
Abstract
Bu-Shen-Ning-Xin Decoction (BSNXD) administration has alleviated the early pathologic damage of atherosclerosis by inhibiting the adhesion molecule expression and upregulating the estrogen receptor (ER) β expression in endothelial cells, and increasing the serum nitric oxide (NO) level without any effect on serum lipid status, endometrium and fat deposition in liver in ovariectomized rabbits. The BSNXD-derived serum increases ER β expression in the human umbilical vein endothelial cells (HUVECs), and decreases malondialdehyde (MDA) production, and upregulates eNOS expression then increases NO synthesis through ERβ-dependent pathway. NO not only suppresses the LPS-induced NF-κB transcription in HUVECs, but also decreases apoptosis of endothelial cells. The BSNXD-derived serum decreases monocyte chemoattractant protein-1 production, and suppresses cell adhesion molecules (ICAM-1, VCAM-1 and E-selectin) expression in HUVECs injured by oxidized low-density lipoproteins (ox-LDL), and these effects can be abolished by ERβ antagonist (R,RTHC) and NO synthase inhibitor (L-NAME). The BSNXD-derived serum-treated HUVECs supernatant reduces CCR2, LFA-1 and VLA-4 expression in monocytes cell line U937 cells, which in turn inhibits adherence of U937 to injured endothelial cells. NO synthesis increases, and MDA production decreases through ERβ-mediated pathway that suppresses apoptosis and NF-κB activity in endothelial cells that downregulates adhesion molecules expression on endothelial cells via ERβ/NO/NF-κB pathway, and in turn leukocyte adhesion, which suggests BSNXD potential value in prophylaxis atherosclerosis.
Similar content being viewed by others


Main
Atherosclerosis is one sort of inflammatory disease.1 An early step in the atherogenic process is transmigration of blood monocytes across an injured or dysfunctional endothelium into the subendothelial space where they differentiate into macrophages.2 The cell adhesion molecules have been implicated as one of the most important factors in atherosclerosis. The cytokine-mediated cell adhesion molecule expression is regulated by the activity of transcription factors, such as NF-κB (NF-κB).3 Nitric oxide (NO) is a widespread signaling molecule in the cardiovascular system, and functions in multiple ways to protect from the initiation and progression of atherosclerosis.4 NO prevents from the adhesion and aggregation of blood cells.5 It has also been shown that the induction and stabilization of I-κ-B-α by NO are important mechanisms by which NO inhibits NF-κB and attenuates atherogenesis.6 NO is synthesized by NO synthase (NOS) with L-arginine as a substrate. The promotion of NO production and prevention from cell adhesion may be an effective approach to control atherosclerosis.
Ovarian dysfunction has been recognized as a major risk factor for the accelerated atherosclerotic vascular disease. A lot of studies in vitro have identified mechanisms by which estrogen exerts beneficial effects on cardiovascular system.7 Epidemiological and experimental evidence indicates several atheroprotective effects of endogenous estrogen, which intervenes from atherosclerosis progression and inflammation.8 Estrogen effects are generally ascribed to transcriptional modulation of target genes through estrogen receptors (ERs), ERα and ERβ. The functional ERs are expressed in vascular endothelial cells.9 Normal coronary arteries of premenopausal women show normal ER expression, whereas in the atherosclerotic vessels of postmenopausal women, ERs are downregulated. ERα plays an important role in mediating estrogen’s vascular protection,9, 10, 11 but the precise mechanisms of ERβ in vascular homeostasis remains elusive. It is reported that the intimal ERβ expression correlates with atherosclerosis in postmenopausal women;12 polymorphisms in ERβ gene are associated with risk of cardiovascular disease in women;13 ERβ is the predominant mRNA transcript in normal human vascular smooth muscle cells and in the media of human arteries.14, 15 The current understanding of ER does not allow one to clearly discern the importance of one isotype receptor over another. Indeed, important concepts are likely yet to be uncovered regarding receptor roles.
Traditional Chinese medicines (TCMs) have been used in Asian countries for over 5000 years to prevent and treat diseases. TCM uses a holistic and synergistic approach to restore the balance of Yin-Yang of body energy so that the body’s normal function and homeostasis is maintained.16 Traditional Chinese herbal medicines often consist of different herbs. The objective of doing this is to form specific formulae aimed to increase therapeutic efficiency and reduce adverse effects.17 According to the hypothesis of TCM, multiple active phytochemical components in the TCM formulae may simultaneously target multiple molecules/pathways, and thus potentially achieve superior effect to single compound.18 Traditional Chinese herbs have long been used for preventing atherosclerosis with less side-effect.19, 20, 21 Bu-Shen-Ning-Xin Decoction (BSNXD) has long been used in the prophylaxis of the atherosclerosis associated with estrogen deficiency, which have already been proved to possess clear prophylactic action on human bodies in the clinical setting, but there is little information about its pharmacological properties and biochemical activities. In this paper, we summarized their anti-inflammatory effects in atherosclerosis animal models and some mechanisms at the cellular and molecular levels here. In order to clarify the effects of BSNXD on atherosclerosis in postmenopause, we use OVX in female rabbits to deplete ovarian function. The present study is to investigate the mechanisms of BSNXD ameliorating atherosclerosis in the OVX rabbits. In vitro, we have demonstrated that the BSNXD-derived serum increases ERβ expression in the human umbilical vein endothelial cells (HUVECs), and decreases malondialdehyde (MDA) production and upregulates eNOS expression then increases NO synthesis through ERβ-dependent pathway.
Results
BSNXD upregulates NO with no effect on E2 and lipid concentration in serum
A significant increase of body weight was observed in the sham group, OVX group and OVX+BSNXD group (ovariectomized rabbits treated with BSNXD) after high cholesterol chow diet. There was no significant difference in final body weight between sham group, OVX group and OVX+BSNXD group (P>0.05, data not shown). We examined whether there was a difference in plasma E2, NO and lipid levels among sham group, OVX and OVX+BSNXD group after 12 weeks of hypercholesterolemic diet. As shown in Figure 1a, the serum E2 concentrations in the OVX group decreased significantly compared with the sham group (P<0.01). BSNXD treatment did not affect the serum E2 level. The ovariectomy resulted in a significantly decreased NO concentration in serum (P<0.01); OVX rabbits with BSNXD treatment exhibited significantly higher serum NO concentration than that of the OVX control (P<0.01, Figure 1b). Since the hypercholesterolemic diet led to hyperlipidemia, lipid concentrations were measured in plasma. As expected, we have not found any change in TC, TG, HDL-C and LDL-C levels in OVX+BSNXD rabbits compared with that of the OVX control or sham (Figure 1c).

