protected against TRAIL-induced apoptosis, there are examples where cellular stress or tissue injury, caused by, for example, spinal cord injury or proteasome inhibition triggers TRAIL sensitivity in non-malignant cells.21, 22, 23 The effect of the plethora of drugs reported to sensitise tumour cells to TRAIL by inducing DNA damage, oxidative stress, endoplasmic reticulum stress, etc., on normal, non-transformed cells is unpredictable.
To date, the mechanism of TRAIL resistance in normal, non-transformed cells is poorly studied. Early reports have found that non-transformed cells expressed higher amounts of DcR1 and DcR2 than cancerous tissues, which may be the means through which they are protected from TRAIL,24, 25 however, there is a lack of sufficient mechanistic in vivo evidence in support of this notion.
The best studied non-transformed cell type for TRAIL resistance are keratinocytes. Keratinocytes, unlike most other non-transformed cells (such as fibroblasts, smooth muscle cells, endothelial cells), display some degree of TRAIL sensitivity. This sensitivity increases by transforming the keratinocytes, which has been linked to reduced cFLIP or XIAP expression in response to the transformation.23, 26, 27 However, our knowledge about the inherent resistance mechanisms in non-transformed cells is currently incomplete.
In the current study, we carried out a systematic analysis of the mechanisms that regulate TRAIL resistance in non-transformed cells. We show that non-transformed cells are protected from TRAIL-induced apoptosis by multiple, redundant pathways. We found that in both human primary fibroblasts and smooth muscle cells cFLIP, anti-apoptotic Bcl-2 proteins and XIAP are independently able to provide resistance to TRAIL. Removal of only one of these proteins is insufficient to induce TRAIL sensitivity. Eliminating the receptor-proximal cFLIP together with either the anti-apoptotic Bcl-2 proteins or XIAP was necessary to relieve TRAIL resistance. We show that resistant tumour cells on the contrary tend to rely on the expression of only one of these anti-apoptotic proteins to maintain TRAIL resistance, which may offer a safe therapeutic window for combination therapies.
Results
DcRs are not essential for resistance to TRAIL
To study the mechanism of TRAIL resistance in non-transformed cells, two common human somatic cell types have been chosen: dermal fibroblasts (hFb) and umbilical artery smooth muscle cells (hUASMCs). Despite the clear advantages of in vivo systems to address our question, they could not be used due to the significant differences in the TRAIL receptor family as well as the regulation of TRAIL sensitivity between human and rodent cells.28, 29 For all key experiments, hFb from two separate donors have been used. The two fibroblast strains gave very similar results in all experiments, thus, results from only one donor (donor 007) are shown here.
To characterise hFb and hUASMC as model systems, surface expression of TRAIL receptors and TRAIL resistance were evaluated. Similarly, to other non-transformed cells,27 the TRAIL receptor predominantly expressed on the surface of both hFb and hUASMC was DR5 (Figure 1a). hFb also expressed DR4 and DcR1, whereas hUASMC expressed DcR1 and DcR2 (Figures 1a and b). The expression of pro-apoptotic proteins was also determined in hFb, and they were found to express all key apoptosis-mediating and executing proteins, such as pro-caspase-8, pro-caspase-9, pro-caspase-3, Bax, Bak, Smac/Diablo (data not shown). To test the sensitivity of hFb and hUASMC to TRAIL, the cells were treated with a dosage of crosslinked TRAIL for 24 h, and cell death (as measured by Annexin V assay) was determined (Figures 1c and d). Neither cell types demonstrated any sensitivity to TRAIL. Because both hFb and hUASMC express DcRs, we decided to bypass their potential ability to compete for TRAIL by selectively activating DR5 using an agonistic antibody (LBY135, Novartis, Figure 1e). Treatment of hFb with LBY135 failed to induce apoptosis, indicating that DcRs are either not involved in TRAIL-resistance, or additional, intracellular regulators exist.

Fibroblasts and smooth muscle cells are not sensitive to TRAIL-induced apoptosis. (a and b) Cell surface expression of TRAIL receptors. Histogram representation of flow cytometric analysis of DR4, DR5, DcR1 and DcR2 expression on the cell surface of (a) hFb and (b) hUASMC. (c and d) Non-transformed cells are resistant to TRAIL-induced apoptosis. (c) hFb and (d) hUASMC were treated with the indicated doses of crosslinked TRAIL (TRAIL-CL) for 24 h and induction of apoptosis was measured with Annexin V analysis. As a positive control, cells were treated with 500 nM staurosporine (STS) for 24 h. (e) Treatment with the agonistic DR5 antibody, LBY135 does not induce apoptosis in fibroblast cells. Cells were treated with indicated doses of crosslinked LBY135, or with 500 ng/ml TRAIL-CL for 24 h and induction of apoptosis was measured with Annexin V analysis. Data shown are mean±S.E.M. (f) Jurkat cells were used as a positive control for TRAIL- and LBY135-induced apoptosis
Cycloheximide (CHX) overcomes the resistance of normal cells to TRAIL-induced apoptosis
To determine whether expression of anti-apoptotic proteins or low expression of pro-apoptotic proteins is responsible for TRAIL resistance, protein synthesis was blocked with CHX (10 μg/ml, for 15 h) before treatment with TRAIL (Figure 2). Pre-treatment with CHX sensitised hFb to TRAIL (Figure 2a), indicating that TRAIL-mediated apoptosis is more likely to be inhibited by anti-apoptotic proteins, rather than being prevented due to the absence of pro-apoptotic proteins.

