
Abstract
Innate immune sensing of dying cells is modulated by several signals. Inflammatory chemokines-guided early recruitment, and pathogen-associated molecular patterns-triggered activation, of major anti-pathogenic innate immune cells like neutrophils distinguishes pathogen-infected stressed/dying cells from sterile dying cells. However, whether certain sterile dying cells stimulate innate immunity by partially mimicking pathogen response-like recruitment/activation of neutrophils remains poorly understood. We reveal that sterile immunogenic dying cancer cells trigger (a cell autonomous) pathogen response-like chemokine (PARC) signature, hallmarked by co-release of CXCL1, CCL2 and CXCL10 (similar to cells infected with bacteria or viruses). This PARC signature recruits preferentially neutrophils as first innate immune responders in vivo (in a cross-species, evolutionarily conserved manner; in mice and zebrafish). Furthermore, key danger signals emanating from these dying cells, that is, surface calreticulin, ATP and nucleic acids stimulate phagocytosis, purinergic receptors and toll-like receptors (TLR) i.e. TLR7/8/9-MyD88 signaling on neutrophil level, respectively. Engagement of purinergic receptors and TLR7/8/9-MyD88 signaling evokes neutrophil activation, which culminates into H2O2 and NO-driven respiratory burst-mediated killing of viable residual cancer cells. Thus sterile immunogenic dying cells perform 'altered-self mimicry' in certain contexts to exploit neutrophils for phagocytic targeting of dead/dying cancer cells and cytotoxic targeting of residual cancer cells.
Similar content being viewed by others

Main
Sensing of dying/dead cells by innate immune cells forms the core of tissue homeostasis and various diseases.1 Thus, the molecular entities governing this interface are of great interest. Over the last decade, three main innate immune-modulatory profiles of sterile cell death (i.e., cell death induced by non-microbial stimuli) have been demarcated, that is, tolerogenic apoptosis, necrosis and damage-associated molecular patterns (DAMPs)-linked apoptosis (or immunogenic apoptosis).2, 3 In general, modulation of the vertebrate innate immunity is explained by two cardinal models, that is, the 'self/non-self model'4 and the 'danger model'.5
Interestingly, these models contradict on cell death immunology. The self/non-self model postulates the activation of innate immunity only by entities of 'non-self' (e.g., pathogens) or 'altered-self' (e.g., pathogen-infected host cell) origins, possessing pathogen-associated molecular patterns (PAMPs) sensed via pattern recognition receptors (PRRs).4 This model maintains that PRR ligands cannot be derived from endogenous sources.6 Conversely, the 'danger model' postulates that non-physiological, sterile, cell death can activate the innate immune system by releasing endogenous DAMPs, a subset of which are potent danger signals and agonists of PRRs like toll-like receptors (TLRs).5 Research from various labs7, 8 including ours3, 9 has credibly validated the danger model and shown that DAMPs or danger signals emanating from dying (cancer) cells indeed accentuate sensing of dying cells by the innate immune cells. Such liberation of DAMPs can either be achieved in an unregulated fashion by (accidental/regulated) necrosis7, 10 or in a spatiotemporally regulated fashion through immunogenic apoptosis.8 Thus, according to the current conceptualizations, although the self/non-self model explains the tolerogenic apoptosis profile yet the danger model alone explains the immunostimulatory profiles of necrosis and immunogenic apoptosis.3, 4, 5 However, the analogy between PAMPs and DAMPs has ignited a long-standing unresolved question, that is, can certain dying cells partially mimic behavior of a pathogen-infected cell? If this would be the case this ‘altered-self mimicry’ could rectify why certain forms of sterile cell death drive innate immune stimulation and reconcile the two models in one paradigm.
At the site of pathogenic invasion (typically peri-/intra-epithelial milieus),11 in parallel with local phagocytic activity by sentinel cells, one of the first inflammatory processes triggered by an altered self cell to limit further damage entails production of specific inflammatory (or dual function) chemokines to recruit major anti-pathogenic innate immune cells, for example, neutrophils.11, 12, 13 Such chemokine-based recruitment eventually paves the way for phagocytosis and direct elimination of (residual) pathogens by innate immune cells.12, 14 To this end, we deemed it necessary to probe whether sterile dying cells, and in particular those undergoing DAMP-linked cell demise, can recruit (via specific chemokines) and activate innate immune cells in a pathogen response-like fashion culminating into cytotoxicity against residual viable cells.
