
Abstract
Vascular deposition of amyloid-β (Aβ) in sporadic and familial Alzheimer’s disease, through poorly understood molecular mechanisms, leads to focal ischemia, alterations in cerebral blood flow, and cerebral micro-/macro-hemorrhages, significantly contributing to cognitive impairment. Here, we show that tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptors DR4 and DR5 specifically mediate oligomeric Aβ induction of extrinsic apoptotic pathways in human microvascular cerebral endothelial cells with activation of both caspase-8 and caspase-9. The caspase-8 inhibitor cellular FLICE-like inhibitory protein (cFLIP) is downregulated, and mitochondrial paths are engaged through BH3-interacting domain death agonist (Bid) cleavage. Upregulation of DR4 and DR5 and colocalization with Aβ at the cell membrane suggests their involvement as initiators of the apoptotic machinery. Direct binding assays using receptor chimeras confirm the specific interaction of oligomeric Aβ with DR4 and DR5 whereas apoptosis protection achieved through RNA silencing of both receptors highlights their active role in downstream apoptotic pathways unveiling new targets for therapeutic intervention.
Similar content being viewed by others


Main
The deposition of amyloid-β (Aβ) in and around cerebral vessels, known as cerebral amyloid angiopathy (CAA), is a condition present in normal aging and in more than 80% of Alzheimer’s disease (AD) patients. CAA compromises medium- and small-size arteries and arterioles as well as capillary endothelium affecting cerebral blood flow and altering the properties of the blood brain barrier, events which in turn cause degeneration of the entire neurovascular unit.1 Therefore, it is not surprising that CAA is now considered a significant contributor to cognitive decline, and dementia being increasingly recognized as a major factor in AD pathogenesis.2 To date, several mechanisms have been proposed as the source of the vascular Aβ deposits including derivation from circulating blood and local production by vessel-wall cells.3, 4 While production of Aβ by vascular cells has been clearly demonstrated, the development of transgenic models with several fold increased levels of plasma Aβ, albeit failing to exhibit brain lesions, argue against the likelihood of a blood origin for brain Aβ. An emerging body of evidence indicates that neuronally originated Aβ is mainly cleared through the blood–brain and brain–cerebrospinal fluid barriers, degraded by resident enzymes, and secreted into the extracellular space entering the perivascular drainage route along with the bulk flow of extracellular fluid, mostly at the level of capillaries or arterioles. This perivascular clearance has a key role in the development of CAA; impaired drainage owing to Aβ accumulation, as well as age-related fibrotic changes in the cerebral vasculature clearly impact the severity of CAA.
Previous data from our lab demonstrated that Aβ induces caspase-mediated mitochondrial apoptosis in cerebral microvascular smooth muscle and endothelial cells (ECs), both with potential to be in direct contact with Aβ vascular deposits in vivo under pathological conditions. This effect is exacerbated by the presence of specific mutations at positions 22 or 34 (E22Q and L34V), known to cause cerebral hemorrhage in familial forms of CAA (human cerebral hemorrhage with amyloidosis Dutch and Piedmont type, respectively).5
Apoptotic cell death typically involves two distinct pathways – the Bcl-2-regulated intrinsic path and the death-receptor-mediated extrinsic mechanism – both translating in morphological and biochemical changes associated with the apoptotic phenotype. The extrinsic pathway classically begins with the binding of an appropriate ligand to a subset of tumor necrosis factor (TNF) receptor superfamily (TNFRSF) members: the death receptors.6 These transmembrane receptors, containing a conserved death domain,7 recruit the intracellular adaptor molecule FADD (Fas-associated protein with death domain), which in turn binds to and activates caspase-8, inducing the formation of DISC (death-inducing signaling complex), to initiate apoptosis. This pathway is regulated by cellular FLICE-like inhibitory protein (cFLIP), crucial for the modulation of death signaling through interaction with procaspase-8 at DISC.6, 8 Caspase activation is generally amplified by engagement of the mitochondrial intrinsic pathway through caspase-8 processing of Bid (BH3-interacting domain death agonist).9 Cleaved/truncated Bid (tBid) interacts with other Bcl-2 family members on the surface of the mitochondria, resulting in mitochondria outer membrane permeabilization, release of cytochrome c, and subsequent caspase-9 activation. Both caspase-8 and -9 cause downstream activation of the effector caspase-3.
Members of the death receptor family include Fas, TNFR1 (tumor necrosis factor receptor 1), the TRAIL (TNF-related apoptosis-inducing ligand) death receptors 4 and 5 (DR4 and DR5), DR3, DR6, and p75/NGFR (nerve growth factor receptor). Our study unveiled for the first time the contribution of DR4 and DR5 to the apoptotic mechanisms elicited by oligomeric Aβ on cerebral microvascular ECs, providing insights into the signaling pathways triggered through their activation and validating their active role in Aβ-mediated apoptosis via RNA silencing. Overall, our results show a direct binding of oligomeric Aβ to DR4 and DR5 leading to their subsequent activation and downstream apoptosis induction, and point out to TRAIL death receptor-mediated paths as novel therapeutic targets for neurovascular degeneration in AD as well as in sporadic and familial CAA.
Results
Caspase-8 activation is an early event in A β -induced EC apoptosis
Challenge of cerebral microvascular ECs with vasculotropic Aβ variants triggers the apoptotic cascade, as evidenced by changes in the cell morphology, DNA fragmentation, release of mitochondrial cytochrome c to the cytoplasm, and phosphatidylserine membrane translocation with annexin V-positive fluorescence (Fossati et al.5, Viana et al.10 and Supplementary Figure 1). The participation of caspase-8 and -9 in Aβ-induced apoptosis was analyzed in cerebral EC challenged 0–24 h with Aβ synthetic homologs. AβE22Q was added to the cells immediately after solubilization, while Aβ-WT and AβL34V, owing to their slower aggregation kinetics, were pre-aggregated (3 days, 37 °C) before addition to the cell cultures to allow the formation of oligomers, considered the species responsible for cell toxicity in our model.5 In all cases, activation of both caspases was evident after only few hours of peptide challenge (Figure 1a) although the time course of activation suggests a slower kinetics or lower intensity for caspase-9. Corroborating the specificity of the cellular responses, the reverse sequence Aβ40-1 peptide, in difference to Aβ-WT, was unable to trigger EC apoptosis or to induce activation of caspase-8 and caspase-9 under the conditions tested (Supplementary Figure 2).