Effect of cycloheximide (CHX) and cFLIP knockdown on TRAIL sensitivity of human fibroblasts. (a) Inhibition of protein synthesis with CHX sensitises hFb to TRAIL-induced apoptosis. Cells were pre-treated for 15 h with 10 μg/ml CHX followed by 24 h treatment with 200 ng/ml TRAIL-CL and induction of apoptosis was quantified with Annexin V analysis. *P<0.01. (b) CHX treatment does not affect cell surface expression of the TRAIL receptors. Histogram representation of flow cytometric analysis of DR4 and DR5 expression on the surface of control- and CHX-treated (10 μg/ml, 15 h) fibroblasts. (c) CHX downregulates cFLIP, Bcl-XL and Mcl-1 expression. The effect of CHX on the expression of anti-apoptotic regulators of the TRAIL apoptotic pathway. Western blot images from whole-cell lysates of hFb treated with 10 μg/ml CHX for 15 h analysed for the expression of cFLIP, XIAP, Bcl-2, Bcl-XL and Mcl-1. (d and e) TRAIL fails to induce considerable pro-caspase-8 processing. (d) hFb were treated with TRAIL-CL for 24 h with or without 15 h pre-treatement with 10 μg/ml CHX and processing of pro-caspase-8 was detected in cell lysates with western blotting. (e) hFb were treated with 200 ng/ml TRAIL-CL for 24 h and 80 μg of cell lysates were analysed by western blotting for pro-caspase-8 cleavage using high-sensitivity chemiluminescent substrate. As a positive control for cleaved pro-caspase-8 fragments, 30 μg of TRAIL-treated (50 ng/ml, 4 h) Colo205 cell lysate was used. (f) Downregulation of cFLIP fails to sensitise hFb to TRAIL-induced apoptosis. hFb were transfected with two different siRNAs against cFLIP or GFP as transfection control (50 nM each). Twenty-four hours after transfection, cells were treated with 200 ng/ml TRAIL-CL for 24 h and induction of apoptosis was measured with Annexin V analysis. Inset: lysates from transfected cells were analysed for cFLIP expression with western blotting. (g) cFLIP knockdown increases pro-caspase-8 processing. hFb were transfected with siRNA against cFLIP (cFLIP1, 50 nM) and treated with 200 ng/ml TRAIL-CL for 24 h. Forty microgram of cell lysates were analysed for pro-caspase-8 processing with western blotting using high-sensitivity chemiluminescent substrate. All graphs show mean values±S.E.M.
In order to find the target of CHX, its effect on the expression of TRAIL receptors and anti-apoptotic proteins was studied in hFb. Although CHX treatment did not alter the cell surface expression of DR4 and DR5 (Figure 2b), nor the expression of XIAP (Figure 2c), it did reduce the expression of cFLIP, B-cell lymphoma-extra large (Bcl-XL) and myeloid cell leukemia sequence 1 (Mcl-1; Figure 2c).
We also noticed that exposure of hFb to TRAIL failed to induce significant processing of pro-caspase-8, it was only apparent when the cells were pre-treated with 10 μg/ml CHX for 15 h (Figure 2d). A low level of caspase-8 processing was only detectable after enhancing detection sensitivity of the western blot by doubling the protein loaded on the gel and using a high-sensitivity chemiluminescent substrate (Immobilon Western HRP substrate, Millipore), indicating that TRAIL signalling is likely to be blocked at the level of the death-inducing signalling complex (DISC; Figure 2e).
cFLIP is a pseudocaspase, structurally related to caspase-8 with three splice variants (long, short and Raji: cFLIPL, S, R, respectively).30 cFLIP inhibits or limits DR-mediated activation of caspase-8 by competing with it for binding to the DISC.31 We found that the dominant isoform of cFLIP in hFb is cFLIPL and that expression of cFLIP was reduced after CHX treatment (Figure 2c). To investigate whether cFLIP is responsible for TRAIL-resistance, it was knocked down in hFb using small interfering RNA (siRNA) targeting the death effector domain region common in all three variants (Figure 2f, inset). Knockdown of cFLIP, however, failed to sensitise hFb to TRAIL-induced apoptosis (Figure 2f), despite the fact that it increased TRAIL-induced pro-caspase-8 processing (Figure 2g). This indicates that cFLIP is likely to have a role in regulating TRAIL sensitivity, but there are additional, downstream inhibitors.
cFLIP and XIAP are together responsible for TRAIL resistance in normal cells
In order to identify what additional factors regulate TRAIL-induced apoptosis in hFb, pro-caspase-3 processing was examined. TRAIL induced processing of pro-caspase-3 to its p20 fragment, but not to the fully processed p17 fragment, detectable in the TRAIL-sensitive Colo205, colon carcinoma cells or in hFb pre-treated with 10 μg/ml CHX for 15 h (Figure 3a lower panel). This p20 caspase-3 fragment was inactive as no detectable level of cleaved poly-ADP ribose polymerase (PARP, a caspase-3 substrate) was present in hFb lysates, only when CHX pre-treatment was applied (Figure 3a upper and lower panels). Similar results were obtained when cFLIP was knocked down. cFLIP knockdown enhanced TRAIL-induced pro-caspase-3 processing, however, it was again only to the p20 fragment, with no associated PARP cleavage (Figure 3b).

Both cFLIP and XIAP provide TRAIL resistance in fibroblasts. (a) TRAIL induces pro-caspase-3 processing to the p20 fragment. hFb were treated with 200 ng/ml TRAIL-CL (3, 6, 12, 24 h) in the absence or presence of a pre-treatment with 10 μg/ml of CHX for 15 h (upper and lower panels, respectively) and 30 μg whole-cell lysate were analysed for pro-caspase-3 and PARP cleavage with western blotting. As a positive control, 30 μg of TRAIL-treated (50 ng/ml, 4 h) Colo205 cell lysate was used. (b) Knockdown of cFLIP increases TRAIL-mediated caspase-3 processing to the p20 form. Cells were transfected with an siRNA (50 nM) against cFLIP (cFLIP1) or GFP as transfection control. Twenty-four hours after transfection, cells were treated with 200 ng/ml TRAIL or TRAIL-CL for 24 h and whole-cell lysates were analysed for pro-caspase-3 processing and PARP cleavage with western blotting. As a positive control, 30 μg of TRAIL-treated (50 ng/ml, 4 h) Colo205 cell lysate was used. (c–e) Cells were transfected with siRNAs (50 nM) against cFLIP (cFLIP1) and/or XIAP or GFP as transfection control. (c) Lysates from transfected cells were analysed for cFLIP and XIAP expression. (d) Knockdown of cFLIP together with XIAP sensitises hFb to TRAIL. Twenty-four hours after transfection, cells were treated with 200 ng/ml TRAIL-CL for 24 h and induction of apoptosis was measured with Annexin V analysis. *P<0.05. (e) cFLIP+XIAP knockdown leads to processing of pro-caspase-3 to its p17 form in response to TRAIL. Twenty-four hours after transfection, cells were treated with 200 ng/ml TRAIL or TRAIL-CL for 24 h and cell lysates were analysed by western blotting for pro-caspase-3 and PARP cleavage. As a positive control, 30 μg of TRAIL-treated (50 ng/ml, 4 h) Colo205 cell lysate was used. (f) Knockdown of cFLIP+XIAP specifically sensitises to TRAIL-induced apoptosis and not to other apoptotic stimuli. Cells were transfected with the indicated siRNAs (50 nM). Twenty-four hours after transfection, cells were treated with 100 μM etoposide or 10 μM doxorubicin for 24 h and induction of apoptosis was measured with Annexin V assay. All graphs show mean values±S.E.M.
XIAP, a known inhibitor of caspase-3, binds to the p20 caspase-3 fragment to block its activity, including the autocatalytic removal of the 3 kDa pro-domain from the p20 subunit.32 To assess the involvement of XIAP in TRAIL resistance, XIAP was knocked down using siRNA (Figure 3c), however, it failed to sensitise hFb to TRAIL-induced apoptosis (Figure 3d).