Results
Immunogenic apoptosis, but not accidental necrosis or tolerogenic apoptosis, causes co-release of CXCL1, CCL2 and CXCL10 chemokines
Initially, we examined the chemokines released during accidental necrosis, tolerogenic apoptosis or immunogenic apoptosis. We assessed the release of 25 major murine chemokines (encompassing key inflammatory/homeostatic/dual-function chemokines;13 Supplementary Figure S1A) in the cell-free-conditioned medium (CM) derived from the low-immunogenic LLC lung epithelial carcinoma cells undergoing tolerogenic apoptosis (induced by tunicamycin (TUN))15, 16 or immunogenic apoptosis (induced by mitoxantrone (MTX))15, 16, 17 and compared them to accidental necrosis (induced by freeze/thawing or F/T).15, 17 Of note, TUN, F/T and MTX are bona fide inducers of these respective cell death immune profiles as published by us15, 17 and others.16, 18 At similar cell death-inducing doses, (~70% cell death; Supplementary Figure S1B) primarily CM derived from MTX-treated cells (but not F/T or TUN) associated with increased co-release of specific chemokines, that is, CXCL1, CCL2 and CXCL10 (Figures 1a and b). A volcano plot based on the same data confirmed that only MTX caused >1.5-fold increase in these chemokines’ release (Figure 1b). This was further substantiated by direct immunoblotting of CM derived from respective dying cells (Figures 1c and d). Beyond MTX, only F/T caused some variable, albeit non-significant, increase in CXCL1/CCL2 release (Figures 1a, b and d).

Immunogenic apoptosis co-releases CXCL1, CCL2 and CXCL10 chemokines, which associate with pathogen response-like pathways. Media from LLCs (F/T, MTX, TUN-treated or untreated/CNTR) was subjected to: (a and b) chemokine antibody array (48 h recovery time point) represented as hierarchically clustered heatmap (fold change over CNTR) (a) or as a Volcano plot (b) for differential release estimation (x axis denotes effect size as log2 scale with baseline correction and y axis denotes multiple t-test analysis; four intra-array replicates across two independent arrays); (c) immunoblotting of concentrated media and corresponding integrated band densitometry (d) (n=3, mean±S.D., unpaired t-test (two-tailed); */**/***P-value versus controls). (e) Immunoblotting of concentrated media from MTX-treated/untreated (CNTR) cells (as indicated), 48 h post treatment. (f and g) Data in (a/b) were analyzed via NMDS statistical analysis to estimate the clustering patterns of various cell death immune profiles based on their specific chemokine signatures (f) and of the specific chemokines across different cell death conditions (g). (h) GO BP enumeration analysis for murine-CXCL1/CCL2/CXCL10 (numbers within Venn diagram denote total significant GO terms enumerated; P<0.05). In (c/e) each chemokine is represented by a separate gel originating from same sample (due to overlapping molecular weights)
Importantly, other known immunogenic apoptosis inducers (radiotherapy>photodynamic therapy/PDT), but not tolerogenic apoptosis inducer (cisplatin),3 also caused increased co-release of CXCL1, CCL2 and CXCL10 (Supplementary Figure S1C). The co-release of CXCL1, CCL2 and CXCL10 following MTX treatment was also evident in other cell lines/cell types, for example, CT2A, ID8, B16 and 4T1 cells (Figure 1e) suggesting a broad cellular association between this signature and immunogenic apoptosis. In general, MTX-induced immunogenic apoptosis exhibited the most divergent chemokine profile, evident from its spatially distinct, non-overlapping, positioning (relative to F/T-treated/TUN-treated/untreated cells) within a non-metric multidimensional scaling 3D environment (Figure 1f). Similarly, in this same statistical environment, CXCL1, CCL2 and CXCL10 formed a spatially distinct cluster – signifying their tendency to be co-released/co-regulated in this context (Figure 1g).