Caspase-8 and caspase-9 activation in Aβ-challenged microvascular ECs. (a) Activation of caspase-8 and -9 was monitored via luminescent assays in ECs challenged up to 24 h with Aβ-E22Q (Q22, 50 μM, freshly solubilized), as well as with Aβ-L34V (V34) and Aβ-WT (50 μM, both pre-aggregated 3 days in EBM-2 medium). Results represent the mean±S.D. of three independent experiments. (b) Downregulation of the endogenous caspase-8 inhibitor cFLIP was observed after 8, 16, and 24 h treatment with Aβ-E22Q (Q22). cFLIP decrease was also observed after 1-day treatment with Aβ-L34V (V34) and 3-day challenge with Aβ-WT. (c) Depletion of 20 kDa full-length Bid was evident after EC treatment with Aβ-E22Q for 8 and 16 h as well as after 3-day challenge with Aβ-L34V or -WT. (d) The pan-caspase inhibitor ZVAD reverted the caspase-8-dependent cleavage of Bid as illustrated by the reduction of the 16-kDa tBid band intensity while not affecting levels of cFLIP. In b, c, and d, immunoreactivity with GAPDH indicates equal protein loading, and lanes labeled C illustrate untreated control cells. In (d), graphs illustrate cFLIP and tBid band intensities – evaluated via ImageJ – normalized to GAPDH
The early detection of caspase-8 activation suggested the involvement of death receptors, known to trigger apoptosis through the engagement of this protease, with downstream activation of caspase-9, subsequent to cytochrome c release to the cytoplasm. Further demonstration of the primary involvement of the extrinsic apoptotic pathway was assessed through evaluation of the expression of c-FLIP and Bid by western blot (WB). Figure 1b illustrates a clear decrease in c-FLIP, a protein inhibiting procaspase-8 processing and known to be downregulated in death receptor activation.7 Downregulation of c-FLIP was evident after 8-h incubation with AβE22Q and was maintained for 24 h. This decrease was also observed in cells challenged 24 h with AβL34V and 3 days with Aβ-WT, consistent with the time frame of apoptosis induction by these peptides.5 The involvement of the receptor-mediated apoptotic pathway was further evaluated through analysis of the cleavage of Bid that, upon processing by caspase-8, engages mitochondrial pathways inducing caspase-9 activation.11 Bid cleavage was demonstrated by the reduction of the 20-kDa full-length Bid in EC treated 8–16 h with AβE22Q, or 3 days with AβL34V and Aβ-WT (Figure 1c). Corroborating caspase participation in Bid cleavage, co-incubation of AβE22Q with the pan-caspase inhibitor ZVAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-fluoromethylketone;100 μM, ENZO Life Sciences, Farmingdale, NY, USA) re-established the levels of the 16-kDa tBid to those of untreated controls (Figure 1d) while not affecting c-FLIP, which can be downregulated independently of caspase activation.8
A β homologs induce overexpression of apoptosis mediators and death receptors in ECs
The differential expression of apoptosis-related genes elicited by challenging EC with AβE22Q was evaluated using pathway-specific PCR arrays. Supplementary Table 1 illustrates all genes analyzed and the pertinent fold change at 6-h challenge; genes that were modified by ≥±1.3-fold in three separate experiments are highlighted in Figure 2a. The changes include translational upregulation of several cytoplasmic apoptosis mediators, like APAF-1, a protein containing a caspase recruitment domain (CARD) that is necessary for the formation of the apoptosome and the activation of caspase-9, the Bcl-2 family member Bcl-2-like protein 2 (also known as Bcl2-W), the apoptosis inducer BIK (also known as Bcl-2-interacting killer), and the CARD-containing microtubule-associated protein CARD6, implicated in the positive modulation of NF-kB signaling. The members of the tumor necrosis factor ligand superfamily found upregulated in the arrays were TNF superfamily member 1 (TNFSF1, also known as TNF-β, a highly inducible cytokine with proinflammatory effects) and CD70 (also known as CD27 ligand or tumor necrosis factor ligand superfamily member 7), a cytokine with homology to TNF classically involved in cell proliferation.

Upregulation of apoptosis mediators and death receptors in microvascular ECs after Aβ challenge. (a) Genes upregulated or downregulated ≥1.3 times following 6 h EC challenge with Aβ-E22Q evaluated via apoptosis-specific PCR arrays (Superarray; SABiosciences). The changes include translational upregulation of several apoptosis mediators (light gray bars), members of the TNF ligand superfamily (dark gray bars), and membrane receptors (black bars); red box highlights TNFRSF members involving activation of caspase-8 with decrease of cFLIP and engaging the adapter protein FADD. (b and c) The expression of TNFRSF10A and 10B (DR4 and DR5, respectively) was measured by quantitative RT-PCR after EC challenge with Aβ-E22Q (Q22) for 3, 6, and 24 h. (d and e) DR4 and DR5 gene expression was evaluated after treatment with Aβ-L34V (V34) and -WT for 2 and 3 days. In (b–e) graphs represent fold of increase of DR4 and DR5 expression compared with untreated controls at the same time points. Results are representative of three separate experiments, each one performed in triplicate. Bars represent mean±S.D. of an independent triplicate experiment
Among the receptors affected by Aβ challenge, and illustrated in Figure 2a by black bars, our results demonstrated upregulation of members of the TNFRSF, including TNFRSF5 (also known as CD40), essential in mediating inflammation-related responses, as well as TNFRSF25, TNFRSF10A, and TNFRSF10B. The first one, also known as DR3, typically activated by the ligand TL1A and signaling through the adapter protein TRADD (TNFR1-associated death domain protein),12 is mainly associated with inflammation-related processes,6 including production of cytokines and inflammatory mediators such as TNFα, MCP-1, and IL-8.13, 14 TNFRSF10A (known as DR4 or TRAILR1) and TNFRSF10B (known as DR5 or TRAILR2), both highlighted by the red box in Figure 2a, are characteristically activated by TRAIL/TNFSF10.
Based on our data illustrated in Figure 1, indicating that Aβ-induced apoptosis involved initial activation of caspase-8 with decrease of c-FLIP, suggestive of primary involvement of death receptors engaging the adaptor molecule FADD, we focused our attention on the upregulation of the TRAIL receptors DR4 and DR5, known to be expressed in EC and throughout the brain.15, 16 Quantitative real-time PCR (RT-PCR) for these genes in EC treated 3–24 h with AβE22Q (Figure 2b) showed that DR4 overexpression was evident after 3-h challenge and continued to increase for 24 h. DR5 overexpression was observed starting at 6 h and the levels, reaching twofold increase over untreated controls, as in the case of DR4, were maintained for the 24 h duration of the experiment (Figure 2c). DR4 and DR5 upregulation was additionally observed after 3-day EC challenge with Aβ-WT and -L34V, albeit reaching lower levels than for AβE22Q, consistent with the attenuated intensity of the apoptotic response induced by these peptides5 (Figures 2d and e).