Effect of BSNXD on serum E2, nitric oxide (NO), lipid, atherosclerotic lesions and estrogen receptor (ER) isotype, VCAM-1 expression in thoracic aortic tissues. The sham rabbits underwent a mock operation and received high cholesterol chow. The ovariectomized rabbits underwent bilateral oophorectomy, and then were randomly divided into two groups: OVX group treated by high cholesterol chow; OVX+BSNXD group received high cholesterol chow plus 5 ml mixed row herbs (BSNXD) per kilogram body weight daily. Blood samples were harvested after 12 weeks treatment. Serum E2 concentration (a), Serum NO levels (b) and plasma lipids such as TC, TG, HDL-C and LDL-C levels (c) were measured. Macroscopic observation of the atheromatous plaque formation on the luminal surface of the aorta (d). Representative micrographs of the intimal lesions (H&E staining) (e); original magnification was × 200. Average percentage of aortic lesion area relative to entire aorta area, intima cross-sectional thickness, intima/media thickness ratio and intimal/medial area ratio (f) was quantified at aortic lesions. S.E.M. of thoracic aorta endothelium, endothelial cells in the OVX group showed irregular in shape, only partially preserved, and the subendothelial surface is visible; aggregated platelets and leukocytes adhering to the endothelium; BSNXD treatment significantly ameliorated the endothelial disruption region; platelets and leukocytes sticking to endothelium were significantly reduced; number of leukocytes per mm2 adhering to the endothelium was decreased by BSNXD treatment (g). Immunohistochemistry staining of ER was performed in artery after BSNXD treatment, and ER expression was much more intense in the artery layer (h). As shown in (i, j), both ERα and ERβ expression in artery wall layer of OVX group were markedly lower than that in Sham animals. In all, 12-week treatment with BSNXD significantly increased ERβ expression, while did not alter the ERα expression. Immunohistochemistry staining for VCAM-1 was performed in artery. The VCAM-1 expression in endothelial cells, subendothelial and smooth muscle cell layers near the endothelium of arterial wall was significantly decreased after BSNXD treatment compared to the OVX control (k); Data are expressed as mean values±S.E.M. (n=6). **P<0.01 compared with the OVX group
BSNXD alleviates atherosclerotic lesions and reduces leukocytes adherent to the injured endothelial cells
High cholesterol diet led to atheromatous plaque formation in aorta luminal surface. A marked lipid deposition accompanied by continuous plaque formation was observed, and marked intimal thickening with focal endothelial injury in the aortas from OVX rabbits. All of the animals subjected to high cholesterol chow developed atheromatous plaque lesion, which consisted of lipid-rich deposits adhering to the aortic wall (Figure 1d). HE staining showed that the atheromatous plaque lesion was characterized by a hyperplasic transformation of the intima that contained elastic fibers, fibroblast-like cells, collagen deposits and foam cells (Figure 1e).
The extent and degree of atherosclerosis were estimated by calculating the percentage of the plaque area to total aorta surface area; intimal hyperplasia in the aortic lesions that expressed as the intima cross-sectional thickness, intima/media thickness ratio and intimal/medial area ratio. It was shown that the OVX rabbit with high cholesterol chow showed severe atherosclerosis. Animals in the sham group showed less atherosclerosis compared with the OVX (P<0.01). The intima cross-sectional thickness, intima/media thickness ratio and intimal/medial area ratio at the site of aortic lesions in the OVX significantly increased when compared with the sham (P<0.01). The average percentage of aortic lesion area in inner surface, intima cross-sectional thickness, intima/media thickness ratio and intimal/medial area ratio in the aortic lesions was significantly decreased after having been treated with BSNXD (all P<0.01) (Figure 1f). The atherosclerosis degree in OVX with BSNXD was significantly decreased compared with the OVX.
S.E.M. analysis showed a wide spectrum of pathological alterations in the luminal surface of thoracic aorta from the OVX. In OVX rabbits, the aorta luminal vessel surface was covered by irregularly orientated endothelial cells; the endothelial cells showed cuboidal appearance protruding into the lumen of the vessel; some endothelial cells contained ‘craters’ disrupting the continuous surface of the endothelium and numerous microvilli; platelets and leukocytes adhering to endothelium were more frequently observed compared with the sham (P<0.01); in regions of the vessel with endothelial denudation, the underlying connective tissue was visible. After BSNXD treatment, vessel surface walls in endothelial disruption region were significantly ameliorated compared with the OVX, and the platelets and leukocytes sticking to endothelium were obviously decreased (P<0.01, Figure 1g).
BSNXD regulates ERβ, VCAM-1 expression in artery wall
Immunohistochemistry staining showed that the staining intensity of ER was significantly lower in each artery wall layer (intima, media or adventitia) in the OVX than that of the sham (P<0.01); OVX with BSNXD administration significantly enhanced ER expression (P<0.01) (Figure 1h), suggesting that ER mediates the effects of BSNXD on atherosclerosis. To provide further evidence regarding which ER isotype involved in the modulation of BSNXD on endothelial cells, we further checked the protein levels of ERα and ERβ expression. Our results showed that both ERα and ERβ expression in artery wall layer of OVX group were markedly lower than that in Sham animals. As shown in Figures 1i and j, BSNXD treatment did not alter the ERα expression, while significantly increased ERβ expression in artery wall layer of OVX animals. Furthermore, selective ERα antagonist (Methyl-piperidino-pyrazole, MPP) and ERβ antagonists (R,R-tetrahydrochrysene, R,RTHC) were used in an additional set of experiments of HUVECs in vitro.
Adhesion molecule such as VCAM-1 plays an important role in the pathogenesis of atherosclerosis. To determine whether VCAM-1 was involved in the effect of BSNXD on atherosclerosis, expression of VCAM-1 in artery wall layer was detected. As shown in Figure 1k, endothelial cell, subendothelial and smooth muscle cell in the arterial wall of the OVX expressing higher level VCAM-1 than that of the sham (P<0.01); OVX with BSNXD treatment significantly decreased VCAM-1 expression than that of the OVX control (P<0.01).
The effects of BSNXD on uterus and liver
Photomicrographs depicting OVX uterine columnar epithelium atrophied, and uterine glands number reduced. Our previous data showed that the relative weights of uterus markedly decreased after ovariectomy.22 Administration of BSNXD for 12 weeks after ovariectomy resulted in no significant change (data not shown) in the relative uterus wet weight and endometrial glands compared with the OVX control.
We found that multifocal depositions of lipids in liver of the OVX rabbits after high cholesterol chow that showed more incidence of fatty liver compared with the sham (P<0.01). OVX with BSNXD treatment were less prone to high cholesterol chow-induced liver steatosis, as revealed by the pathological and histological analysis (data not shown). Moreover, BSNXD administration presented less incidence of fatty liver on the high-fat diet compared with the OVX control (Figure 2).

Effect of BSNXD on morphology of liver. (a) Representative liver images are shown. (b) fatty liver incidence of different group are represented. As evaluated by the pathological and morphological analysis of the liver from each group, OVX+BSNXD rabbits showed less incidence of fatty liver on the high-fat diet compared with OVX rabbits. Data are expressed as mean values±S.E.M. (n=6). **P<0.01 compared with the OVX group
BSNXD has prophylactic effect but not therapeutic effect on established atherosclerosis associated with estrogen deficiency
The other in vivo experiment (treatment experiments) is to treat ovariectomized rabbit with established atherosclerotic plaques to see whether the BSNXD could revert the phenotype and not only prevent it. Determinations of lesion area by enface preparation of the aorta showed extensive lesion formation by the end of the inductive phase. There was a little reduction of lesions when animals were switched to a normal chow with or without BSNXD. Also, BSNXD group had similar aortic lesions when compared with the saline group (data not shown). So from our study, we can see BSNXD has prophylactic effect but not therapeutic effect on established atherosclerosis associated with estrogen deficiency.
BSNXD-derived serum increases ERβ expression in HUVECs
Real-time PCR and western blot were used to analyze the transcription and translation levels of ERα and ERβ in HUVECs. The results showed that ERα and ERβ were steadily expressed in HUVECs. We tried to add the BSNXD drug directly to the culture medium of HUVEC in our in vitro experiments, it has no effect on ERα or ERβ expression (data not shown); while BSNXD-derived serum increased significantly the transcription and translation of ERβ, but not ERα in HUVECs (P<0.01, Figures 3a and b).