Co-release of CXCL1, CCL2 and CXCL10 represents a pathogen response-like chemokine signature
Next, we analyzed whether the CXCL1-CCL2-CXCL10 cluster represents a pathogen response-like chemokine (PARC) signature. Using Gene Ontology (GO) bioinformatics analysis we observed that these three chemokines, statistically significantly, enumerated biological process (BP) terms (highlighted in red, Figure 1h) associated with responses to pathogens; especially bacteria, molecules of bacterial origin (e.g., lipopolysaccharide (LPS)) or viruses (Figure 1h). To further test the validity of CXCL1-CCL2-CXCL10 cluster as a bona fide PARC signature, we first performed a comprehensive meta-analysis of Gene Expression Omnibus (GEO) data sets consisting of experiments involving mammalian cells/tissues treated with various pathogens (or pathogen-derived products) including bacteria, viruses and protozoan parasites (Figure 2a). Indeed, many pathogens or pathogen-origin products upregulated CXCL1-/CCL2-/CXCL10-coding genes (Figure 2a) wherein these exhibited significant co-upregulation (Figure 2b). Interestingly, CXCL1/CCL2/CXCL10’s tendency toward co-upregulation was highest in response to bacteria/bacterial products (Figure 2c) and viruses (Figure 2d), but least to protozoan parasites (Figure 2e).

Immunogenic apoptosis is associated with a PARC signature. (a) GEO expression data sets' meta-analysis (GDS accession numbers indicated) of human (Hu)/mice (Mu)/rat (Ra) cells/organs/tissues treated with indicated pathogens/left untreated (CNTR) (hierarchically clustered heatmap of fold change over CNTR). (b–e) Radar plots of Spearman correlations between gene expressions of data in (a). (f and g) Immunoblotting of concentrated media from LLCs, untreated/CNTR or treated with bacterial products (f) or oncolytic viruses (g). (h) Mean integrated band densitometries of immunoblots in Figure 1c (48 h)/f (48 h)/g (48 h for others but 24 h for NDV)/Supplementary Figure S1C (48 h) were pooled and represented as hierarchically clustered heatmap (fold change over CNTR). In (f/g) each chemokine is represented by a separate gel originating from same sample (due to overlapping molecular weights)
Certain bacterial-origin products (e.g., LPS) or known oncolytic viruses14 also upregulated CXCL1/CCL2/CXCL10-coding genes (highlighted in red, Figure 2a). Henceforth, we experimentally challenged LLC cells with bacterial-origin products (i.e., LPS and flagellin) or well-established oncolytic viruses (reovirus, parvovirus and Newcastle disease virus (NDV)14, 19 (Supplementary Figure S2). Remarkably, these bacterial products (Figure 2f) and oncolytic viruses (Figure 2g) caused increased co-release of CXCL1/CCL2/CXCL10 in a temporally defined manner (although bacterial products sustained the chemokine co-release better, thereby substantiating the GEO meta-analysis; Figures 2b–e). Finally pooled analysis of the results in Figure 1c/Figures 2f–g/Supplementary Figure S1C revealed the tendency of immunogenic apoptosis inducers (MTX/radiotherapy/PDT) to cluster together with bacterial-origin products and oncolytic viruses in terms of increased co-release of CXCL1/CCL2/CXCL10; whereas necrosis (F/T) and tolerogenic apoptosis (TUN/cisplatin) clustered apart (Figure 2h). These data collectively indicate that immunogenic apoptosis, but not accidental necrosis or tolerogenic apoptosis, associates with a 'putative' PARC signature.