A β colocalization with DR4 and DR5 on EC membranes
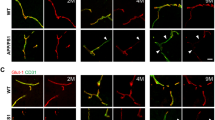
Confirmation of DR4 and DR5 overexpression and assessment of possible colocalization with Aβ peptides was performed by immunocytochemistry and confocal microscopy. AβE22Q 24-h treatment induced a clear overexpression of DR4 and its colocalization on the plasma membrane with Aβ, as indicated in Figure 3 by overlapping of the respective fluorescent signals. DR4 upregulation was also observed after a more prolonged 3-day challenge with Aβ-WT and -L34V albeit with a fluorescent signal of lower intensity than the one elicited by AβE22Q, in agreement with the qPCR data for DR4 mRNA (Figure 2). For both Aβ-WT and -L34V treatments, colocalization of DR4 with the respective amyloid peptides was also observed (Figure 3, central and lower panels). Immunostaining of DR5 after 24-h challenge with E22Q showed increase in the fluorescent signal, consistent with the qPCR data, and partial colocalization with the peptide (Figure 4). In the case of Aβ-WT and L34V peptides, a 3-day challenge did not result in a clear increase of the receptor fluorescent signal, consistent with the minimal ∼1.25-fold increase expression evaluated by qRT-PCR (Figure 2). Likewise, colocalization of DR5 with the respective peptides was not evident (Figure 4 central and lower panel).

Confocal microscopy analysis of DR4 localization in Aβ-challenged cerebral ECs. EC cultures were treated with Aβ-E22Q (Q22) for 1 day and with Aβ-L34V (V34) and Aβ-WT for 3 days. Localization of the respective Aβ homologs and DR4 was evaluated by immunofluorescence and analyzed by confocal microscopy. Green staining represents DR4, red highlights Aβ, and blue signal illustrates nuclei counterstained with TO-PRO. Bar represents 50 μm

Confocal microscopy analysis of DR5 localization in Aβ-challenged cerebral ECs. EC cultures were treated for 1 day with Aβ-E22Q (Q22) and for 3 days with Aβ-L34V (V34) and Aβ-WT as in Figure 3. Localization of Aβ peptides and DR5 was evaluated by immunofluorescence and analyzed by confocal microscopy. Green staining highlights DR5, red staining indicates Aβ, and blue illustrates nuclei counterstained with TO-PRO. Bar represents 50 μm
Further elucidation of the relationship between DR4 upregulation at the cell membrane and Aβ colocalization, as well as confirmation of the time frame required for the receptor’s increased expression, was assessed by immunostaining after a short-time challenge (2–8 h). Notably, the receptor colocalized at all time points with the peptide. Furthermore, DR4 expression increased in parallel with the peptide membrane localization (Figure 5), suggesting a direct binding of Aβ to the receptor. To rule out the possibility that the observed DR4 and DR5 upregulation was driven by their canonical ligand, TRAIL, this protein was quantitated by ELISA in EC culture supernatants and lysates after challenge with AβE22Q, AβL34V, and Aβ-WT. In all cases, the levels of soluble TRAIL in the media or in the cell lysates were below the 2.8 pg/ml minimum detectable level of the assay (data not shown), confirming that, in our system, DR4 and DR5 upregulation was independent from the pathways elicited by their classical ligand.

Analysis of DR4 upregulation and colocalization with Aβ after short-term treatment with Aβ-E22Q. ECs were treated with AβE22Q (50 μM) for 2, 4, or 8 h, and colocalization of the peptide with the DR4 receptor was analyzed by immunofluorescence and confocal microscopy. Green signal indicates DR4 and Aβ is highlighted in red. Blue represents nuclei visualized with TO-PRO. In all cases, bar represents 25 μm
A β 40 variants bind DR4 and DR5 in vitro
The direct binding of Aβ to TRAIL death receptors was evaluated by immunoprecipitation (IP) using chimeras constituted by the extracellular portion of DR4 and DR5 fused to the immunoglobulin G (IgG) Fc fragment (DR4/Fc and DR5/Fc). The Fc fragment within the chimera allowed immobilization onto paramagnetic beads coupled with Protein-G, and subsequent IP of the peptide species exhibiting specific binding for the respective receptors. Binding of AβE22Q was performed using freshly solubilized peptide, whereas binding of Aβ-WT and Aβ-L34V, exhibiting slower oligomerization, was carried out with freshly solubilized peptides as well as with 3-day pre-aggregated preparations. In all cases, after non-denaturing elution of the material bound to the receptor-coated beads, both the eluted components and the original Aβ preparations incubated with the beads (input) were separated by native-gel electrophoresis, followed by WB. As illustrated in Figure 6a left panel, consistent with previous studies,5 freshly solubilized E22Q exhibited the presence of high molecular mass (HMM) oligomers, in addition to the abundant monomeric, dimeric, and tetrameric components. Figure 6a right panel illustrates that in spite of the high abundance of low molecular mass components, only dimers and HMM aggregates of ∼150–350 KDa (as estimated by Fergusson plot analysis) displayed specific binding to DR4 and, to a lower extent, DR5, in agreement with the immunofluorescence data (Figures 3 and 4). Notably, the DR4/DR5-bound fraction is enriched in ∼350-KDa HMM oligomers, which are minor components in the peptide preparation, as evidenced by the absence of immunoreactivity in this molecular mass range in the input fraction. No binding was observed to Fas chimeras or to Fc, used as nonspecific binding control.