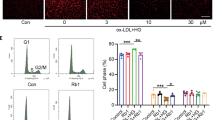
BSNXD regulates estrogen receptor β mediating nitric oxide production and NF-κB suppression in human umbilical vein endothelial cells (HUVECs). The primary HUVECs were exposed to control serum or 10% BSNXD-derived serum for 48 h; in the final 24 h culture the ox-LDL was added. The supernatants were collected, and ERα/ERβ mRNA (a) and protein (b) expression were analyzed; eNOS expression (c), NO (d) and malondialdehyde (MDA) (e) concentrations were determined; eNOS expression, NO production was enhanced and MDA was downregulated by the drug-derived serum. The primary HUVECs were collected, dual-stained with Annexin V-FITC/PI, and then analyzed by FCM (f); the 10% drug-derived serum could inhibit the ox-LDL-induced apoptosis. BSNXD-derived serum inhibited the LPS-induced NF-κB transcription in HUVECs (g). BSNXD treatment increased ERβ transcription and translation in HUVECs. Pretreatment of selective ERβ antagonist (R, R-tetrahydrochrysene, R, RTHC) but not ERα antagonist (methyl-piperidino pyrazole, MPP) could block these effects on (eNOS, NO, MDA expression, apoptosis, NF-κB transcription, etc.), which is induced by the drug-derived serum. Meanwhile, pretreatment with the NO synthase inhibitor (NG-nitro-L-arginine methyl ester, L-NAME) significantly decreased the NO production, blocked the anti-apoptosis and NF-κB activity inhibition effect of BSNXD in HUVECs. Data are expressed as mean values±S.E.M. (n=6). *P<0.05, **P<0.01
BSNXD-derived serum upregulates eNOS expression, increases NO and downregulates MDA production via ERβ-dependent pathway in HUVECs
We further analyzed the modulation of the BSNXD drug directly on eNOS expression, NO and MDA production in HUVECs induced by ox-LDL. eNOS expression, NO and MDA production was not changed by the drug directly (data not shown); but eNOS expression, NO production was enhanced, and MDA was decreased by the drug-derived serum (P<0.05 or P<0.01, respectively). Meanwhile, we pretreated, respectively, the HUVECs with 10−6 M MPP or 10 nM R,RTHC, which revealed that the ERβ other than ERα antagonist could block these effects induced by the drug-derived serum (P<0.05) (Figures 3c–e). Moreover, pretreatment with the NOS inhibitor (NG-nitro-L-arginine methyl ester, L-NAME) also significantly decreased the NO production in HUVECs (P<0.05) (Figure 3d). Therefore, BSNXD-derived serum upregulates eNOS expression, increases NO and decreases MDA production via ERβ other than α pathway in endothelial cells.
BSNXD-derived serum inhibits apoptosis and NF-κB activity in HUVECs through NO and ERβ pathway
To investigate the protective effects of BSNXD on HUVECs apoptosis induced by ox-LDL, we analyzed the percentage of the early apoptotic cells with the Annexin V-FITC/PI dual-labeling assay. The 10% drug-derived serum could relieve the ox-LDL-induced apoptosis (P<0.01). The anti-apoptosis effect of BSNXD could be blocked by NOS inhibitor (P<0.01) or ERβ antagonist (P<0.05), but not ERα antagonist (Figure 3f), which suggests that BSNXD inhibits the endothelial cells apoptosis in an ERβ and NO-dependent manner.
The activity of the transcription factor NF-κB in the LPS-stimulated HUVECs was also examined. It was found that NF-κB-luciferase activity decreased significantly after 10% drug-derived serum treatment (P<0.01); this effect of BSNXD-derived serum was significantly inhibited by ERβ antagonist or NOS inhibitor (P<0.01) (Figure 3g). It may be proposed that BSNXD exerts NF-κB activity suppression by increasing NO production via ERβ mediating pathway in endothelial cells.
BSNXD-derived serum suppresses adhesion molecules expression in HUVECs through ERβ/NOS/NF-κB pathway
We further investigated the effect of BSNXD on chemotactic factors such as monocyte chemoattractant protein-1 (MCP-1), adhesion molecules such as ICAM-1, VCAM-1 and E-selectin expression in HUVECs. The MCP-1 (Figure 4a), ICAM-1 (Figure 4b), VCAM-1 (Figure 4c) and E-selectin (Figure 4d) expression were greatly decreased after treatment with the 10% drug-derived serum (P<0.01); and ERβ antagonist (P<0.01) other than ERα antagonist (P>0.05) could block these effects of BSNXD-derived serum. Meanwhile, NOS inhibitor could also abolish these effects of BSNXD-derived serum (P<0.01). Our results suggest that BSNXD-derived serum downregulates chemotactic factor MCP-1 and adhesion molecules expression via ERβ/NO/NF-κB pathway in endothelial cells.

BSNXD suppresses adhesion molecules expression on HUVECs through ERβ/NO/NF-κB pathway. The primary HUVECs were exposed to control serum or 10% BSNXD-derived serum for 48 h; in the final 24 h culture the ox-LDL was added. The supernatants were collected, and MCP-1 concentrations were determined. The MCP-1 expression of HUVECs was determined by western blot analysis. The mRNA and protein expression levels of cell adhesion molecules (ICAM-1, VCAM-1 and E-selectin) were assessed by RT-PCR, western blot and FACS. The MCP-1(a), ICAM-1 (b), VCAM-1 (c) and E-selectin (d) expression was greatly decreased after treatment with the 10% drug-derived serum. NOS inhibitor (L-NAME), ERβ antagonist (R, RTHC) other than ERα antagonist (MPP) could block these effects induced by the drug-derived serum. Data are expressed as mean values±S.E.M. (n=6). *P<0.05, **P<0.01
BSNXD-derived serum-treated HUVECs supernatant inhibits CCR2, LFA-1, VLA-4 expression in U937 and U937 cell adhesion
We further examined the effects of HUVECs supernatant under different treatments on CCR2 (Figure 5a), LFA-1 (Figure 5b) and VLA-4 (Figure 5c) expression in U937. Our previous results showed that the supernatant from ox-LDL-treated HUVECs induced a significant increase of CCR2, LFA-1 and VLA-4 expression in U937;22 the ox-LDL-induced increasing could be inhibited by the 10% drug-derived serum (P<0.01). Thereafter, we investigated the BSNXD effect on U937 cell adhesion to HUVECs. The 10% BSNXD-derived serum could suppress U937 cell adhesion (P<0.01); ERβ antagonist (P< 0.05) but not ERα antagonist (P>0.05) could block the effect of the 10% drug-derived serum. Simultaneously, NOS inhibitor (P<0.01) could also abolish this effect of BSNXD-derived serum (Figure 5d). Our results suggest that the anti-atherogenic effect of BSNXD on U937 cells adhesion is also via ERβ/NO/NF-κB pathway.