Immunogenic apoptosis selectively recruits neutrophils as first innate immune responders
Next, we tested whether immunogenic apoptotic cells can attract innate immune cells in a manner reminiscent of an altered self cell, for example, rapid, early recruitment of neutrophils.11, 12, 16 We used a model (Figure 3a), in which the necrotic or (tolerogenic/immunogenic) apoptotic LLC cells were injected intra-dermally into ear pinna of syngeneic mice (Supplementary Figure S3A); followed by estimation of early chemotactic recruitment of various (CD45+) leukocytes (Figure 3b) like (CD11b+Ly6G+) neutrophils (Figure 3c), (CD11b+F4/80+) macrophages (Figure 3d), (CD11c+) monocytes/dendritic cells (DCs) (Figure 3e), (CD3+) T cells (Figure 3g) and (B220+) B cells/plasmacytoid DCs (pDCs)/lymphokine-activated cells (Figure 3f). Immunogenic apoptosis triggered increased early recruitment of leukocytes in comparison to PBS/CNTR (Figure 3b). Strikingly, only immunogenic apoptosis but not necrosis/tolerogenic apoptosis, caused a significantly sustained, rapid, recruitment of neutrophils (Figure 3c), which was preferred over other major leukocytic populations (Figures 3d–g). This is in contrast to previous reports where monocytes/macrophages, rather than neutrophils, have been demonstrated to preferentially respond to apoptosis.20 Although macrophages/monocytes/DCs did not exhibit early recruitment, they can still be (co-)involved in local sentinel immunological activity.1 We further confirmed the increased recruitment of neutrophils in ear pinna injected with MTX-induced dying cells through immunohistochemistry (Figures 3h and i) and by documenting typical polymorphonuclear neutrophils (i.e., PMNs with ⩾3-lobed nucleus) (Figure 3j).

Neutrophils are recruited by immunogenic dying cells as first innate immune responders via the PARC signature. (a) Mice ear pinna model for in vivo chemotaxis of immune cells toward dead/dying cells (recovered 24 h post treatment) or PBS/CNTR constituted immunophenotyping for CD45+leukocytes (b), CD11b+Ly6G+neutrophils (c), CD11b+F4/80+macrophages (d), CD11c+monocytes/DCs (e), B220+cells (f) and CD3+T cells (g) (n=4–8 ear pinna across four mice, mean±S.E.M., two-way ANOVA; */**/***P-value/not signficant (NS) versus controls). Also, PBS/CNTR or MTX-treated (for 24 h) LLCs-injected mice ear pinna were processed for immunohistochemistry (h) (scale bar=100 μm; Ly6G+ area quantification (i), n=5 mice, mean±S.E.M., Mann–Whitney t-test) or cytospin analysis (scale bars: CNTR=50 μm; MTX=20 μm, neutrophil's margin indicated manually; polymorphonuclear cells' quantification as pie charts; n=5 mice, Fischer’s exact test) (j). (k) Hierarchically clustered heatmap represents correlation between Figure 1a versus c (fold change over CNTR). Ear pinna immunophenotyping for neutrophils for conditions (PBS/CNTR or MTX) where chemokines were blocked via antibodies (l) (n=6–8 ear pinna across 3–4 mice, mean±S.E.M., two-way ANOVA (Bonferroni post tests), */**/***P-value/NS as indicated); or chemokines were shRNA-targeted (m) (n=4 mice, mean±S.E.M., two-way ANOVA, */**/***P-value as indicated)
Immunogenic apoptosis-triggered neutrophil recruitment depends on pathogen response-like chemokine signature
Neutrophils are typically the first innate immune responders to infections12 and, under PAMPs’ influence they perform crucial functions like phagocytic clearance and direct pathogen elimination.12, 14 Our results raised precedence to investigate whether the PARC signature is responsible for this neutrophil recruitment. GO bioinformatics analysis indicated an association between CXCL1-CCL2-CXCL10 and ‘neutrophil chemotaxis’ (Supplementary Figure S3B). Also an unbiased correlation analysis (involving chemokines exhibiting >1.2-fold increase in release by at least one cell death pathway, Figure 1a) revealed that release of CXCL1/CCL2/CXCL10 was maximally positively correlated (⩾0.80) with the levels of neutrophil chemotaxis (Figure 3k), across both time points. To establish a cause–effect relationship, the dead/dying cells were deprived of the PARC signature via specific blocking antibodies before injection. This simultaneous blockade of CXCL1-CCL2-CXCL10 markedly decreased neutrophils' recruitment toward MTX-treated cells (Figure 3l). Above observations were not specific for LLC cells; as MTX-treated B16 cells also significantly chemo-attracted neutrophils and simultaneous blockade of CXCL1-CCL2-CXCL10 significantly reduced this neutrophil recruitment (Supplementary Figure S3C).