To examine a potential two-point control of TRAIL-induced apoptosis, cFLIP and XIAP (cFLIP+XIAP) were knocked down together. cFLIP+XIAP knockdown sensitised hFb to TRAIL-induced apoptosis (Figure 3d), which was associated with full processing of pro-caspase-3 to its p17 active fragment and intracellular cleavage of PARP (Figure 3e). Knockdown of cFLIP+XIAP sensitised hFb only to TRAIL, but not to compounds which predominantly target the intrinsic apoptosis pathway (doxorubicin and etoposide; Figure 3f), highlighting that these proteins represent bona fide resistance mechanisms against TRAIL rather than being generic pro-survival factors.
Study of hUASMCs identified the same mechanism of resistance. Similar to hFb, hUASMC cells became sensitive to TRAIL after treatment with CHX, and this effect could be replicated by simultaneous knockdown of cFLIP+XIAP, but not by knockdown of either protein alone (Supplementary Figure 1).
The Bcl-2 family of proteins inhibit activation of the mitochondrial pathway
XIAP inhibits pro-caspase-9 and -3 activation and/or activity. However, the activity of XIAP is controlled by Smac/DIABLO and Omi/HtrA2, released from mitochondria upon outer mitochondrial membrane permeabilisation in apoptotic cells.33, 34, 35 As CHX treatment failed to reduce XIAP expression (Figure 2c), we questioned whether XIAP is the actual protein that inhibits TRAIL-induced apoptosis, or instead, it is the anti-apoptotic Bcl-2 proteins that block caspase-8-mediated activation of Bax/Bak and consequent Smac/DIABLO- and/or Omi/HtrA2-release. Supporting this theory, we found that activation of Bax coincided with, rather than occurred after caspase-3 activation in CHX+TRAIL treated hFb, similar to that where the intrinsic apoptotic pathway has directly been activated with staurosporine (STS) (Supplementary Figure 2A, 2B). Moreover, overexpression of Bcl-XL was able to protect fibroblasts from CHX+TRAIL-induced apoptosis (Supplementary Figure 2C).
To dissect the contribution of anti-apoptotic Bcl-2 proteins in maintaining TRAIL resistance, we blocked the function of anti-apoptotic Bcl-2 proteins by knocking down Mcl-1 (Figure 4a) and/or inhibiting Bcl-2 and Bcl-XL with a high dose of ABT737 (10 μ M), a BH3-mimetic compound.36 Expression of cFLIP was also knocked down in these experiments to remove the receptor-proximal inhibition of the TRAIL-apoptosis pathway. Inhibition of anti-apoptotic Bcl-2 function with ABT737, but not knockdown of Mcl-1, sensitised a small proportion of hFb to TRAIL (Figure 4b). Of note, the effect of combined knockdown/inhibition of Mcl-1+Bcl-2+Bcl-XL on TRAIL sensitivity could not be studied as it compromised hFb basal viability. More importantly, knockdown of cFLIP before inhibition of anti-apoptotic Bcl-2 proteins sensitised hFb to TRAIL. Knockdown of Mcl-1 had no effect on TRAIL sensitisation (Figure 4c), which may be due to the redundancy among anti-apoptotic Bcl-2 proteins, where upon loss of Mcl-1, Bcl-2 and Bcl-XL can fulfil its function.

The Bcl-2 family of proteins inhibit TRAIL-induced activation of the mitochondrial pathway. (a) Efficacy of Mcl-1 and cFLIP knockdown analysed with western blotting from lysates of hFb transfected with siRNAs (50 nM) against Mcl-1 and/or cFLIP (cFLIP1) or GFP (control siRNA). (b) Effect of ABT737 and Mcl-1 knockdown on TRAIL-sensitivity of hFb. Control- or Mcl-1 siRNA-transfected hFb were pre-treated with 10 μM ABT737 for 15 h followed by a treatment with 125 ng/ml TRAIL-CL for 24 h and induction of apoptosis was measured with Annexin V analysis. (c) Inhibition of Bcl-2 and Bcl-XL after knockdown of cFLIP leads to TRAIL sensitisation. hFb were transfected with siRNAs (50 nM) against Mcl-1 and/or cFLIP (cFLIP1) or GFP (control siRNA). Twenty-four hours after transfection, cells were pre-treated for 15 h with 10 μM ABT737 followed by 125 ng/ml TRAIL-CL for 24 h and induction of apoptosis was measured with Annexin V analysis. (d) The Smac mimetic BV6 sensitises fibroblasts to TRAIL-induced apoptosis. cFLIP was knocked down in hFb before treatment with 2 or 5 μM BV6 for 3 h followed by 125 ng/ml TRAIL-CL for 24 h. Induction of apoptosis was measured with Annexin V assay. All graphs show mean values+S.E.M.; *P<0.05, **P<0.01
These findings suggest that in hFb activation of the mitochondrial pathway is required to overcome XIAP-mediated caspase inhibition by either the release of Smac/DIABLO or cytochrome c. The Smac-mimetic, embelin binds to the BIR3 domain of XIAP and blocks binding of caspase-9, but not caspase-3, to XIAP.37 We found that embelin together with knockdown of cFLIP could not sensitise hFb to TRAIL (Supplementary Figure 3), indicating that relieving XIAP-mediated caspase-9 inhibition is not sufficient to trigger TRAIL sensitivity. A second Smac-mimetic, BV6 binds to the BIR2 and BIR3 domains of XIAP and disrupts both caspase-9 and -3 binding to XIAP.38 Treatment of hFb with BV6 where cFLIP has been knocked down resulted in a profound sensitisation to TRAIL (Figure 4d), showing that induction of TRAIL sensitivity in non-transformed hFb would require Smac/DIABLO-release from the mitochondria as a mechanism to overcome XIAP-mediated inhibition of caspase-3, and not only of caspase-9.
Non-redundant mechanism of TRAIL resistance in transformed cells
During their development, cancer cells accumulate losses in various pathways, whereas others can become hyper-activated. Thus, cancer cells become dependent on a smaller ensemble of signalling pathways, and the intensity of these pathways is enhanced. This loss of redundancy or robustness has been reported, for example, in certain protein kinase pathways.39 There are an increasing number of studies reporting that knocking down a single regulatory protein can re-establish TRAIL sensitivity, indicating that cancerous cells may lose redundancy in the regulation of the extrinsic apoptotic pathway.