BSNXD-treated HUVECs supernatant inhibits CCR2, LFA-1, VLA-4 expression in U937 and attenuates U937 cell adhesion. After HUVECs were treated as above, the supernatant was collected and used to treat U937 cells. CCR2 (a), LFA-1 (b) and VLA-4 (c) mRNA and protein expression levels in treated U937 cells were assessed by RT-PCR, flow cytometry and western blot. The ox-LDL-induced increase (Control group) could be inhibited by the 10% drug-derived serum. Furthermore, 10% BSNXD could suppress U937 cell adhesion; NOS inhibitor (L-NAME) and ERβ antagonist (R, RTHC) but not ERα antagonist (MPP) could block the anti-adhesion effect of the 10% drug-derived serum (d). Data are expressed as mean values±S.E.M. (n=6). *P<0.05, **P<0.01
Discussion
The development of an atherosclerotic lesion requires a complex interplay between mononuclear cells and endothelia. Our histopathological analysis has demonstrated that BSNXD significantly relieves the extent and degree of atherosclerosis; S.E.M. examination also shows that BSNXD ameliorates vessel wall injury, and the platelets and leukocytes adhering to the endothelium are obviously decreased. BSNXD reduces lipids deposition in liver and consequently the incidence of fatty liver, and has no stimulating effects on the uterus. Disruptions of ovarian function are associated with increased risk of metabolic disease, it causes ectopic lipid deposition. In our experiments, there was no significant difference in final body weight and serum lipid levels between sham, OVX and OVX+BSNXD group. Meanwhile, multifocal depositions of lipids in the liver of the OVX rabbits showed more incidence of fatty liver compared with the sham. OVX with BSNXD treatment were less prone to liver steatosis and fatty liver. There is strong difference in liver steatosis, without relevant differences in circulating fatty acid; suggesting that E2 deficiency induced hepatic lipid accumulation. The present findings support a role for disruptions of ovarian function in the development of visceral adiposity,23 which in humans is known to precede the development of the metabolic syndrome. BSNXD could reduce E2 deficiency associated with ectopic lipid deposition. Some anti-atherosclerotic drugs can prevent from atherosclerosis by protecting LDL from oxidation and anti-hypercholesterolemic effect.24, 25 Estrogen treatment prevents from the development of fatty streaks.11 Our data show that lipid parameters (TC, LDL-C, HDL-C) have not been changed by BSNXD. As other studies have showed, the changes in lipid parameters are too minor to explain the atheroprotective effect of the hormone.11 There are plasma lipid-dependent and -independent effects of anti-atherosclerotic drugs. Our results show that the effects of BSNXD on atheroma regression occur completely independent of systemic cholesterol levels. This effect seems to be a direct anti-inflammatory effect of BSNXD on the arterial wall. Previous research also showed the crucial role of intact endothelium, as the anti-atherogenic effect of E2 was abolished.26
It is important to conclude whether the effects of BSNXD are specific or represent a combined effect with estrogen generated from BSNXD. The present study shows that BSNXD treatment does not change serum E2 concentration and ERα expression in thoracic aortic tissues, but does increase ERβ expression. BSNXD may exert roles directly through ERβ. BSNXD may enhance proliferation of estrogen-sensitive cells, and thereby increase endothelial reactivity to E2.27 Also, we can see that BSNXD has prophylactic effect but not therapeutic effect on established atherosclerosis associated with estrogen deficiency. In postmenopausal women with established atherosclerosis, E2 had no effect on the progression of atherosclerosis.28 Cumulated data support a ‘window-of-opportunity’ for maximal reduction of CHD and minimization of risks with HRT initiation before age 60 and/or within 10 years of menopause.29, 30
Atherosclerosis is an inflammatory disease characterized by endothelial dysfunction, impairment of NO production,4, 31 etc. NO is a crucial mediator in endothelial vasodilator function. It is synthesized from the terminal guanidine nitrogen of L-arginine by NOS enzymes.32 The bioavailability of NO is a representation of endothelial function. Our results show that eNOS expression and NO is increased by BSNXD, and the atheroprotective effect may be dependent on NO production. Adhesion molecule such as VCAM-1 plays an important role in the pathogenesis of atherosclerosis. Our results showed that VCAM-1 expression in artery wall is significantly decreased by BSNXD treatment, which suggests that BSNXD plays direct anti-inflammatory effects on arterial wall.33
Our results showed that BSNXD-derived serum but not BSNXD raw herbs itself has effective action on HUVECs In vitro. After oral administration of BSNXD, compounds absorbed into the bloodstream then become active compounds. Compounds may be functional by oxidation, hydrolysis or reduction; primary metabolites may also undergo conjugation reactions to form secondary metabolites.34, 35 Our results show that the BSNXD-derived serum increases ERβ expression in HUVECs, and results in an enhanced NO and reduced MDA production. ERβ other than ERα antagonist can block these effects induced by the drug-derived serum; meanwhile, NOS inhibitor can significantly decrease the NO production in HUVECs. BSNXD appears to increase NO and decrease MDA production via ERβ-dependent pathway. Furthermore, BSNXD treatment can inhibit the apoptosis of HUVECs in the ERβ and NO-dependent manner.
Nuclear transcription factor NF-κB is known to be the key regulator for modulate functional genes expression including adhesion molecule. We have investigated whether NF-κB activity is involved in the inhibitory mechanism of BSNXD on adhesion molecule expression. As expected, BSNXD significantly inhibits NF-κB-luciferase transcription activity, which suggests that BSNXD attenuates the inflammatory reaction by inhibition of NF-κB in the vascular endothelium. It is reported that the induction and stabilization of I-κB-α by NO are important mechanisms by which NO inhibits NF-κB and attenuate atherogenesis.6 Our data also confirm that the effect of BSNXD-derived serum on NF-κB activity in endothelial cells is significantly inhibited by ERβ antagonist or NOS inhibitor. It is proposed that BSNXD exerts NF-κB activity downregulation by increasing NO production via ERβ-dependent pathway in endothelial cells, which subsequently attenuates atherogenesis.
Inflammatory cells play a crucial role in the pathogenesis of atherosclerosis.36, 37 Penetration of atherogenic lipoproteins is the first step of the atheromatous process.38 Adhesion molecules, such as ICAM-1, VCAM-1 and E-selectin, mediate the binding of monocytes and lymphocytes to vascular endothelial cells through interactions with the counter-receptor LFA-1, VLA-4, etc. Chemokine MCP-1 may recruit monocytes to migrate into the intima of the arterial wall, and enhance the progression of the atherosclerotic lesions.39 Our results demonstrated that BSNXD decreases VCAM-1 expression in thoracic aortic tissues, inhibits the chemotactic factor such as MCP-1, adhesion molecules such as ICAM-1, VCAM-1 and E-selectin expression in HUVECs, and in turn attenuates the ox-LDL-induced inflammation in the endothelial cells. Inhibition of ERβ or NOS with selective antagonist or inhibitor in HUVECs can block these effects of BSNXD, which suggests that the anti-inflammatory and anti-atherogenic effect of BSNXD on endothelial cells is dependent on ERβ/NO/NF-κB pathway. Furthermore, supernatant from the BSNXD-treated HUVECs can inhibit CCR2, LFA-1 and VLA-4 expression in U937 cells, and efficiently suppress the increased adhesion of U937 cells to HUVECs, suggesting that the effects of BSNXD on monocytes might participate in the anti-atherogenic effects. The NOS inhibitor or ERβ antagonist could block the effect of BSNXD on U937 cell adhesion, suggesting that the anti-atherogenic effect of BSNXD on U937 cells adhesion is also through ERβ/NO/NF-κB pathway.
Taken together, the effect of BSNXD on endothelial cells seems to increase ERβ expression, NO production, and decrease MDA production through ERβ-mediated pathway. The newly formed NO suppresses apoptosis and NF-κB activity of endothelial cells; thus, BSNXD suppresses adhesion molecules expression on HUVECs through ERβ/NO/NF-κB pathway, and attenuates leukocyte adhesion (Figure 6), which provides evidence that BSNXD can alleviate inflammation in endothelial cells, prevent from the migration and adherence of monocytes to endothelial cells, and postpone the progression of atherosclerosis. Consequently, BSNXD presents potential in clinical prophylaxis for atherosclerosis.

Summary of anti-inflammatory roles of BSNXD in atherosclerosis. BSNXD upregulates estrogen receptor β (ERβ) pathway mediating nitric oxide synthesis and downregulates malondialdehyde (MDA) production. NO in turn suppresses apoptosis and NF-κB activity in endothelial cells. BSNXD suppresses monocyte chemoattractant protein-1 (MCP-1), and cell adhesion molecules expression in HUVECs via ERβ/NO/NF-κB pathway. Moreover, BSNXD can induce HUVECs to downregulate CCR2, LFA-1 and VLA-4 expression in U937 cells, and in turn inhibit adherence of U937 to injured endothelial cells, which postpones the progression of atherosclerosis
Materials and Methods
Chinese medicinal formula
Herbal formula BSNXD is composed of eight crude herbs that is prepared as seen in Table 1. The rule of compositions comes from Traditional Chinese Medicinal theory, and the compositions are due to our clinical experience.