Binding of Aβ variants to death receptor-Fc chimeras. Aβ homologs – either freshly solubilized (F) or pre-aggregated for 3 days (3d) – were immunoreacted with paramagnetic beads coupled to DR4/Fc (DR4), DR5/Fc (DR5), and Fas/Fc (Fas) chimeras, as well as to the IgG-Fc fragment employed as negative control for nonspecific binding. Immunoprecipitated material was eluted under non-denaturing conditions and analyzed by WB after native 5–30% gradient gel electrophoresis. In all cases, bound panels represent the immunoprecipitated material specifically retained by the respective immobilized receptor chimeras; input panels illustrate the oligomerization state of the starting material incubated with the respective immobilized chimeras. Results are representative of at least three experiments. (a) Aβ-E22Q (Q22), (b) Aβ-L34V (V34), (c) Aβ–WT, and (d) reverse Aβ40-1 used as negative control and previously conjugated to biotin to allow WB detection as anti-Aβ 4G8 and 6E10 antibodies are not immunoreactive with the reverse sequence peptide. This panel also illustrates – for control purposes – binding of biotin-conjugated Aβ-WT to DR4 and DR5; both biotinylated peptides were pre-aggregated for 3 days before binding to the receptor chimeras. In (d), WB was probed with streptavidin-HRP as detector reagent. In all cases, fluorograms were developed by chemiluminiscence
For Aβ-WT and -L34V, no high-order oligomerization was observed in fresh preparations, whereas 3-day incubation resulted in the formation of HMM oligomers in addition to the abundant low molecular mass species (Figures 6b and c, left panels). Analysis of the material retained by the beads demonstrated lack of binding to DR4 or DR5 of any of the species present in the freshly solubilized preparations (Figures 6b and c, right panels). Pre-aggregation resulted in a strong and specific binding of Aβ-L34V to DR4 and a lower interaction with DR5 (Figure 6b, right panel). As for E22Q, the Aβ species interacting with both receptors corresponded to HMM oligomers (∼150–350 KDa). Comparable HMM species of Aβ-WT bound DR4 (Figure 6c), but not DR5 chimeras, likely reflecting the lower affinity interaction with this receptor. Confirming the specificity of Aβ binding to DR4/DR5, the receptor chimeras failed to bind 3-day pre-aggregated reverse sequence Aβ40-1 (Figure 6d).
DR4 and DR5 silencing protects ECs from A β -induced apoptosis
Silencing RNA experiments were designed to demonstrate the requirement of DR4 and DR5 as initiators of the Aβ-induced apoptotic machinery in EC. Transcripts of the housekeeping genes GAPDH and Cyclophilin-B, used to determine efficacy and specificity of the silencing in the difficult to transfect EC, were reduced by 90% and 80%, respectively, after 3-day silencing by the respective siRNAs (Figure 7a), with no effect on the expression of the DRs, as assessed by qRT-PCR. Expression of DR4 and DR5 mRNAs was also successfully reduced by ∼75% after 3-day incubation with the respective siRNAs with no noticeable crossreactivity between both receptors. To determine the effect of DR4 and DR5 silencing on Aβ-induced apoptosis, we measured the extent of nucleosome formation by cell death ELISA after Aβ-E22Q treatment in cells in which DR4 and DR5 were previously silenced. E22Q-accelerated EC toxicity17 allowed the evaluation of apoptosis in a time frame consistent with the persistency of the transient receptor silencing. Knocking down the expression of DR4, DR5, or both, resulted in 60–70% protection from Aβ-induced apoptosis, in comparison with cells transfected with non-silencing siRNA control (Figure 7b), confirming the significant role played by DR4 and DR5 activation. Whether the lack of total inhibition achieved by knocking down both receptors reflects the absence of their total silencing in our system or the involvement of additional receptors remains to be elucidated. In this sense, it should be pointed out that silencing of DR3 – the TNF receptor exhibiting comparable upregulation in the PCR arrays (Supplementary Table 1 and Figure 2a) – failed to prevent AβE22Q-mediated apoptosis (Supplementary Figure S3), further strengthening the relevance of DR4/DR5 pathways. Silencing the expression of DR4 and DR5 additionally translated in significant inhibition of caspase-8 activation (Figure7c) once more highlighting the relevance of these receptors in the Aβ-elicited apoptotic pathways.

Protection from Aβ-induced apoptosis by DR4 and DR5 gene silencing. (a) qRT-PCR performed after siRNA delivery of two control genes (GAPDH and Cyclophilin B; top panel) as well as DR4 and DR5 (bottom panel) demonstrate the efficiency and specificity of the EC-silencing approach. (b) Quantitation of EC apoptosis following challenge with Aβ-E22Q (Q22, 1 day, 25 μM) was evaluated by cell death ELISA, after silencing the expression of DR4, DR5, or both receptors. (c) Quantitation of caspase-8 activation (Caspase Glo-8) following EC challenge with Aβ-E22Q (25 μM; 6 h) after silencing the expression of DR4 and DR5. In both (b and c), siNC represents the non-targeting siRNA used as a negative control and results are expressed as fold of increase compared with controls in absence of E22Q. Graphs in (b and c) represent the mean±S.D. of two separate experiments, each one in duplicate. *,** indicate statistically significant differences, P≤0.05 and P<0.01, respectively, for amyloid-challenged cells in the presence of DR4 and DR5 siRNA compared with siNC
Discussion
Aβ accumulation around cerebral vessels is known to induce degeneration of the entire neurovascular unit.18 Previous studies from our laboratory monitored the Aβ-elicited apoptotic events in cerebral microvascular cells demonstrating induction of caspase-mediated mitochondrial pathways with Bax (Bcl-2-associated X protein) translocation and cytochrome c release to the cytosol.5, 10 Prevention of vessel-wall cell death through inhibition of cytochrome c release or by the action of pan- and pathway-specific caspase inhibitors provided a clear indication of the engagement of both extrinsic and intrinsic apoptotic pathways either independently or synergistically.
The results presented herein unveiled the participation of death receptor-mediated pathways in Aβ-induced EC apoptosis (illustrated in the diagram of Figure 8), as evidenced by caspase-8 activation – for all variants tested – as well as by the involvement of cFLIP and Bid signaling molecules characteristic of the extrinsic apoptotic pathway. The overexpression of DR4 and DR5 after Aβ challenge together with the membrane colocalization of the receptors with Aβ homologs and the protective effect achieved after knocking down DR4 and DR5 unquestionably established the participation of both receptors in EC Aβ-induced apoptosis. Death receptors of the TNFR family have been previously implicated in Aβ-mediated neuronal cell responses. Both the p75 neurotrophin receptor and TNF-α receptors mediated Aβ42-induced neuritic dystrophy and neuronal death19, 20 while antibody-mediated blockade of DR5 prevented the toxicity on neuronal SHSY5Y cells of a non-physiological Aβ fragment.21 Supporting a mechanistic involvement of apoptotic extrinsic pathways in AD pathogenesis, both Aβ and APP were shown to activate neuronal death receptor signaling through direct binding to p75 and DR6.22, 23 Our data unveils, for the first time, the involvement of death receptors on the oligomeric Aβ-induced apoptosis in brain vascular cells pinpointing to DR4 and DR5 as major players in CAA-related dysfunction.