To test this hypothesis the mRNA expression levels of cFLIP, XIAP, Mcl-1, Bcl-2 or Bcl-XL have been extracted from the expression profiles of 78 TRAIL-resistant cancer cell lines (Supplementary Figure 4).40 The cell lines that ‘overexpressed’ any of the above anti-apoptotic genes were selected as described in Materials and Methods. Using Boolean expressions, we determined what percentage of the cell lines overexpresses only one of the above anti-apoptotic genes. If a cell line was present in only one ‘overexpression’ list, the hypothesis of non-redundant resistance pathways was considered to be supported. We found that 40 out of the 78 cell lines (51%) fulfilled this criterion (Supplementary Table 1). We have also determined the expression of the above genes at the protein level in 17 TRAIL-resistant cancer cell lines (Supplementary Table 2) and the same analysis was carried out. We found that the hypothesis was true for 13 cell lines (76%), indicating that these cell types may only possess a single mechanism of resistance (Table 1).
We then tested whether these tumour cells in fact rely on only one resistance mechanism, provided by the ‘overexpressed’ protein. Representative cell lines that expressed high levels of cFLIP (HCA7 colon carcinoma cells), or XIAP (Panc-1 pancreatic cancer cells) or anti-apoptotic Bcl-2 proteins (HL-60, Oci-AML2 acute myeloid leukemia cells, RKO colon carcinoma cells), were selected and the ‘overexpressed’ protein was inhibited or knocked down. In each case, removal of the ‘overexpressed’ protein was sufficient to induce a profound sensitisation to TRAIL (Figure 5), providing evidence that all these TRAIL-resistant cancer cells are resistant due to the expression of only one anti-apoptotic protein and that they have lost the redundancy in resistance mechanisms seen in non-transformed cells. This is further supported as inhibition of anti-apoptotic Bcl-2 proteins via ABT737 or IAP inhibition with BV6 had no additional effect on TRAIL sensitivity of HCA-7 cells. Equally, no sensitisation of OCI-AML2 cells in response to BV6 could be detected (Supplementary Figure 5).

Tumour cells rely on only one mechanism of resistance. (a) Knockdown of cFLIP sensitises HCA7 cells to TRAIL-induced apoptosis. HCA7 cells were transduced with Ad5shcFLIP (MOI 200) or Ad5eGFP (MOI 200) as a transduction control. Twenty-four hours after transduction, cells were treated with 200 ng/ml TRAIL for 24 h and induction of cell death was quantified. Data shown are mean±S.E.M. (b) Inhibition of XIAP sensitises Panc-1 cells to TRAIL-induced apoptosis. Cells were treated with the indicated doses of the Smac mimetic BV6 for 1 h followed by 200 ng/ml of TRAIL for 24 h after which induction of cell death was quantified. Data shown are mean±S.E.M. (c–e) Inhibition of Bcl-2 and Bcl-XL sensitises RKO (c), Oci-AML2 (d) and HL60 (e) to TRAIL-induced apoptosis. Cells were treated with the indicated doses of the BH3 mimetic ABT737 for 1 h followed by 200 ng/ml of TRAIL for 24 h after which induction of cell death was quantified. Induction of apoptosis was quantified by determining the percentage of cells with lost mitochondrial membrane potential using tetramethylrhodamine ethyl ester perchlorate in all experiments. The graphs show mean values±S.E.M.
Similar results were gained with Panc-1 cells. These cells express high levels of XIAP and could be profoundly sensitised to TRAIL by inhibition or knockdown of XIAP. On the other hand, inhibition of anti-apoptotic Bcl-2 proteins with ABT737 induced a minimal, approximately 5% increase in TRAIL sensitivity, whereas knockdown of cFLIP had no sensitising effect (Supplementary Figure 5).
Discussion
Because of the tumour-selective cytotoxicity of TRAIL,20, 41 there is effort to identify drugs that can sensitise resistant cancer cells to TRAIL and thus increase the range of tumours targetable with a TRAIL-based therapy. Sensitisation of resistant tumour cells, in vitro and in mouse xenograft models appears to be achievable denoted by a large number of studies reporting that agents that (1) induce various forms of cellular stress (DNA damage, oxidative stress, protein folding/ER stress, etc.), (2) influence epigenetic alterations (e.g., histone deacetylase inhibitors) or (3) inhibit oncogenes or oncogene-driven survival pathways could all sensitise different TRAIL-resistant tumour cells.20, 42, 43 Although these results are very promising, it is currently unpredictable whether the effect of these identified sensitisers is selective to tumour cells or if they would also sensitise non-transformed cells to TRAIL and thus cause toxicity.
In order to determine non-toxic TRAIL-based drug combinations for clinical use, a better understanding of the mechanisms protecting non-transformed cells from TRAIL is necessary. This study addressed this question using two non-transformed cell types abundant in the body, fibroblasts and smooth muscle cells. These cell types were chosen despite the drawbacks and limitations of the in vitro culture system over an in vivo rodent model, because the TRAIL receptor family is significantly different between rodents and humans.28, 29 The studies presented here identified the same mechanism of TRAIL resistance in the two cell types. It was found that the components of the TRAIL-apoptotic machinery are present in both cell types; but its activation is blocked by anti-apoptotic proteins. Treatment of hFb with TRAIL could induce a low level of pro-caspase-8 processing resulting in processing of pro-caspase-3 to its p20 fragment but no associated cleavage of cellular caspase-3 substrate proteins, such as PARP, could be found, indicating that there is more than one point where the apoptotic pathway is inhibited. We show that the first point of control in hFb and hUASMC is at the level of pro-caspase-8 activation, mediated by cFLIP. In line with our findings, oncogenic Ras transformation has been found to trigger TRAIL-sensitisation in immortalised fibroblasts. The mechanism involved enhanced recruitment of pro-caspase-8 to the TRAIL receptor DISC corroborating that a checkpoint for TRAIL-induced apoptosis in non-transformed cells exist at the level of the DRs.44 The second point of control is the regulation of caspase-3 activity. Full processing of caspase-3 and consequent cell death required either blocking of XIAP or anti-apoptotic Bcl-2 protein function. Blocking XIAP function could be achieved by gene knockdown, but also by using a Smac mimetic that is able to block the binding of XIAP to caspase-3 and -9. This indicates that XIAP-mediated inhibition of effector caspase activation is a key feature in maintaining TRAIL resistance and release of Smac/DIABLO and/or Omi/HtrA2 from the mitochondria is likely to be more important than release of cytochrome c for execution of apoptosis.