To analyze the neutrophil-attracting properties of individual chemokines, we expressed in LLC cells, shRNAs targeting CXCL1 or CCL2 or CXCL10, thereby severely suppressing their release following MTX treatment (Supplementary Figure S4). This individual abrogation of each chemokine significantly affected the in vivo recruitment of neutrophils by MTX-treated cells in a hierarchical, chemokine-dependent fashion (CXCL1≈CXCL10»CCL2) (Figure 3m). Thus PARC signature sustains a dominant neutrophil-attracting phenotype, which allows immunogenic apoptosis to recruit neutrophils as first innate immune responders.
To further validate the CXCL1/CCL2/CXCL10 co-regulation as a PARC signature, we performed a ‘reverse-enumeration’ bioinformatics analysis wherein murine chemokines were selected (from the entire ‘chemokinome’) in an unbiased manner using the major pathogen response and neutrophil chemotaxis related GO BP terms (highlighted in red, Figure 4a). This analysis strongly enumerated for CXCL1/CCL2/CXCL10 (Figure 4b) and some of their closely homologous chemokines (Figures 4b and c) within the uppermost quartile (Q3). This further positions CXCL1-CCL2-CXCL10 as constituting a dominant, albeit non-exclusive, neutrophil-recruiting PARC signature.11, 13

‘Reverse enumeration’ bioinformatics analysis related to pathogen response-like neutrophil chemotaxis enumerates CXCL1, CCL2 and CXCL10 and their close homologs. (a) GO terms related to pathogen responses identified in Figure 1h and ‘neutrophil chemotaxis’ were used for creating a directed acyclic graph (DAG). (b) Murine chemokines enumerated through AmiGO2 web application using GO terms highlighted in red boxes in a were used to plot a frequency distribution graph, and relevant statistical percentiles and quartiles are indicated. (c) Literature-based analysis was used to create a manual Venn diagram indicating existence of close murine homologs of CCL2, CXCL1 and CXCL10 (relevant references are indicated in the figure)
Pathogen response-like chemokine signature-driven neutrophil recruitment by immunogenic apoptosis is evolutionarily conserved in a cross-species manner
In principle, the PARC signature-based neutrophil recruitment should be evolutionarily conserved across vertebrates since chemokines evolved ~650 million years ago in fish – a quintessential feature of vertebrate immunity21 (for instance zebrafish, but not fruitfly/roundworms, possess homologs of several murine chemokines21 including Ccl2/Cxcl10, Supplementary Figure S5A). Similarly, as per self/non-self model,4 our results obtained in a ‘self set-up’ (murine dying cells in syngeneic mice) are expected to maintain at least some of their specificity in a ‘non-self setup’ (in a cross-species manner, e.g., murine dying cells in zebrafish).
Henceforth, we injected F/T (negative control) or MTX-treated LLC cells in the yolk sac of zebrafish larvae (Supplementary Figures S5B and C) transgenic for GFP+neutrophils (mpx:GFP) and mCherry+macrophages (fms:nfsB.mCherry)22 (larval zebrafish, at this developmental stage, have a simple innate immune system dominated by these two cell types)22, 23 (Supplementary Figure S6A). Similar to the ‘self set-up’, MTX-induced dying cells (but not F/T-induced) specifically recruited zebrafish neutrophils (Supplementary Figures S6B–D). Conversely, while MTX-treated cells did not chemoattract macrophages yet F/T-treated cells did (Supplementary Figures S6B and C). Next, we injected MTX-treated cells deprived of CXCL1-CCL2-CXCL10 via antibody-based blockade into the zebrafish larvae's yolk sac. This markedly decreased neutrophils' recruitment into the yolk sac (Supplementary Figures S7A and B). We also injected different zebrafish larvae with MTX-treated cells with or without individual shRNAs against CXCL1 or CCL2, or CXCL10 (Supplementary Figure S4). Interestingly, CXCL10 and CCL2 depletion (CXCL10≈CCL2) but not CXCL1 depletion, affected the in vivo recruitment of neutrophils (Supplementary Figures S7C and D). These results are partially dissimilar to mice setup (CXCL1≈CXCL10»CCL2; Figure 3m) but in line with the bioinformatics analysis showing the presence of Ccl2/Cxcl10 homologs but not Cxcl1 homolog in zebrafish (Supplementary Figure S5A). These results demonstrate the ability of immunogenic apoptosis-associated PARC signature to recruit neutrophils in a cross-species (evolutionarily conserved) fashion.