Proposed apoptotic changes elicited in cerebral ECs by Aβ oligomers. The diagram summarizes the postulated apoptotic transformations induced by Aβ oligomeric assemblies in cerebral EC. Aβ oligomers, interacting with membrane receptors DR4 and DR5, trigger activation of caspase-8, which operates cleavage of Bid into tBid. The truncated form of Bid, interfering in the equilibrium between Bcl-2 and Bax, induces the release of CytC from the mithochondria. Cytosolic CytC stimulates the activation of caspase-9 that, in concurrence with caspase-8, induces downstream activation of caspase-3 promoting the final execution of apoptosis
The unique role of TRAIL death receptors in the nervous system is highlighted by the widespread expression of DR4 and DR5 in many cell types in the human brain, including neurons, astrocytes, oligodendrocytes, and EC, as well as by DR5 overexpression in AD.21 In contrast to the ample distribution of DR4 and DR5, their physiological ligand, TRAIL, is not constitutively produced in the brain, and its release has been only reported during neuroinflammation.16 The current findings reveal a novel mechanism by which brain cells – through the engagement of the receptors upon binding to Aβ oligomers – become susceptible to DR4- and DR5-mediated apoptosis, in absence of their typical ligand. Physiologically, death receptors are activated by binding to their specific ligands either previously released into the extracellular space or expressed as membrane components in the same or in different cells. It is, however, not uncommon for certain death receptors that the activation of downstream signaling cascades takes place independently of their canonical ligand mainly through upregulation and clustering of the receptors.24, 25, 26 Indeed, receptor density, distribution, and clustering are key spatial features that influence effective physical and biochemical cellular responses. In this sense, Fas–Fas bridge formation, stabilized upon clustering, leads to a sufficient number of Fas molecules in close proximity, as occurring after traditional Fas–ligand binding.27 Particular attention has been given lately to the generation of high receptor densities involving plasma membrane microdomains known as lipid rafts. These platforms are postulated to have important roles in membrane protein trafficking and receptor-specific signaling.28, 29 In the case of AD, lipid rafts appear to serve as a stage for various processes related to the disease pathogenesis including secretase activity and APP processing as well as Aβ fibrillization, neuronal internalization, and downstream toxicity.30 In the case of DR4 and DR5, their redistribution into lipid rafts has been shown to facilitate the formation of DISCs and downstream caspase activation.31 Whether the observed membrane colocalization of Aβ with DR4 and DR5 in our system reflects the interaction with lipid components or relies on the association with lipid raft microdomains remain to be further elucidated.
Direct binding to multivalent ligands is another mechanism capable of causing receptor clustering and initiating downstream signaling. In fact, multivalent presentation typically enhances the affinity and specificity of a wide array of interactions. Through specific structural features including high density and a particular arrangement of binding sites, multivalent ligands exhibit unique capabilities to cluster receptors.32 In the case of Aβ, oligomerization confers the peptide potent effects for neuronal cells – inducing synaptic disruption and neurotoxicity 33 – and for vessel-wall cells – initiating rapid apoptotic pathways.5, 10, 34, 35 Aβ multimerization is also important for receptor activation. Aβ oligomers act as an extracellular scaffold for the group 1 metabotropic glutamate receptor 5 (mGluR5), resulting in the receptor clustering and stabilization and inducing synaptic failure.36 Binding to multivalent molecules such as specific antibodies or multivalent peptides is also a well-established way to trigger DR4 and DR5, a feature that is currently being explored for therapeutic potential in cancer treatment clinical trials.37 Our data bridge together the aggregation potential of Aβ and the capability of Aβ oligomers to specifically bind DR4 and DR5 with the requirement of both receptors for clustering to induce downstream signaling pathways. By delineating the molecular mechanisms elicited by Aβ oligomers in cerebral EC, our work unveils new targets for pharmacological intervention in amyloid-induced vessel-wall apoptosis.
Materials and methods
Aβ peptides
Synthetic homologs of the amyloid subunits present in the vascular deposits in sporadic and familial Dutch- and Piedmont-AD cases – wild-type (WT) Aβ40 and the genetic variants containing the E22Q and L34V substitutions, respectively, as well as the reverse sequence Aβ40-1 peptide – were synthesized using N-tert-butyloxycarbonyl chemistry by James I Elliott at Yale University. Aβ homologs were dissolved to 1 mM in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; Sigma, St. Louis, MI, USA), incubated for 12 h to breakdown pre-existing β-sheet structures,5 and lyophilized. Peptides were subsequently dissolved in DMSO to a 10-mM concentration, followed by the addition of deionized water to 1-mM concentration, and further dilution into culture media to the required concentrations for the different experiments.
Biotinylation of Aβ-WT and reverse Aβ40-1 peptides for the receptor-chimera-binding studies was performed after HFIP pre-treatment using sulfo-NHS-biotin (Thermo Fisher Scientific, Rockford, IL, USA) as previously described in our laboratory.38 Biotin-labeled peptides were concentrated and separated from free biotin using 3-kDa cutoff Centricon centrifugal filter devices (Millipore, Billerica, MA, USA).
Cell cultures
Immortalized human brain microvascular ECs HCMEC/D3 were obtained from Babette Weksler39 and maintained in complete EBM-2 medium (Lonza, Allendale, NJ, USA) added of growth supplements and 2.5% fetal bovine serum (FBS). This cell line retains the morphological characteristics of primary brain ECs and expresses specific brain endothelial markers and cell surface adhesion molecules.
Caspase-8 and -9 activity assays
Caspase activation was measured by luminescent assays (Caspase-Glo 8 and Caspase-Glo 9, Promega, Madison, WI, USA), in cells treated with the Aβ variants in EBM-2/1% FBS. Briefly, 10 000 cells/well were plated in 96-well plates and incubated for 0–24 h with the Aβ peptides either freshly solubilized in the case of E22Q or pre-aggregated at 37 °C for 3 days for L34V and Aβ40-WT. Caspase-Glo reagent was added to the cell cultures resulting in cell lysis, followed by caspase cleavage of the substrate and generation of a luminescent signal produced by the luciferase reaction. After 40-min incubation the signal, proportional to the amount of caspase activity present, was evaluated in a plate-reading luminometer (Synergy HT Multi-Mode Microplate Reader, Biotek, Winooski, VT, USA). To inhibit nonspecific background activity, the proteasome inhibitor MG-132 was added to the Caspase-Glo reagent before the experiment as indicated by the manufacturer. In all cases, results are expressed as fold change compared with untreated control cells.