It also has to be noted that although this study found that inhibition/knockdown of anti-apoptotic proteins is sufficient to induce TRAIL sensitivity, it is also possible that increased expression of pro-apoptotic proteins would generate the same effect. Supporting this theory, Nieminen and colleagues have reported that oncogenic transformation of fibroblasts led to induction of the pro-apoptotic Bcl-2 protein, Bak, which enabled TRAIL-mediated activation of the mitochondrial death pathway and switched the cells’ phenotype from resistant to sensitive.45
The studies presented here highlight that the regulators of TRAIL resistance in non-transformed cells act in a redundant manner, that is, removal of one inhibitor is not sufficient to sensitise non-transformed cells to TRAIL. Only simultaneous loss of cFLIP and XIAP expression or loss of cFLIP and anti-apoptotic Bcl-2 protein-function could induce TRAIL-sensitivity. Further proving this redundancy, CHX-induced sensitisation (which was associated with reduced expression of cFLIP, Bcl-2 and Bcl-XL) could be fully reverted by overexpressing only one anti-apoptotic factor (Bcl-XL).
In line with our findings, it has been shown that increased TRAIL sensitivity in transformed keratinocytes was associated with reduced cFLIP and XIAP expression and overexpression of these proteins could restore TRAIL resistance.23, 26 Other studies also suggest redundant mechanisms of resistance in non-transformed cells. Astrocytes lack DR expression; however, upon restoring DR expression cells are still resistant to TRAIL-induced apoptosis, unless cFLIP is knocked down.46 Furthermore, sensitisation to TRAIL in HUVECs has been shown to correlate with a reduction of both cFLIPS and Bcl-2 expression.47 Unfortunately, no knockdown studies were performed to test whether these proteins are sufficient for TRAIL resistance.
In contrast to non-transformed cells, many tumour cells appear to have lost this redundancy in resistance mechanisms. We demonstrated that various tumour cell types rely on the expression of a single anti-apoptotic protein and inhibition or removal of this pivotal block was sufficient to restore TRAIL sensitivity. This is in line with other reports that in several types of cancer, knockdown or inhibition of cFLIP alone48, 49 or XIAP50, 51, 52 was sufficient to restore sensitivity to TRAIL in resistant tumour cells. Likewise, knockdown or inhibition of the Bcl-2 block, whether Bcl-2 itself,53, 54 Bcl-XL17, 55 or Mcl-156 was sufficient to sensitise several cancer cell types to TRAIL-induced apoptosis.
The observation that transformed cells have lost the redundancy in resistance pathways offers a potential therapeutic window. A combination of TRAIL and an agent that targets one of the anti-apoptotic proteins is likely to be non-toxic to non-transformed cells. However, we still require biomarkers or gene expression signatures that can identify which is the dominating resistance pathway in a patient in order to find an effective and safe combination therapy.
Materials and Methods
Cell culture and treatments
hFbs from two different healthy donors (donor 006 and donor 007) were a kind gift of Dr. Linda Howard (Regenerative Medicine Institute (REMEDI), National Centre for Biomedical Engineering Science (NCBES), National University of Ireland (NUI) Galway) and were cultured in low-glucose concentration (1000 mg/l) Dulbecco’s modified Eagle’s medium (Sigma, St Louis, MO, USA) supplemented with 10% fetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin. Human umbilical artery smooth muscle cells (Lonza, Basel, Switzerland) were cultured in Smooth Muscle Cell Growth Medium 2 (PromoCell, Heidelberg, Germany) supplemented with 50 U/ml penicillin and 50 μg/ml streptomycin. HCA-7, PANC-1 and RKO cells were cultured in high-glucose concentration (4500 mg/l) Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 10% fetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin. OCI-AML2 cells were cultured in Minimum Essential Medium Eagle (Sigma) supplemented with 10% foetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin, and HL-60 cells were cultured RPMI-1640 medium (Sigma) supplemented with 10% foetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin. Cells were maintained at 37 °C in a humidified, 5% CO2 atmosphere.
hFb and hUASMCs were treated with recombinant human TRAIL (rhTRAIL, non-tagged, fragment of amino acids 114–281, kind gift from Wim J Quax, University of Groningen, the Netherlands), crosslinked FLAG-TRAIL95 (TRAIL-CL; aa 95–281, gift from Wim J Quax of University of Groningen, crosslinked with enhancer (anti-FLAG antibody) from Enzo Life Sciences, (Farmingdale, NY, USA), cat. no. ALX-804-034-C050). As both non-crosslinked and crosslinked or trimerisation-enforcing formulation (e.g., leucine-zipper tagged TRAIL) of TRAIL may be relevant in clinical use, both non-tagged and crosslinked TRAIL has been tested in most experiments.
To selectively activate DR5, crosslinked agonistic DR5 antibody (LBY135, Novartis, Basel, Switzerland) was used. To crosslink LBY135, LBY135 was incubated with threefold molar excess of anti-human Fab-fragment antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA, cat. no. 109-006-098) for 30 min at room temperature. To test the role of regulatory mechanisms, cells were treated with CHX (Sigma), ABT737 (Selleck Chemicals, Houston, TX, USA), Etoposide (Sigma), Doxorubicin (Sigma), STS (Sigma), Embelin (Sigma) or BV6 (Genentech, San Francisco, CA, USA) at the concentration and times specified in the figure legends.
Annexin V staining
Cell death was monitored by labelling phosphatidyl serine externalised on the surface of apoptotic cells with Annexin-V-FITC (Immunotools Ltd, Friesoythe, Germany). Following treatment, cells were collected by gentle trypsinisation and incubated for 10 min at 37 °C to allow membrane recovery, after which cells were pelleted by centrifugation and incubated with Annexin-V-FITC in calcium buffer (10 mM HEPES/NaOH, pH 7.5, 140 mM NaCl and 2.5 mM CaCl2) for 15 min on ice in the dark. Cells were washed in calcium buffer before acquisition on a FACSCanto I or II flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Analysis was performed using FACSDiva software (Becton Dickinson).
Measurement of mitochondrial transmembrane potential (ΔΨm)
Changes in ΔΨm were detected using the fluorescent dye tetramethylrhodamine ethyl ester perchlorate (Molecular Probes, Life Technologies, Carlsbad, CA, USA). Following treatment, cells were collected by gentle trypsinisation and tetramethylrhodamine ethyl ester perchlorate was added to the cells at a final concentration of 100 nM. Cells were incubated for 30 min at room temperature in the dark followed by immediate analysis by flow cytometry (FACSCanto II flow cytometer, Becton Dickinson). As a positive control for mitochondrial depolarisation, cells were treated with 10 μM carbonyl cyanide 3-chlorophenylhydrazone (Sigma) for 2 h.