Reagents
The 0.2% cholesterol-enriched rabbit diet was obtained from the Laboratory Animal Facility of Chinese Academy of Sciences (Shanghai, China). Sudan III, LPS and L-NAME were provided by Sigma-Aldrich (St. Louis, MO, USA). MPP and R,RTHC were purchased from Tocris Cookson Inc. (Ellisville, MO, USA). The malondialdehyde (MDA) and NO assay kit were supplied by Institute of Nanjing Jiancheng Biology Engineering (Nanjing, PR China). Triglycerides (TG), total cholesterols (TC), low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) were analyzed by using Boehringer Mannheim reagent kits (Mannheim, Germany). The human CCL2/MCP-1 Quantikine ELISA Kit was from R&D Systems, Minneapolis, MN, USA). The estradiol (E2) EIA kit was a product of BioCheck Inc. (Burlingame, CA, USA). All other antibodies were commercially obtained: annexin V-fluorescein isothiocyanate (FITC) (Bender Med Systems, Vienna, Austria); goat anti-mouse IgG1-FITC (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA); phycoerythrin (PE) anti-human CD54 (ICAM-1) (mouse IgG1, κ) and PE anti-human CD106 (VCAM) (mouse IgG1, κ) (eBioscience, San Diego, CA, USA); FITC-conjugated mouse anti-human CD11a (LFA-1) and FITC-conjugated mouse anti-human CD49d (VLA-4) antibodies (Becton Dickinson, Palo Alto, CA, USA); FITC labeled anti-CCR2 (clone 48607) antibody (Dakocytomation, Kyoto, Japan); ER Ab-17 (NeoMarkers Inc. Fremont, CA, USA); Rabbit polyclonal to ERα antibody (Abcam, Cambridge, UK); Rabbit polyclonal to ERβ antibody (Upstate Biotechnology, Lake Placid, NY, USA); murine monoclonal anti-E-selectin (CD62E) antibody (Sigma-Aldrich); mouse monoclonal antibody against rabbit VCAM-1(Santa Cruz Biotechnology).
Experiment and drug administration
The experimentation of animals was carried out according to the Principles of Laboratory Animal Care (NIH publication number 85-23, revised 1985). Twelve female New Zealand rabbits underwent bilateral oophorectomy, which were purchased from the Laboratory Animal Facility of Chinese Academy of Sciences (Shanghai, China). In 2 weeks after OVX, the rabbits were then randomly divided into two groups (OVX and OVX+BSNXD) of six rabbits each. The OVX group serving as controls received saline treatment and high cholesterol chow made of the standard rabbit chow and additives of 0.5% cholesterol, 15% egg yolk and 5% pork lard.40 Those from group OVX+BSNXD received high cholesterol chow plus 5 ml mixed row herbs (BSNXD) per kilogram body weight daily, a dosage equivalent to the human adult dose based on an established formula for human–rabbit drug conversion.41 The sham group underwent a mock operation and received high cholesterol chow daily (n=6). After 12 weeks of treatment, all rabbits were weighed; the animals were killed after the last treatment, and the blood samples and tissues were harvested for further investigation.
The other in vivo experiment (treatment experiments) is to treat ovariectomized rabbit with established atherosclerotic plaques to see whether the BSNXD could revert the phenotype and not only prevent it. Twelve female New Zealand rabbits underwent bilateral oophorectomy. In 2 weeks after OVX, the rabbits were fed an atherogenic high fat, high cholesterol diet for 3 months. At the end of the 3 months (inductive phase), four rabbits were killed, and surface area of the lesions in the aorta were measured. The remaining rabbits (n=8) were divided into two groups, and placed on a normal chow diet and saline (n=4), or normal chow and BSNXD (n=4) for >3 months (regressive phase). The lesion areas were measured in these animals.
The drug-derived serum preparation
At 1 h after the last intragastric administration of BSNXD, the serum was acquired from auricular arteriae of rabbits, inactivated at 56°C for 30 min, and filtrated by 0.2 μm of filtrator and put into use with different concentrations, or followed by storage at −70°C until application.
Serum lipids, NO and E2 determination
Blood samples were acquired from rabbits’ ear marginal vein in the morning after an overnight fast. Serum lipids (including TC, TG, HDL-C and LDL-C) were measured once before beginning the hypercholesterolemic diet again after treatment for 12 weeks, through standard enzymatic techniques on an automated analyzer (Hitachi 911, Tokyo, Japan) by using Boehringer Mannheim reagent kits according to the manufacturer’s protocol. NO was determined as NO2−/NO3− concentrations by using NO assay kit following the manufacturer’s protocol. The absorbance was measured at 550 nm by microplate reader (Molecular Devices, Sunnyvale, CA, USA). The NO concentration was quantified by extrapolation from potassium nitrate standard curve in each experiment. All the assays were done in triplicate. Serum E2 concentrations were measured by using an E2 enzyme immunoassay kit according to the manufacturer’s protocol.
Characterization and quantification of aortic atherosclerotic lesions
Rabbits were euthanized after blood sampling, and the thoracic aortas (including aortic arch) were excised, cut longitudinally, and fixed in 4% paraformaldehyde for 48 h. The aortas were then washed briefly in 70% alcohol and immersed in a solution of Sudan III (4% w-v in 70% alcohol) for 45 min. The images of the aorta inner surface segment were processed with an image analysis program (Olympus Microimage 3.0; Olympus Optical Co, LTD, Tokyo, Japan). The severity of aortic atherosclerosis was evaluated as the lesion area relative to the inner surface.42
The aortas were embedded in paraffin blocks. Serial 3 μm thick slices of the artery were provided for histological and immunohistochemical analysis. Serial sections with 500 μm intervals were stained with hematoxylin–eosin for histological analysis. We analyzed five slides with three to four sections on each slide. The medial and intimal thicknesses and areas were measured by image processing software, and the intima/media thicknesses and area ratios were determined.43
For scanning electron microscopy (S.E.M.) analysis, thoracic aorta was fixed overnight at 4°C in a buffered glutaraldehyde solution. The samples were then post-fixed in osmium tetroxide for 1 h, rinsed, dehydrated in a graded ethanol series, immersed in amyl acetate and dried in a critical point dryer (Bio-Rad E3000; Polaron, West Sussex, UK). All tissue samples lumens were opened, and coated with gold. Examination was performed by using S.E.M. (JEOL JSM 5200, Tokyo, Japan).
Immunohistochemical characterization and quantitative analysis are as described previously.44 For immunohistochemical staining, paraffin sections were cleared in xylene, rehydrated in graded ethanol (100–70%), and subjected to antigen retrieval by microwave (320W, 11 min). Thereafter, the sections were incubated in 3% hydrogen peroxide (H2O2) for 30 min to abolish endogenous peroxidase activity. The specimens were then blocked for 1 h in blocking serum, and incubated overnight at 4°C in a humidified chamber with and without mouse monoclonal antibody against rabbit VCAM-1, ER Ab-17, rabbit polyclonal to ERα antibody or rabbit polyclonal to ERβ antibody. The samples were then rinsed in PBS and incubated with biotinylated secondary antibody for 30 min at room temperature followed by peroxidase-conjugated streptavidin for 30 min, and developed with 3,3′-diamino-benzidine. Sections were counterstained with Mayer’s hematoxylin for 1 min and mounted. Negative controls were obtained by substituting the primary antibodies with isotype. The aortic expressions of VCAM-1, ER, ERα and ERβ were determined by quantitative immunohistochemistry by using ImagePro Plus 4.5 software (Media Cybernetics, Silver Spring, MD, USA).44
Uterus and liver histomorphometry
After euthanasia, the uterus and liver were removed from the rabbits. The wet weights of the organs were measured. Macroscopic examination followed, and representative sections were taken. Samples were fixed in 4% formalin, embedded in paraffin. Routine histochemistry was performed on sections 4 μm in thickness, including staining with H&E.