Western blot
Evaluation of cFLIP as well as full-length and tBid expression was performed using WB analysis after electrophoretic separation on 12% SDS polyacrylamide gels. Proteins were electrotransferred to PVDF membranes (0.45 μm pore size; Immobilon, Millipore) at 400 mA for 1.5 h, using 10 mM 3-cyclohexylamino-1-propanesulfonic acid (CAPS, Sigma) buffer, pH 11.0, containing 10% (v/v) methanol. Membranes were blocked with 5% non-fat milk in PBS containing 0.1% Tween 20, and subsequently immunoreacted with polyclonal rabbit antibodies recognizing cFLIP and human Bid (1 : 2000 and 1 : 1000, respectively; Cell Signaling/Millipore, Billerica, MA, USA), as well as monoclonal antibody against GAPDH (Imgenex, San Diego, CA, USA) as a loading control, followed by the pertinent horseradish peroxidase (HRP)-labeled anti-rabbit or anti-mouse IgG (1/5000 or 1/10 000, respectively; GE Healthcare Life Sciences, Pittsburgh, PA, USA). Fluorograms were developed by enhanced chemiluminescence (ECL) with ECL western blotting detection reagent (GE Healthcare Life Sciences), and exposed to Hyperfilm ECL (GE Healthcare Life Sciences).
For the assessment of Aβ binding to DR4-Fc, DR5-Fc, and Fas-Fc chimeras (R&D Systems, Minneapolis, MN, USA), proteins were electrophoretically separated under native conditions using 5–30% gradient polyacrylamide gels, in the absence of SDS, employing 25 mM Tris/glycine buffer, pH 8.8, as running buffer. Molecular mass markers for native gel electrophoresis consisted of proteins with acidic isoelectric point: human albumin, ovoalbumin, soybean trypsin inhibitor, lactoglobulin, and insulin. After transferring for 2.5 h at 400 mA, as above, membranes were incubated with a combination of mouse monoclonal anti-Aβ antibodies 4G8 (epitope: residues Aβ18-22) and 6E10 (epitope: residues Aβ3-8), both from Covance (Princeton, NJ, USA), at 1 : 3000 dilution each, followed by HRP-labeled anti-mouse IgG and ECL chemistry, as above. Molecular masses of Aβ oligomeric assemblies were estimated based in their relative mobility compared with standards following linear regression and Ferguson plot analysis with the aid of GraphPad Prism software (GraphPad, La Jolla, CA, USA), as we previously described.40
PCR arrays and quantitative RT-PCR
Human apoptosis-specific PCR arrays (SABiosciences, Qiagen, Valencia, CA, USA) were performed in EC challenged with Aβ40-E22Q peptide for 6 h. After isolation of the RNA and retrotrascription, RT-PCR of the 384-well arrays was performed using an ABI 7900 SDS system (Applied Biosystems, Carlsbad, CA, USA) at New York University Genomics Facility. Data analysis was performed using RT2 Profiler PCR Array Data Analysis software (SABiosciences). Quantitative RT-PCR for evaluation of TNFRSF10A (DR4) and TNFRSF10B (DR5) receptors, as well as the internal controls β-Actin, GAPDH, and cyclophilin B, was performed with the same instrument using the oligonucleotide primers described below custom synthesized at Gene Link (Hawthorne, NY, USA). DR4: 5′-TCCAGCAAATGGTGCTGAC-3′ (F) and 5′-GAGTCAAAGGGCACGATGTT-3′ (R); DR5: 5′-CCAGCAAATGAAGGTGATCC-3′ (F) and 5′-GCACCAAGTCTGCAAAGTCA-3′ (R); β -actin 5′-TGGACATCCGCAAAGACCTG-3′ (F) and 5′-CCGATCCACACGGAGTACTT-3′ (R); GAPDH: 5′-TGCACCACCAACTGCTTAG-3′ (F) and 5′-GGATGCAGGGATGATGTT-3′ (R); cyclophilin-B 5′-GATGGCACAGGAGGAAAGAG-3′ (F) and 5′-AGCCAGGCTGTCTTGACTGT-3′ (R).
Immunocytochemical evaluation of DR4 and DR5 expression
EC were plated on glass chamber slides (Thermo Fisher Scientific), precoated with type-I collagen (Sigma), and allowed to rest for 1 day before treatment with the different peptides, as above, for 2 h to 3 days. Cells were fixed with 4% paraformaldehyde, incubated with monoclonal antibodies recognizing TRAIL-R1 (DR4) and TRAIL-R2 (DR5) (Alexis Biochemicals, San Diego, CA, USA ; 1 : 200 in PBS containing 0.1% Tween 20 and 5 mg/ml BSA (PBST/BSA)) followed by Alexa Fluor 488-conjugated anti-mouse IgG (Invitrogen, Carlsbad, CA, USA; 1 : 200 in PBST/BSA). Aβ peptides were stained using rabbit polyclonal anti-Aβ40 (Invitrogen, 1 : 200 in PBST/BSA), followed by Alexa Fluor 568-conjugated anti-rabbit IgG. Nuclei were counterstained with To-pro (Invitrogen). Specificity of immune detection was corroborated by omission of the primary antibody in the immunostaining procedure. In all cases, imaging was performed in a Zeiss LSM 510 laser scanning confocal/Confocor2 microscope using a 40 × DIC oil immersion objective and LSM 510 software; acquired images were imported into ImageJ (National Institute of Health; http://rsbweb.nih.gov/ij/).
Quantitative evaluation of TRAIL by ELISA
Production of TRAIL following treatment with the different Aβ peptides was evaluated using a specific ELISA (Quantikine Human TRAIL Immunoassay; R&D Systems). Briefly, 106 cells/well were seeded on 6-well plates and cultured for 1 day, as above, before treatment with the peptides (50 μM in 1 ml of EBM-2 containing 1% FBS). After 1- and 3-day incubation, conditioned media was collected, cells lysed, and both supernatants and lysates tested for TRAIL expression by capture ELISA, following the manufacturer’s specifications. Final color was evaluated at 450 nm, with reference wavelength set at 540 nm, on a Spectracount microplate reader (Packard, Meriden, CT, USA). Concentration of TRAIL was determined through the use of a standard curve generated with recombinant protein provided with the kit.