TRAIL receptor immunocytochemistry
Following treatment, cells were collected by gentle trypsinisation and incubated for 10 min at 37 °C to allow membrane recovery. Cells were pelleted by centrifugation and incubated with mouse monoclonal antibodies against TRAIL-R1 (ALX-804-297-C100) or TRAIL-R2 (ALX-804-298-C100; Enzo Life Sciences) or goat polyclonal antibodies against TRAIL R3/TNFRSF10C (AF630) or TRAIL R4/TNFRSF10D antibodies (AF633; R&D Systems, Minneapolis, MN, USA) for 1 h on ice followed by either goat anti-mouse IgG-FITC (F2012; Sigma) for DR4 and DR5 or rabbit anti-goat-FITC (305-096-045; Jackson ImmunoResearch) for DcR1 or DcR2 for 30 min on ice. As negative controls, isotype control antibodies were used (goat IgG whole molecule (005-000-003; Jackson ImmunoResearch) and mouse IgG1 clone 11 711 (MAB002; R&D Systems)). After staining, cells were fixed in 1% formaldehyde in PBS and receptor expression was measured within 24 h on a FACSCanto I flow cytometer (Becton Dickinson). Analysis was performed using FlowJo software.
Intracellular flow cytometric analysis for active Bax
Following treatment, cells were collected by gentle trypsinisation and incubated for 10 min at 37 °C to allow membrane recovery. Cells were fixed with 4% formaldehyde in PBS for 10 min at room temperature followed by incubation with 2 μg/ml mouse monoclonal antibody against active Bax (Clone 6A7; 556 467; BD Pharmingen, San Diego, CA, USA) diluted in permeabilisation buffer (0.5% BSA and 0.1% saponin in PBS) for 1 h on ice followed by goat anti-mouse IgG-FITC (F2012; Sigma) for 1 h on ice. After staining, cells were resuspended in 4% formaldehyde in PBS and active Bax expression was measured on a FACSCanto II flow cytometer (Becton Dickinson).
Protein lysate preparation and western blot analysis
Cells were harvested by centrifugation and lysed in buffer containing 1% Igepal-630, 20 mM HEPES pH 7.5, 350 mM NaCl, 1 mM MgCl2, 0.5 mM EDTA, 0.1 mM EGTA, 0.5 mM dithiothreitol, 1 μM phenylmethylsulphonyl fluoride, 1.0 μg/ml pepstatin, 10 μM leupeptin, 2.5 μg/ml aprotinin, 2.5 μg/ml leupeptin and 250 μM N-acetyl-leucyl-leucyl-norleucinal after being washed two times in ice-cold PBS. Cellular proteins (30 μg) were subjected to electrophoresis on 10 or 12% SDS polyacrylamide gel and transferred onto Protran(R) nitrocellulose membrane (Sigma). After blocking in 5% non-fat milk and 0.05% Tween-20 in PBS, blots were incubated with rabbit polyclonal antibodies against caspase-3 (1:1000; #9662; Cell Signaling Technologies, Danvers, MA, USA), PARP (1:1000; #9542; Cell Signaling Technologies), Mcl-1 (1:1000; #4572; Cell Signaling Technologies) or actin (1:500; A2103; Sigma) and mouse monoclonal antibodies against caspase-8 1C12 (1:1000; Cell Signaling Technology), XIAP (1:5000; Assay Design, Enzo Life Sciences), cFLIP (1:200; Enzo Life Sciences), Bcl-XL (H-5) (1:200; Santa Cruz, CA, USA) and Bcl-2 (100) (1:200; Santa Cruz). For detection, appropriate horseradish peroxidase-conjugated goat secondary antibodies were used. Protein bands were visualised with SuperSignal West Pico Chemiluminescent Substrate (Pierce, Thermo Fisher Scientific, Rockford, IL, USA) or Immobilon western HRP substrate (Millipore, Billerica, MA, USA) on X-ray film (Agfa, Mortsel, Belgium).
Transfections and siRNAs
Fibroblast cells (5–6 × 105) were pelleted and resuspended in 100 μl NHDF Nucleofector solution (Amaxa, Lonza) and PBS containing 50 nM siRNA against GFP, cFLIP, XIAP or Mcl-1. Cells were transfected by nucleofection using programme P-022 as per manufacturer’s protocol (Amaxa). Twenty-four hour post-transfection, fresh medium was added to the cells and cells were treated as specified in figure legends. Smooth muscle cells were transfected with Lipofectamine 2000 transfection reagent as per manufacturer’s protocol (Invitrogen, Life Technologies) with 50 nM siRNA against GFP, cFLIP or XIAP. Twenty-four hour post-transfection, cells were treated as specified in figure legends. The following c-FLIP, XIAP or Mcl-1 sequences were targeted: cFLIPS/L1 (cFLIP1) 5′-GGAGCAGGGACAAGUUACA-3′, cFLIPS/L2 (cFLIP2) 5′-GCAAGGAGAAGAGUUUCUU,57 XIAP 5′-GAAGCUAGAUUAAAGUCCU-3′ (Sigma), Mcl-1 5′-GAAUUGAUUACCCGCCGAA-3′ (Sigma). The GFP target sequence was 5′-GGCUACGUCCAGGAGCGCACC-3′.
Transduction and viral vectors
Cells were transduced with a recombinant replication-incompetent E1-E3-deleted adenoviral vector (Ad5 serotype) encoding Bcl-XL (Ad5Bcl-XL; kind gift of Dr. Thomas Ritter, REMEDI, NCBES, NUI Galway), an adenoviral vector expressing cFLIP shRNA (Ad5shFLIP; kind gift of Dr. Ralf Zwacka, NCBES, NUI Galway),58 an adenoviral vector expressing EGFP (Ad5EGFP; kind gift of Dr. Ralf Zwacka, NCBES, NUI Galway) or an empty adenoviral vector (Ad5Null; kind gift of Dr. Thomas Ritter, REMEDI, NCBES, NUI Galway). hFb cells were transduced by spin centrifugation; the virus was added to the cells and the plate was spun at 2000 × g for 90 min at 37 °C after which fresh medium was added. To transduce HCA7 cells, the cells were seeded at 1.5 × 105 cells per ml the day before transduction. The virus was added in complete growth medium for 3 h after which the transduction medium was replaced with fresh growth medium and the experiments were carried out the following day. The optimal multiplicity of infection (MOI) was determined at 250 for hFb and 200 for HCA7 cells, at which ≥80% of the cells were transduced after 24–48 h with Ad5GFP. Cells were fixed in 1% PFA before further analyses.