Immunogenic apoptosis triggers neutrophil-driven phagocytosis and pro-inflammatory stimulation
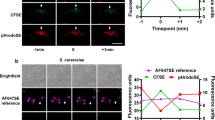
Next, we investigated the neutrophils-dying cells interaction in ex vivo co-cultures. Phagocytosis analysis was done with LLC cells stained through pH-sensitive dye (pHrodo) that emits heightened fluorescence upon phagosome contact (pH≈5), thereby distinguishing engulfed from un-engulfed cells. A fraction of MTX-treated LLC cells underwent significant phagocytic clearance by neutrophils (Figures 5a and b). MTX-treated cells surface exposed (ecto-) two important 'eat me' signals, that is, phosphatidylserine (ecto-PtdSer) (Supplementary Figure S1B) and calreticulin (ecto-CRT), but not heat-shock protein90 (HSP90) (Figure 5c).24 Disruption of these pro-phagocytic signals (PtdSer by annexin-V and CRT by antibody) significantly decreased engulfment by neutrophils (Figures 5a and b). Neutrophils tended to prefer ecto-CRT as an 'eat me' signal over PtdSer (Figure 5b).

Immunogenic apoptotic cells induce neutrophil-driven phagocytosis and pro-inflammatory activation. Phagocytosis assay utilized untreated (CNTR)/MTX-treated LLC cancer cells (CCs) versus neutrophils, with/without indicated blockers (a and b) (n=3, mean±S.D., two-way ANOVA; */**/***P-value/not signficant (NS) versus controls or as indicated). (c) Ecto-CRT/HSP90 on LLCs (24 h recovery) (n=3, mean±S.E.M., unpaired t-test (two-tailed)). LLCs (24 h recovery) were co-cultured with neutrophils for indicated time points, and latter's maturation estimated (d); n=3, mean±S.E.M., two-way ANOVA (Tukey's multiple comparison test); P-value as in legend; Cytokines in co-culture-media of (d). (e) (n=3, mean±S.E.M., two-way ANOVA (uncorrected Fisher's LSD); */**/***P-value/NS versus controls or as indicated (#)). (f) Ear pinna injected with MTX-treated LLCs (expressing specific shRNAs) were immunophenotyped (n=4 mice, mean±S.E.M., two-way ANOVA; */**/***P-value as indicated; #/##/###P-value/NS versus CO-shRNA)
Moreover, neutrophils interacting with MTX-treated cells underwent strong phenotypic (CD86high/MHC-IIhigh) and functional (IL6highIL1βhighIL10low) maturation (Figures 5d and e). Neutrophils interacting with F/T-treated cells (negative control) remained largely immature (Figures 5d and e). Importantly MTX-treated LLC (Figure 5f) or B16 cells (Supplementary Figure S8A) caused increased accumulation of CD86high/MHC-IIhighneutrophils in vivo thus confirming the physiological relevance of this neutrophil-based sensing. Disruption of neutrophil recruitment through shRNA-based depletion of CXCL1 or CCL2, or CXCL10 also resulted in proportionately reduced accumulation of CD86high/MHC-IIhigh neutrophils in vivo (Figure 5f) (CXCL1≈CXCL10»CCL2). In fact there was a strong positive correlation between neutrophil recruitment and accumulation of activated neutrophils in vivo (Supplementary Figure S8B) – indicating that disruption of ‘upstream’ neutrophil recruitment also compromises ‘downstream’ neutrophil maturation/sensing.