Binding of Aβ40 variants to death receptor-Fc chimeras
Direct binding of Aβ homologs to the death receptors DR4, DR5, and Fas was evaluated through incubation of the different peptides with protein chimeras of the respective receptors with the IgG Fc fragment immobilized onto Protein-G-coated paramagnetic beads. Briefly, Protein-G-coated Dynabeads (Invitrogen; 50 μl) were separately incubated for 20 min with 3 μg each of DR4-Fc, DR5-Fc, or Fas-Fc protein chimeras (R&D systems), as well as human IgG-Fc (Bethyl Laboratories, Montgomery, TX, USA), which was used as negative control for nonspecific binding. After washing, the receptor-coated magnetic beads were further incubated for 30 min at RT with Aβ-E22Q, -L34V, or -WT (either fresh, in the case of E22Q, or pre-aggregated for 3 days, for the other two peptides; all at 50 μM concentration in EBM-2 medium). After magnetic separation of the beads, supernatants were discarded, and the beads washed three times with PBS containing 0.1% Tween 20. The IP material was eluted from the paramagnetic beads by short incubation (2 min) with a non-denaturing elution buffer (Glycine pH 2.8). Both the peptides used as starting material (input), and fractions bound to the immobilized receptors (bound) were separated in native 5–30% gradient gel electrophoresis and analyzed by WB probed with a combination of 4G8 and 6E10 anti-Aβ antibodies, as described above. To confirm the specificity of Aβ binding to DR4/DR5, the immobilized receptor chimeras were incubated with biotin-conjugated reverse Aβ40-1, pre-aggregated for 3 days. Both the starting material (input) and bound fractions were analyzed by WB probed with streptavidin-HRP (Life Technologies/Invitrogen, Carlsbad, CA, USA) followed by ECL as above.
DR4 and DR5 gene silencing
Silencing of DR4 and DR5 was performed through passive siRNA delivery via Accell SMART pool siRNA (Thermo Scientific-Dharmacon, Rockford, IL, USA), following the manufacturer’s protocol. Briefly, 2000 cells/well – plated in 96-well dishes – were treated separately with the respective siRNA solutions constituted by each siRNA pool as well as a fluorescent non-targeting siRNA, employed as negative control, diluted in Accell delivery media to a 1 μM final concentration. The efficiency and specificity of the DR4 and DR5 silencing as well as the GAPDH and Cyclophilin B controls were tested by RT-PCR. To assess the effect of DR4 and DR5 silencing on Aβ-induced toxicity, cells were pre-incubated 64 h in the siRNA-containing media and subsequently challenged with Aβ-E22Q (25 μM in the same culture media containing siRNA); caspase-8 activation and apoptosis induction were evaluated after 6 h and 24 h, respectively. The level of DR4/DR5 receptor silencing after E22Q challenge was monitored by qRT-PCR on a parallel set of control cells, as described above.
Cell death ELISA
The extent of apoptosis caused by Aβ in the presence or absence of the respective siRNAs was assessed by quantitation of nucleosome formation using Cell Death ELISA (Roche Applied Science, Indianapolis, IN, USA). Briefly, after siRNA and incubation with E22Q peptide, plates were centrifuged in a Beckman J-6B centrifuge(10 min;1000 r.p.m.), cells lysed, and DNA-histone complexes (nucleosomes) quantitated by Cell Death ELISA, as previously described.5, 34
Statistical analysis
Analysis of variance for multiple comparison tests and Tuckey’s post-hoc test were performed using GraphPad InStat (GraphPad Software). Statistical significance was set at P≤0.05.
Abbreviations
- Aβ:
-
amyloid-β
- Bid:
-
BH3-interacting domain death agonist
- CAA:
-
cerebral amyloid angiopathy
- cFLIP:
-
cellular FLICE-like inhibitory protein
- DR4:
-
death receptor 4
- DR5:
-
death receptor 5
- ECs:
-
endothelial cells
- FADD:
-
Fas associated protein with death domain
- HMM:
-
high molecular mass
- IP:
-
immunoprecipitation
- TNF:
-
tumor necrosis factor
- TNFR:
-
tumor necrosis factor receptor
- TNFSF:
-
tumor necrosis factor superfamily
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- WB:
-
western blot
- WT:
-
wild type
References
Rostagno A, Holton JL, Lashley T, Revesz T, Ghiso J . Cerebral amyloidosis: amyloid subunits, mutants and phenotypes. Cell Mol Life Sci 2010; 67: 581–600.
Keage HA, Carare RO, Friedland RP, Ince PG, Love S, Nicoll JA et al. Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol 2009; 9: 3.
Attems J, Jellinger K, Thal DR, Van Nostrand W . Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2011; 37: 75–93.
Nicoll JAR, Yamada M, Frackowiak J, Mazur-Kolecka B, Weller RO . Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer's disease. Neurobiol Aging 2004; 25: 589–597.
Fossati S, Cam J, Meyerson J, Mezhericher E, Romero IA, Couraud P-O et al. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-β variants in cells composing the cerebral vessel walls. Faseb J 2010; 24: 229–241.
Wilson NS, Dixit V, Ashkenazi A . Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol 2009; 10: 348–355.
Haase G, Pettmann B, Raoul C, Henderson CE . Signaling by death receptors in the nervous system. Curr Opin Neurobiol 2008; 18: 284–291.
Son YG, Kim EH, Kim JY, Kim SU, Kwon TK, Yoon AR et al. Silibinin sensitizes human glioma cells to TRAIL-mediated apoptosis via DR5 up-regulation and down-regulation of c-FLIP and survivin. Cancer Res 2007; 67: 8274–8284.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Viana RJ, Nunes AF, Castro RE, Ramalho RM, Meyerson J, Fossati S et al. Tauroursodeoxycholic acid prevents E22Q Alzheimer's Abeta toxicity in human cerebral endothelial cells. Cell Mol Life Sci 2009; 66: 1094–1104.
Werner AB, de Vries E, Tait SW, Bontjer I, Borst J . TRAIL receptor and CD95 signal to mitochondria via FADD, caspase-8/10, Bid, and Bax but differentially regulate events downstream from truncated Bid. J Biol Chem 2002; 277: 40760–40767.
Pobezinskaya YL, Choksi S, Morgan MJ, Cao X, Liu ZG . The adaptor protein TRADD Is essential for TNF-like ligand 1A/death receptor 3 signaling. J Immunol 2011; 186: 5212–5216.