Study of anti-apoptotic gene expression profiles and Boolean testing
Raw microarray data of 78 tumour cell lines were obtained from the publicly available GEO microarray data (GSE8332).40 Background correction and normalisation of the dataset have been performed using the Affy package of Bioconductor (RMA algorithm) in the statistical environment, R (version 2.10.1).59 The microarray probeset best representing the genes of interest has been selected using the JetSet method designed for scoring and ranking probe sets for specificity, splice isoform coverage and robustness against the degeneration of the transcript developed by Li et al.60 The selected representative mRNA probesets for each gene of interest was extracted from the normalised microarray data. Based on the range of expression of each gene across the 78 cell lines (Supplementary Figure 4), an arbitrary threshold for ‘overexpression’ was set at the level of ≥1.2-fold of the mean expression across all cell lines and it included approximately the top 20% (highest expressing) cell lines for each gene of interest. The hypothesis was considered correct if a cell line ‘overexpressed’ only one of the genes, that is, a cell line was only present in one overexpression list. The hypothesis was proven true if one of the following boolean expressions was true: (i) a given cell line is present in the cFLIP-overexpressing cell line list but it is not present in the overexpression lists for any other genes studied; (ii) if the cell line is present in the XIAP-overexpression list but it is not present in the overexpression lists for any other genes studied; (iii) if the cell line is present in either one or more overexpression lists for Bcl-2, Bcl-XL, Mcl1; but it is not included in either the XIAP- or cFLIP-overexpression lists.
Statistical analysis
Differences in Annexin V staining between the treatment groups were analysed using a paired Student’s t-test, with a significance of P<0.05. Error bars are shown as standard error of mean (S.E.M). All statistical analysis was performed using Minitab 16.
Abbreviations
- ΔΨm:
-
mitochondrial membrane potential
- TRAIL:
-
tumour necrosis factor-related apoptosis-inducing ligand
- DR:
-
death receptor
- DcR:
-
decoy receptor
- cFLIP:
-
cellular FLICE-like inhibitory protein
- XIAP:
-
X-linked inhibitor of apoptosis protein
- hFb:
-
human dermal fibroblasts
- hUASMC:
-
human umbilical artery smooth muscle cells
- CHX:
-
cycloheximide
- DISC:
-
death-inducing signalling complex
- PARP:
-
poly-ADP ribose polymerase
- siRNA:
-
small interfering RNA
- STS:
-
staurosporine
- Mcl-1:
-
myeloid cell leukemia sequence 1
- Bcl-2:
-
B-cell lymphoma 2
- Bcl-XL:
-
B-cell lymphoma-extra large
- Bax:
-
Bcl-2-associated X protein
- Bak:
-
Bcl-2 homologous antagonist/killer
- MOI:
-
multiplicity of infection
References
Ashkenazi A, Dixit VM . Death receptors: signaling and modulation. Science 1998; 281: 1305–1308.
Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A . Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000; 12: 611–620.
Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000; 12: 599–609.
Manzo F, Nebbioso A, Miceli M, Conte M, De Bellis F, Carafa V et al. TNF-related apoptosis-inducing ligand: signalling of a ‘smart’ molecule. Int J Biochem Cell Biol 2009; 41: 460–466.
Yamada H, Tada-Oikawa S, Uchida A, Kawanishi S . TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells. Biochem Biophys Res Commun 1999; 265: 130–133.
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 1999; 104: 155–162.
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 1999; 5: 157–163.
Dyer MJ, MacFarlane M, Cohen GM . Barriers to effective TRAIL-targeted therapy of malignancy. J Clin Oncol 2007; 25: 4505–4506.
Koschny R, Walczak H, Ganten TM . The promise of TRAIL--potential and risks of a novel anticancer therapy. J Mol Med (Berl) 2007; 85: 923–935.
Sanlioglu AD, Dirice E, Aydin C, Erin N, Koksoy S, Sanlioglu S . Surface TRAIL decoy receptor-4 expression is correlated with TRAIL resistance in MCF7 breast cancer cells. BMC Cancer 2005; 5: 54.
Aydin C, Sanlioglu AD, Karacay B, Ozbilim G, Dertsiz L, Ozbudak O et al. Decoy receptor-2 small interfering RNA (siRNA) strategy employing three different siRNA constructs in combination defeats adenovirus-transferred tumor necrosis factor-related apoptosis-inducing ligand resistance in lung cancer cells. Hum Gene Ther 2007; 18: 39–50.
Sanlioglu AD, Karacay B, Koksal IT, Griffith TS, Sanlioglu S . DcR2 (TRAIL-R4) siRNA and adenovirus delivery of TRAIL (Ad5hTRAIL) break down in vitro tumorigenic potential of prostate carcinoma cells. Cancer Gene Ther 2007; 14: 976–984.
Mulherkar N, Prasad KV, Prabhakar BS . MADD/DENN splice variant of the IG20 gene is a negative regulator of caspase-8 activation. Knockdown enhances TRAIL-induced apoptosis of cancer cells. J Biol Chem 2007; 282: 11715–11721.
Li P, Jayarama S, Ganesh L, Mordi D, Carr R, Kanteti P et al. Akt-phosphorylated mitogen-activated kinase-activating death domain protein (MADD) inhibits TRAIL-induced apoptosis by blocking Fas-associated death domain (FADD) association with death receptor 4. J Biol Chem 2010; 285: 22713–22722.
Sun M, Song L, Li Y, Zhou T, Jope RS . Identification of an antiapoptotic protein complex at death receptors. Cell Death Differ 2008; 15: 1887–1900.
Roth W, Reed JC . FLIP protein and TRAIL-induced apoptosis. Vitam Horm 2004; 67: 189–206.
Hinz S, Trauzold A, Boenicke L, Sandberg C, Beckmann S, Bayer E et al. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene 2000; 19: 5477–5486.
Fulda S, Meyer E, Debatin KM . Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002; 21: 2283–2294.
Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 2004; 5: 25–35.
Mahalingam D, Szegezdi E, Keane M, de Jong S, Samali A . TRAIL receptor signalling and modulation: Are we on the right TRAIL? Cancer Treat Rev 2009; 35: 280–288.
Cantarella G, Di Benedetto G, Scollo M, Paterniti I, Cuzzocrea S, Bosco P et al. Neutralization of tumor necrosis factor-related apoptosis-inducing ligand reduces spinal cord injury damage in mice. Neuropsychopharmacology 2010; 35: 1302–1314.
Koschny R, Ganten TM, Sykora J, Haas TL, Sprick MR, Kolb A et al. TRAIL/bortezomib cotreatment is potentially hepatotoxic but induces cancer-specific apoptosis within a therapeutic window. Hepatology (Baltimore, MD) 2007; 45: 649–658.
Leverkus M, Sprick MR, Wachter T, Mengling T, Baumann B, Serfling E et al. Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance-mediating block of effector caspase maturation. Mol Cell Biol 2003; 23: 777–790.
Degli-Esposti MA, Smolak PJ, Walczak H, Waugh J, Huang CP, DuBose RF et al. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med 1997; 186: 1165–1170.
Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997; 277: 818–821.
Leverkus M, Neumann M, Mengling T, Rauch CT, Brocker EB, Krammer PH et al. Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res 2000; 60: 553–559.