HUVECs culture and the drug-derived serum treatment in vitro
Ox-LDL was prepared as described previously.22 The primary HUVECs were isolated, grown and identified as described previously.45 The passage 3 of the cells in EBM was for experiments. When cells were at 75% confluency, changed culture medium to phenol red-free EBM-2 medium supplemented with 10% charcoal/dextran-treated FBS, and maintained for 24 h. The starved cells in serum-free EBM for further 24 h, then exposed to control serum from the ovariectomized rabbits treated with saline, or 10% BSNXD drug-derived serum for 48 h. To determine if NO or ER subtype is involved in the effects of the BSNXD drug-derived serum, NOS inhibitor, selective ERα and ERβ antagonists were used in an additional set of experiments. In all, 0.1 mM L-NAME (NOS inhibitor),46 10−6 M MPP (ERα antagonist) or 10 nM R,RTHC (ERβ antagonist) was administered 1 h before exposure to BSNXD drug-derived serum, and continued during the subsequent 48 h in combination with BSNXD drug-derived serum.47, 48 Finally, the cells were treated with ox-LDL for 24 h.
The supernatant was collected as conditioned medium (CM) or for measurement of NO, MDA and MCP-1. NO and MDA production were determined by using commercially available kits. NO was detected as described above; MDA was measured at a wavelength of 532 nm by reacting with thiobarbituric acid (TBA) to form a stable chromophoric product. MCP-1 was quantified with a sandwich ELISA, and a curve calibrated from MCP-1 standards according to the manufacturer’s instructions.
The HUVECs were collected for the analysis of apoptosis, ERα/ERβ, chemotactic factor MCP-1 or adhesion molecules (ICAM-1, VCAM-1 and E-selectin) expression. U937 cells were treated with the different CM from HUVECs for 24 h prior to analyzing CCR2, LFA-1 and VLA-4 mRNA and protein expression.
Cell apoptosis analysis
For cell apoptosis assays, according to the manufacturer’s instructions, HUVECs were washed with PBS, stained with Annexin V-FITC and propidium iodide, and then analyzed by FACS Calibur (Becton Dickinson, Palo Alto, CA, USA). In every sample, 1 × 105 cells were counted.
Cell adhesion assay
U937 cells were labeled with the fluorescent dye 2,7-bis(2-carboxyethyl)-5(6)-carboxyfluorescenin acetoxymethylester (BCECF-AM, Acros, Geel, Belgium) at a 10 μM final concentration in RPMI1640 complete medium containing 10% FBS at 37°C for 1 h. After HUVECs were stimulated and washed, the labeled U937 cells were added to each well, and allowed to interact for 1 h at 37°C. U937 cells bound to HUVECs were lysed with 50 mM Tris-HCl, pH8.0, containing 0.1% SDS. The quantitative results were obtained by using a Fluoroscan ELISA plate reader at 485 nm excitation and 535 nm emission wavelengths. The detailed methodology was previously reported.22
RT-PCR
RT-PCR was carried out to evaluate HUVECs adhesion molecules (ICAM-1, VCAM-1, E-selectin) and U937 cells adhesion molecules (CCR2, LFA-1, VLA-4) mRNA expression. Total RNA was isolated from cells by using TRIzol reagent. The forward and reverse primers for these genes are as our previous reports.22 All the reactions were normalized using β-actin as control. PCR products were detected by electrophoresis on a 1.5% agarose gel with ethidium bromide. The annealing temperature was for ICAM-1 was 62°C, for VCAM-1 and E-selectin was 55°C, for CCR2, LFA-1, VLA-4 and β-actin was 57°C. The number of PCR cycles was 30–35 for each reaction.
Quantitative real-time PCR
Real-time PCR was carried out to evaluate ERα and ERβ mRNA in HUVECs by using the method described in the previous study.49, 50, 51 RNA samples were extracted by using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). ERα and ERβ expression were analyzed by using TaqMan probes (Hs00174860 and Hs00230957, respectively; Applied Biosystems, Warrington, UK) according to the manufacturer’s description. GAPDH was used as endogenous reference gene (Hs00266705). Standard curves for all analyzed genes were run on each plate, by using serial diluted cDNA to normalize the runs. The data obtained from GAPDH were used to normalize the sample variation in the amount of input cDNA.
Flow cytometry
HUVECs adhesion molecules (ICAM-1, VCAM-1, E-selectin) and U937 cells adhesion molecules (CCR2, LFA-1, VLA-4) protein expression were analyzed by flow cytometry. HUVECs were incubated with PE-conjugated monoclonal antibodies against human ICAM-1 and VCAM-1, and U937 cells were incubated with FITC-labeled monoclonal antibodies against human CCR2, CD11a and CD49d for 30 min at 4°C. E-selectin expression on HUVECs was detected by indirect immunofluorescence. The cells were incubated with mouse monoclonal antibody against human E-selectin. The cells were then washed twice with ice-cold PBS-1%FCS. A goat anti-mouse IgG FITC-conjugated secondary antibody was added to the cells, and incubated for 30 min in the dark before being washed twice as above. The cells were analyzed by flow cytometry on a FACS Calibur (Becton Dickinson) by using the (Becton Dickinson).
Western blot
Western blot analysis was carried out to evaluate ERα, ERβ, eNOS,52 MCP-1, ICAM-1, VCAM-1 and E-selectin protein levels in HUVECs, also adhesion molecules (CCR2, LFA-1, VLA-4) protein expression in U937 cells. A volume of 150 ml of lysis buffer (0.1% Triton X-100, 0.5% sodium deoxicholate acid, 0.1% SDS, 0.1% PMSF, in 100 ml of PBS containing protease inhibitors: 1 mg/ml leupeptin, 0.5 mg/ml pepstatin and 1 mg/ml bestatin) was added and maintained at 4°C for 30 min. Thereafter, the cells were collected, boiled for 5 min and sonicated for 10 s. Protein content was measured by using Bradford protein assay. Equal amounts of proteins were then separated by 10% of SDS-polyacrylamide gel electrophoresis, and the proteins were transferred to a PVDF membrane by electrotransfer for 1.5 h. The membranes were blocked with 5% non-fat-milk in TBS-T, and then incubated with anti-ERα (sc-8002; Santa Cruz Biotechnology), anti- ERβ (sc-8974; Santa Cruz Biotechnology), anti-eNOS (Cell Signaling Technology Inc., Beverly, MA, USA), anti-MCP-1 (Santa Cruz Biotechnology), anti-ICAM-1 (Cell Signaling Technology), anti-VCAM-1 (Santa Cruz Biotechnology), anti-E-selectin (Santa Cruz Biotechnology), mouse monoclonal antibodies for human CCR2 (Abcam), anti-CD11a antibody (LFA-1) (Abcam), Anti-Integrin α 4 antibody (VLA-4) (Abcam) or anti-β-actin (Cell Signaling Technology) overnight at 4°C followed by secondary horseradish peroxidase-conjugated antibody (Rockland Co., Gilbertsville, PA, USA). The proteins of interest were identified by detection of HRP-labeled antibody complexes with chemiluminescence with ECL Western Blotting Detection Kit (GE Healthcare, Buckinghamshire, UK). The intensity of the band was scanned, and analyzed with (Alpha Innotech Corporation, San Leandro, CA, USA). Data were presented as a ratio of ERα, ERβ or eNOS versus β-actin, respectively.