Su WB, Chang YH, Lin WW, Hsieh SL . Differential regulation of interleukin-8 gene transcription by death receptor 3 (DR3) and type I TNF receptor (TNFRI). Exp Cell Res 2006; 312: 266–277.
Croft M . The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol 2009; 9: 271–285.
Tsai AC, Pan SL, Sun HL, Wang CY, Peng CY, Wang SW et al. A new vascular targeting agent, induces apoptosis of human umbilical vein endothelial cells via p53-mediated death receptor 5 up-regulation. J Biol Chem 2010; 285: 5497–5506.
Dorr J, Bechmann I, Waiczies S, Aktas O, Walczak H, Krammer PH et al. Lack of tumor necrosis factor-related apoptosis-inducing ligand but presence of its receptors in the human brain. J Neurosci 2002; 22: RC209.
Fossati S, Cam J, Meyerson J, Mezhericher E, Romero IA, Couraud PO et al. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-beta variants in cells composing the cerebral vessel walls. FASEB J 2010; 24: 229–241.
Zlokovic BV . The blood-brain barrier in ealth and chronic neurodegenerative disorders. Neuron 2008; 57: 178–201.
Knowles JK, Rajadas J, Nguyen TV, Yang T, LeMieux MC, Vander Griend L et al. The p75 neurotrophin receptor promotes amyloid-beta(1-42)-induced neuritic dystrophy in vitro and in vivo. J Neurosci 2009; 29: 10627–10637.
Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ . Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci 2008; 28: 3941–3946.
Uberti D, Ferrari-Toninelli G, Bonini SA, Sarnico I, Benarese M, Pizzi M et al. Blockade of the tumor necrosis factor-related apoptosis inducing ligand death receptor DR5 prevents beta-amyloid neurotoxicity. Neuropsychopharmacology 2007; 32: 872–880.
Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M . APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009; 457: 981–989.
Yaar M, Zhai S, Fine RE, Eisenhauer PB, Arble BL, Stewart KB et al. Amyloid beta binds trimers as well as monomers of the 75-kDa neurotrophin receptor and activates receptor signaling. J Biol Chem 2002; 277: 7720–7725.
Micheau O, Solary E, Hammann A, Dimanche-Boitrel MT . Fas ligand-independent, FADD-mediated activation of the Fas death pathway by anticancer drugs. J Biol Chem 1999; 274: 7987–7992.
Poh TW, Huang S, Hirpara JL, Pervaiz S . LY303511 amplifies TRAIL-induced apoptosis in tumor cells by enhancing DR5 oligomerization, DISC assembly, and mitochondrial permeabilization. Cell Death Differ 2007; 14: 1813–1825.
Rossin A, Derouet M, Abdel-Sater F, Hueber AO . Palmitoylation of the TRAIL receptor DR4 confers an efficient TRAIL-induced cell death signalling. Biochem J 2009; 419: 185–192 2 p following 192.
Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E et al. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009; 457: 1019–1022.
Xu L, Qu X, Luo Y, Zhang Y, Liu J, Qu J et al. Epirubicin enhances TRAIL-induced apoptosis in gastric cancer cells by promoting death receptor clustering in lipid rafts. Mol Med Report 2011; 4: 407–411.
Korade Z, Kenworthy AK . Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008; 55: 1265–1273.
Rushworth JV, Hooper NM . Lipid rafts: linking alzheimer's amyloid-beta production, aggregation, and toxicity at neuronal membranes. Int J Alzheimers Dis 2010; 2011 603052.
Psahoulia FH, Drosopoulos KG, Doubravska L, Andera L, Pintzas A . Quercetin enhances TRAIL-mediated apoptosis in colon cancer cells by inducing the accumulation of death receptors in lipid rafts. Mol Cancer Ther 2007; 6: 2591–2599.
Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL . Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J Am Chem Soc 2002; 124: 14922–14933.
Walsh DM, Selkoe DJ . A beta oligomers - a decade of discovery. J Neurochem 2007; 101: 1172–1184.
Fossati S, Ghiso J, Rostagno A . Insights into caspase-mediated apoptotic pathways induced by amyloid-β in cerebral microvascular endothelial cells. Neurodegener Dis 2012; 10: 324–328.
Davis J, Cribbs DH, Cotman CW, Van Nostrand WE . Pathogenic amyloid beta-protein induces apoptosis in cultured human cerebrovascular smooth muscle cells. Amyloid 1999; 6: 157–164.
Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010; 66: 739–754.
Pavet V, Beyrath J, Pardin C, Morizot A, Lechner MC, Briand JP et al. Multivalent DR5 peptides activate the TRAIL death pathway and exert tumoricidal activity. Cancer Res 2010; 70: 1101–1110.
Matsubara E, Soto C, Governale S, Frangione B, Ghiso J . Apolipoprotein J and Alzheimer's amyloid beta solubility. Biochem J. 1996; 316: 671–679.
Weksler BB, Subileau EA, Perrière N, Charneau P, Holloway K, Leveque M et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb J. 2005; 19: 1872–1874.
Rostagno A, Lashley T, Ng D, Meyerson J, Braendgaard H, Plant G et al. Preferential association of serum amyloid P component with fibrillar deposits in familial British and Danish dementias: similarities with Alzheimer's disease. J Neurol Sci 2007; 257: 88–96.
Acknowledgements
This work was supported by NIH Grants AG030539, NS051715, the Alzheimer’s Association, and the American Heart Association.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by D Bano
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Fossati, S., Ghiso, J. & Rostagno, A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Aβ. Cell Death Dis 3, e321 (2012). https://doi.org/10.1038/cddis.2012.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.55
Keywords
This article is cited by
-
Meloxicam Inhibits Apoptosis in Neurons by Deactivating Tumor Necrosis Factor Receptor Superfamily Member 25, Leading to the Decreased Cleavage of DNA Fragmentation Factor Subunit α in Alzheimer’s Disease
Molecular Neurobiology (2023)
-
Caspases in Alzheimer’s Disease: Mechanism of Activation, Role, and Potential Treatment
Molecular Neurobiology (2023)
-
Understanding the role of key amino acids in regulation of proline dehydrogenase/proline oxidase (prodh/pox)-dependent apoptosis/autophagy as an approach to targeted cancer therapy
Molecular and Cellular Biochemistry (2020)
-
Modulation of blood-brain barrier function by a heteroduplex oligonucleotide in vivo
Scientific Reports (2018)
-
Clinical perspectives of TRAIL: insights into central nervous system disorders
Cellular and Molecular Life Sciences (2016)