Zhang XD, Nguyen T, Thomas WD, Sanders JE, Hersey P . Mechanisms of resistance of normal cells to TRAIL induced apoptosis vary between different cell types. FEBS Lett 2000; 482: 193–199.
Schneider P, Olson D, Tardivel A, Browning B, Lugovskoy A, Gong D et al. Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Biol Chem 2003; 278: 5444–5454.
Wu GS, Burns TF, Zhan Y, Alnemri ES, El-Deiry WS . Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res 1999; 59: 2770–2775.
Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN . c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem 2005; 280: 14507–14513.
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190–195.
Deveraux QL, Takahashi R, Salvesen GS, Reed JC . X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997; 388: 300–304.
Du C, Fang M, Li Y, Li L, Wang X . Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102: 33–42.
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102: 43–53.
Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM et al. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem 2002; 277: 445–454.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435: 677–681.
Nikolovska-Coleska Z, Xu L, Hu Z, Tomita Y, Li P, Roller PP et al. Discovery of embelin as a cell-permeable, small-molecular weight inhibitor of XIAP through structure-based computational screening of a traditional herbal medicine three-dimensional structure database. J Med Chem 2004; 47: 2430–2440.
Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007; 131: 669–681.
Klein S, Levitzki A . Signal Transduction Therapy for Cancer – Whither Now? Curr Signal Trans Ther 2006; 1: 1–12.
Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 2007; 13: 1070–1077.
Sayers TJ . Targeting the extrinsic apoptosis signaling pathway for cancer therapy. Cancer Immunol Immunother 2011; 60: 1173–1180.
Fulda S . Histone deacetylase (HDAC) inhibitors and regulation of TRAIL-induced apoptosis. Exp Cell Res 2012; 318: 1208–1212.
Mellier G, Huang S, Shenoy K, Pervaiz S . TRAILing death in cancer. Mol Aspects Med 2010; 31: 93–112.
Nesterov A, Nikrad M, Johnson T, Kraft AS . Oncogenic Ras sensitizes normal human cells to tumor necrosis factor-alpha-related apoptosis-inducing ligand-induced apoptosis. Cancer Res 2004; 64: 3922–3927.
Klefstrom J, Verschuren EW, Evan G . c-Myc augments the apoptotic activity of cytosolic death receptor signaling proteins by engaging the mitochondrial apoptotic pathway. J Biol Chem 2002; 277: 43224–43232.
Song JH, Bellail A, Tse MC, Yong VW, Hao C . Human astrocytes are resistant to Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. J Neurosci 2006; 26: 3299–3308.
Alladina SJ, Song JH, Davidge ST, Hao C, Easton AS . TRAIL-induced apoptosis in human vascular endothelium is regulated by phosphatidylinositol 3-kinase/Akt through the short form of cellular FLIP and Bcl-2. J Vasc Res 2005; 42: 337–347.
Mitsiades N, Mitsiades CS, Poulaki V, Anderson KC, Treon SP . Intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human multiple myeloma cells. Blood 2002; 99: 2162–2171.
Geserick P, Drewniok C, Hupe M, Haas TL, Diessenbacher P, Sprick MR et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL- and CD95L-mediated apoptosis. Oncogene 2008; 27: 3211–3220.
Fakler M, Loeder S, Vogler M, Schneider K, Jeremias I, Debatin KM et al. Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance. Blood 2009; 113: 1710–1722.
Ndozangue-Touriguine O, Sebbagh M, Merino D, Micheau O, Bertoglio J, Breard J . A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene 2008; 27: 6012–6022.
Vogler M, Walczak H, Stadel D, Haas TL, Genze F, Jovanovic M et al. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res 2008; 68: 7956–7965.
Sinicrope FA, Penington RC, Tang XM . Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis is inhibited by Bcl-2 but restored by the small molecule Bcl-2 inhibitor, HA 14-1, in human colon cancer cells. Clin Cancer Res 2004; 10: 8284–8292.
Hetschko H, Voss V, Horn S, Seifert V, Prehn JH, Kogel D . Pharmacological inhibition of Bcl-2 family members reactivates TRAIL-induced apoptosis in malignant glioma. J Neurooncol 2008; 86: 265–272.
Ray S, Bucur O, Almasan A . Sensitization of prostate carcinoma cells to Apo2L/TRAIL by a Bcl-2 family protein inhibitor. Apoptosis 2005; 10: 1411–1418.
Clohessy JG, Zhuang J, de Boer J, Gil-Gomez G, Brady HJ . Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J Biol Chem 2006; 281: 5750–5759.
Nakajima A, Kojima Y, Nakayama M, Yagita H, Okumura K, Nakano H . Downregulation of c-FLIP promotes caspase-dependent JNK activation and reactive oxygen species accumulation in tumor cells. Oncogene 2008; 27: 76–84.
Buneker CK, Yu R, Deedigan L, Mohr A, Zwacka RM . IFN-gamma combined with targeting of XIAP leads to increased apoptosis-sensitisation of TRAIL resistant pancreatic carcinoma cells. Cancer Lett 2012; 316: 168–177.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5: R80.
Li Q, Birkbak NJ, Gyorffy B, Szallasi Z, Eklund AC . Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics 2011; 12: 474.
Acknowledgements
MvD was supported by an EMBARK fellowship from IRCSET and by the Thomas Crawford Hayes Fund, TS is supported by a Fellowship from the College of Science of NUIG. The presented study was supported by grants from the Irish Cancer Society (research fellowship to ES, CRF09SZE) and from Science Foundation Ireland (Starting Investigator Research Grant to ES, 09/SIRG/B1575). AS is a scientific founding member and director of Aquila Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no competing interests.
Additional information
Edited by D Aberdam
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
van Dijk, M., Halpin-McCormick, A., Sessler, T. et al. Resistance to TRAIL in non-transformed cells is due to multiple redundant pathways. Cell Death Dis 4, e702 (2013). https://doi.org/10.1038/cddis.2013.214
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.214
Keywords
This article is cited by
-
Targeting of dermal myofibroblasts through death receptor 5 arrests fibrosis in mouse models of scleroderma
Nature Communications (2019)
-
Secretion of tumoricidal human tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by recombinant Lactococcus lactis: optimization of in vitro synthesis conditions
Microbial Cell Factories (2018)
-
Repression of Mcl-1 expression by the CDC7/CDK9 inhibitor PHA-767491 overcomes bone marrow stroma-mediated drug resistance in AML
Scientific Reports (2018)
-
Decoy receptors block TRAIL sensitivity at a supracellular level: the role of stromal cells in controlling tumour TRAIL sensitivity
Oncogene (2016)
-
NGF FLIPs TrkA onto the death TRAIL in neuroblastoma cells
Cell Death & Disease (2016)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}