Transfection and luciferase assay
Transient transfection with NF-κB-luciferase was constructed by using Lipofectamin (Gibco BRL, Gaithersburg, MD, USA). HUVECs cell cultures, luciferase transfection and control experiments, LPS stimulation, luciferase activity assay by using dual luciferase kit (Promega, Madison, WI, USA) and TD-20/20 luminometer (TurnerDesigns, Sunnyvale, CA, USA) that have been extensively described.22 Briefly, cells were transfected with 2 μg of NF-κB-luciferase, allowed to recover for 24 h, and then 0.1 mM L-NAME (NOS inhibitor), 10-6 M MPP (ERα antagonist) or 10 nM R,RTHC (ERβ antagonist) was administered 1 h before stimulated with LPS (10 μg/ml) in the presence of the drug-derived serum or the control. The cells were harvested 8 h after treatment, and luciferase activity was determined.
Statistical analysis
All values are expressed as the mean±S.E.M. Data were analyzed with aid of SPSS database. The difference between experimental groups of equal variance was analyzed by using Student’s t-test with P<0.05 being considered significant.
Abbreviations
- BSNXD:
-
Bu-Shen-Ning-Xin Decoction
- CCR2:
-
C–C motif chemokine receptor-2
- CHD:
-
coronary heart disease
- CM:
-
conditioned medium
- ER:
-
estrogen receptor
- HRT:
-
hormone replacement therapy
- HUVECs:
-
human umbilical vein endothelial cells
- ICAM-1:
-
intercellular adhesion molecule-1
- LFA-1:
-
lymphocyte function-associated antigen 1
- L-NAME:
-
NG-nitro-L-arginine methyl ester
- MCP-1:
-
monocyte chemoattractant protein-1
- MDA:
-
malondialdehyde
- MPP:
-
Methyl-piperidino-pyrazole
- NF-κB:
-
nuclear factor-κB
- NO:
-
nitric oxide
- NOS:
-
nitric oxide synthase
- OVX:
-
ovariectomy
- ox-LDL:
-
oxidized low-density lipoproteins
- R,RTHC:
-
R,R-tetrahydrochrysene
- S.E.M.:
-
scanning electron microscopy
- TCMs:
-
traditional Chinese medicines
- VCAM-1:
-
vascular cell adhesion molecule-1
- VLA-4:
-
Integrin α4β1 (Very Late Antigen-4)
References
Tousoulis D, Kampoli AM, Papageorgiou N, Androulakis E, Antoniades C, Toutouzas K et al. Pathophysiology of atherosclerosis: the role of inflammation. Curr Pharm Des 2011; 17: 4089–4110.
Libby P . Inflammation in atherosclerosis. Nature 2002; 420: 868–874.
Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, Evans PC . Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond) 2010; 118: 593–605.
Ignarro LJ, Napoli C . Novel features of nitric oxide, endothelial nitric oxide synthase, and atherosclerosis. Curr Atheroscler Rep 2004; 6: 281–287.
Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P . Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA 2001; 98: 4202–4208.
Demer L, Tintut Y . The roles of lipid oxidation products and receptor activator of nuclear factor-kappaB signaling in atherosclerotic calcification. Circ Res 2011; 108: 1482–1493.
Mendelsohn ME . Protective effects of estrogen on the cardiovascular system. Am J Cardiol 2002; 89: 12E–17E discussion 17E-18E.
Meyer MR, Haas E, Barton M . Gender differences of cardiovascular disease: new perspectives for estrogen receptor signaling. Hypertension 2006; 47: 1019–1026.
Osako MK, Nakagami H, Koibuchi N, Shimizu H, Nakagami F, Koriyama H et al. Estrogen inhibits vascular calcification via vascular RANKL system: common mechanism of osteoporosis and vascular calcification. Circ Res 2010; 107: 466–475.
Mendelsohn ME, Karas RH . The protective effects of estrogen on the cardiovascular system. N Engl J Med 1999; 340: 1801–1811.
Hodgin JB, Maeda N . Minireview: estrogen and mouse models of atherosclerosis. Endocrinology 2002; 143: 4495–4501.
Christian RC, Liu PY, Harrington S, Ruan M, Miller VM, Fitzpatrick LA . Intimal estrogen receptor (ER)beta, but not ERalpha expression, is correlated with coronary calcification and atherosclerosis in pre- and postmenopausal women. J Clin Endocrinol Metab 2006; 91: 2713–2720.
Rexrode KM, Ridker PM, Hegener HH, Buring JE, Manson JE, Zee RY . Polymorphisms and haplotypes of the estrogen receptor-beta gene (ESR2) and cardiovascular disease in men and women. Clin Chem 2007; 53: 1749–1756.
Hodges YK, Tung L, Yan XD, Graham JD, Horwitz KB, Horwitz LD . Estrogen receptors alpha and beta: prevalence of estrogen receptor beta mRNA in human vascular smooth muscle and transcriptional effects. Circulation 2000; 101: 1792–1798.
Liu PY, Christian RC, Ruan M, Miller VM, Fitzpatrick LA . Correlating androgen and estrogen steroid receptor expression with coronary calcification and atherosclerosis in men without known coronary artery disease. J Clin Endocrinol Metab 2005; 90: 1041–1046.
Efferth T, Li PC, Konkimalla VS, Kaina B . From traditional Chinese medicine to rational cancer therapy. Trends Mol Med 2007; 13: 353–361.
Wang L, Zhou GB, Liu P, Song JH, Liang Y, Yan XJ et al. Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc Natl Acad Sci USA 2008; 105: 4826–4831.
Chow MS, Huang Y . Utilizing Chinese medicines to improve cancer therapy—fiction or reality? Curr Drug Discov Technol 2010; 7: 1.
Park WH, Hong MY, Chung KH, Kim HM, Lee YC, Kim CH . Effects of traditional herbal medicine, Hwaotang, on atherosclerosis using the spontaneous familial hypercholesterolemia model, Kurosawa and Kusanagi-hypercholesterolemic rabbits and the venous thrombosis rats. Phytother Res 2005; 19: 846–853.
Sieveking DP, Woo KS, Fung KP, Lundman P, Nakhla S, Celermajer DS . Chinese herbs Danshen and Gegen modulate key early atherogenic events in vitro. Int J Cardiol 2005; 105: 40–45.
Lee S, Lim HJ, Park HY, Lee KS, Park JH, Jang Y . Berberine inhibits rat vascular smooth muscle cell proliferation and migration in vitro and improves neointima formation after balloon injury in vivo. Berberine improves neointima formation in a rat model. Atherosclerosis 2006; 186: 29–37.
Wang L, Hao Q, Wang YD, Wang WJ, Li DJ . Protective effects of dehydroepiandrosterone on atherosclerosis in ovariectomized rabbits via alleviating inflammatory injury in endothelial cells. Atherosclerosis 2011; 214: 47–57.
Jackson KC, Wohlers LM, Lovering RM, Schuh RA, Maher AC, Bonen A et al. Ectopic lipid deposition and the metabolic profile of skeletal muscle in ovariectomized mice. Am J Physiol Regul Integr Comp Physiol 2012; 304: R206–R217.
Chen MF, Hsu HC, Liau CS, Lee YT . The role of vitamin E on the anti-atherosclerotic effect of fish oil in diet-induced hypercholesterolemic rabbits. Prostaglandins Other Lipid Mediat 1999; 57: 99–111.
Freyschuss A, Al-Schurbaji A, Bjorkhem I, Babiker A, Diczfalusy U, Berglund L et al. On the anti-atherogenic effect of the antioxidant BHT in cholesterol-fed rabbits: inverse relation between serum triglycerides and atheromatous lesions. Biochim Biophys Acta 2001; 1534: 129–138.
Holm P, Andersen HL, Andersen MR, Erhardtsen E, Stender S . The direct antiatherogenic effect of estrogen is present, absent, or reversed, depending on the state of the arterial endothelium. A time course study in cholesterol-clamped rabbits. Circulation 1999; 100: 1727–1733.
Chen F, Knecht K, Birzin E, Fisher J, Wilkinson H, Mojena M et al. Direct agonist/antagonist functions of dehydroepiandrosterone. Endocrinology 2005; 146: 4568–4576.
Hodis HN, Mack WJ, Azen SP, Lobo RA, Shoupe D, Mahrer PR et al. Hormone therapy and the progression of coronary-artery atherosclerosis in postmenopausal women. N Engl J Med 2003; 349: 535–545.
Hodis HN, Collins P, Mack WJ, Schierbeck LL . The timing hypothesis for coronary heart disease prevention with hormone therapy: past, present and future in perspective. Climacteric 2012; 15: 217–228.
Whayne TF . Atherosclerosis: current status of prevention and treatment. Int J Angiol 2011; 20: 213–222.
Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding QF et al. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci USA 2006; 103: 17018–17023.
Scalera F, Martens-Lobenhoffer J, Tager M, Bukowska A, Lendeckel U, Bode-Böger SM . Effect of L-arginine on asymmetric dimethylarginine (ADMA) or homocysteine-accelerated endothelial cell aging. Biochem Biophys Res Commun 2006; 345: 1075–1082.
Arnal JF, Gourdy P, Elhage R, Garmy-Susini B, Delmas E, Brouchet L et al. Estrogens and atherosclerosis. Eur J Endocrinol 2004; 150: 113–117.
Su S, Guo J, Duan JA, Wang T, Qian D, Shang E et al. Ultra-performance liquid chromatography-tandem mass spectrometry analysis of the bioactive components and their metabolites of Shaofu Zhuyu decoction active extract in rat plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2010; 878: 355–362.
Dong W, Wang P, Meng X, Sun H, Zhang A, Wang W et al. Ultra-performance liquid chromatography-high-definition mass spectrometry analysis of constituents in the root of Radix Stemonae and those absorbed in blood after oral administration of the extract of the crude drug. Phytochem Anal 2012; 23: 657–667.
Bjorkbacka H, Fredrikson GN, Nilsson J . Emerging biomarkers and intervention targets for immune-modulation of atherosclerosis - A review of the experimental evidence. Atherosclerosis 2012; 227: 9–17.
Van-Assche T, Huygelen V, Crabtree MJ, Antoniades C . Gene therapy targeting inflammation in atherosclerosis. Curr Pharm Des 2011; 17: 4210–4223.
Hodgin JB, Krege JH, Reddick RL, Korach KS, Smithies O, Maeda N . Estrogen receptor alpha is a major mediator of 17beta-estradiol’s atheroprotective effects on lesion size in Apoe−/− mice. J Clin Invest 2001; 107: 333–340.
Register TC . Primate models in women’s health: inflammation and atherogenesis in female cynomolgus macaques (Macaca fascicularis). Am J Primatol 2009; 71: 766–775.
Dornas WC, Oliveira TT, Augusto LE, Nagem TJ . Experimental atherosclerosis in rabbits. Arq Bras Cardiol 2010; 95: 272–278.
Sun J (eds). Methodology of Animal Experiments. People’s Health Publishing House: Beijing, 356–357 p 2001.
Blumel JE, Castelo-Branco C, Gonzalez P, Moyano C, Iturriaga M, Videla L et al. Transdermal estrogens do not appear to modify the extent of lesional areas of aortic atherosclerosis in oophorectomized rabbits on a cholesterol-rich diet. Atherosclerosis 2000; 148: 303–308.
Ducasse E, Chevalier J, Cosset JM, Creusy C, Eschwege F, Speziale F et al. Ionizing radiation to prevent arterial intimal hyperplasia at the edges of the stent: induces necrosis and fibrosis. J Surg Res 2006; 135: 331–336.
Liu XD, Chen HB, Tong Q, Li XY, Zhu MJ, Wu ZF et al. Molecular characterization of caveolin-1 in pigs infected with Haemophilus parasuis. J Immunol 2011; 186: 3031–3046.
Huang FY, Mei WL, Li YN, Tan GH, Dai HF, Guo JL et al. Toxicarioside A inhibits tumor growth and angiogenesis: involvement of TGF-beta/Endoglin signaling. PLoS One 2012; 7: e50351.
Zhong W, Zou G, Gu J, Zhang J . L-arginine attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Diabetes Res Clin Pract 2010; 89: 38–45.
Chen YJ, Lee MT, Yao HC, Hsiao PW, Ke FC, Hwang JJ . Crucial role of estrogen receptor-alpha interaction with transcription coregulators in follicle-stimulating hormone and transforming growth factor beta1 up-regulation of steroidogenesis in rat ovarian granulosa cells. Endocrinology 2008; 149: 4658–4668.
Somponpun S, Sladek CD . Role of estrogen receptor-beta in regulation of vasopressin and oxytocin release in vitro. Endocrinology 2002; 143: 2899–2904.
Yu X, Ling W, Mi M . Relationship of impairment induced by intracellular S-adenosylhomocysteine accumulation with DNA methylation in human umbilical vein endothelial cells treated with 3-deazaadenosine. Int J Exp Pathol 2009; 90: 638–648.
Wang L, Wang YD, Wang WJ, Li DJ . Differential regulation of dehydroepiandrosterone and estrogen on bone and uterus in ovariectomized mice. Osteoporos Int 2009; 20: 79–92.
Toth B, Saadat G, Geller A, Scholz C, Schulze S, Friese K et al. Human umbilical vascular endothelial cells express estrogen receptor beta (ERbeta) and progesterone receptor A (PR-A), but not ERalpha and PR-B. Histochem Cell Biol 2008; 130: 399–405.
Sahara M, Sata M, Morita T, Hirata Y, Nagai R . Nicorandil attenuates monocrotaline-induced vascular endothelial damage and pulmonary arterial hypertension. PLoS One 2012; 7: e33367.
Acknowledgements
This work was supported by National Natural Science Foundation of China No. 30801502 (to L Wang), Shanghai Pujiang Program No. 11PJ1401900 (to L Wang), by National Key Research Program of China 2006CB944007, National and Shanghai Leading Academic Discipline Project 211XK22, and Program for Outstanding Medical Academic Leader (to D-J Li).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Federici
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Wang, L., Qiu, XM., Hao, Q. et al. Anti-inflammatory effects of a Chinese herbal medicine in atherosclerosis via estrogen receptor β mediating nitric oxide production and NF-κB suppression in endothelial cells. Cell Death Dis 4, e551 (2013). https://doi.org/10.1038/cddis.2013.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.66
Keywords
This article is cited by
-
PRMT5 critically mediates TMAO-induced inflammatory response in vascular smooth muscle cells
Cell Death & Disease (2022)
-
12-Hydroxyeicosapentaenoic acid inhibits foam cell formation and ameliorates high-fat diet-induced pathology of atherosclerosis in mice
Scientific Reports (2021)
-
The histone demethylase LSD1 promotes renal inflammation by mediating TLR4 signaling in hepatitis B virus-associated glomerulonephritis
Cell Death & Disease (2019)
-
Bitter melon (Momordica charantia) attenuates atherosclerosis in apo-E knock-out mice possibly through reducing triglyceride and anti-inflammation
Lipids in Health and Disease (2018)
-
Total tanshinones exhibits anti-inflammatory effects through blocking TLR4 dimerization via the MyD88 pathway
Cell Death & Disease (2017)