Immunogenic apoptosis stimulates neutrophils by engaging extracellular nucleic acids–MyD88-TLR7/8/9 and extracellular ATP-purinergic receptor signaling axes
PARC signature did not seem to directly modulate neutrophil-based sensing, as blocking it didn’t negatively affect neutrophil activation (Supplementary Figure S8C), thereby implying presence of other immunostimulatory factors. Immunogenic apoptotic cells emit DAMPs-like ATP (agonist of purinergic-2/P2 receptors/P2Rs), high-mobility group box 1 (HMGB1) protein or HSPs-70/90 (agonists of TLRs myeloid differentiation primary response 88/MyD88 pathway) or annexin-A1 (AnxA1),3, 7 known to operate at the dying cell-DC/macrophage interface.3, 24 However, not much clarity exists on dying cell-derived neutrophil-activating factors/danger signals. In line with the previous studies,2, 17, 25 in response to MTX, immunogenic apoptotic LLCs released ATP (Figure 6a), HMGB1 and AnxA1 (but not HSP70/90) (Figure 6b). Interestingly, blocking strategies blunting these DAMP’s presence/function revealed that only ATP degradation (via apyrase/Apy enzyme), but not HMGB1-blockade (Figure 6c) abrogated neutrophil stimulation. Conversely, AnxA1 acted as a partial ‘anti-inflammatory DAMP’ as blocking it increased neutrophil's CD86 levels (Figure 6c).

Nucleic acids–TLR7/8/9-MyD88 and extracellular ATP-P2Rs axes facilitate activated neutrophil-driven cytotoxicity against residual cells. Extracellular ATP (24 h recovery; n=6) (a), HMGB1/AnxA1/HSPs (b), nucleic acids (n=3) (g), double-stranded DNA (dsDNA) (n=3) (h) were analyzed in conditioned media of MTX-treated LLC cancer cells (CCs) (a, g and h: mean±S.E.M., unpaired t-test (two-tailed)). LLCs (24 h recovery) were co-cultured with neutrophils and latter's maturation estimated after 48 h co-culture; here effects of receptor inhibitors (d and e), Myd88−/−neutrophils (f), HMGB1/AnxA1-blocking antibodies, apyrase/Apy (c), DNase/RNase (i), TLR3 inhibitors or TLR7/8/9 antagonists (j) were analyzed (e, f, c, i and j; n=3, mean±S.E.M., two-way ANOVA (Tukey's multiple-comparison test); P-value as in legend). Cytokines in co-culture-media of (e and f/c/i and j). (k) (n=3, mean±S.E.M., two-way ANOVA (uncorrected Fisher's LSD); */**/***P-value/not signficant versus controls or as indicated (#)). (n) H2O2 and NO/nitrite in specific co-culture-media of e and f/c/i and j. Co-cultures from Figure 5d or (e and f/c/i and j) analyzed for %cell death in LLCs (Zombie-yellow+CD11b−Ly6G−) (l and m); this extended to H2O2 inhibitors (apocynin/Apo; catalase)/NO inhibitor (L-NMMA)/caspases inhibitor (zVAD) (o) (n, l, m and o: n=3, mean±S.D., unpaired t-test (two-tailed); black */**/***P-value as indicated and red */**/***P-value versus MTX CCs+neutrophils)
Next, we blocked the three most important neutrophil PRRs/receptor–adaptor systems,12, 26 that is, formyl peptide receptor 1 (FPR1), P2Rs family and MyD88 (TIR adaptor protein relevant for most TLRs, except TLR3)7 in neutrophil–LLC co-cultures (Figure 6d). Blocking P2Rs or MyD88 activity (but not FPR1) compromised neutrophil maturation (Figure 6e). However, blocking MyD88 exerted higher negative effect on neutrophil stimulation (CD86↓/MHC-II↓) than P2Rs inhibition (MHC-II↓) (Figure 6e). Furthermore, neutrophils derived from Myd88−/−mice phenocopied this dominant effect of MyD88 peptide antagonist, on neutrophil stimulation (Figure 6f), thereby inferring an important role for TLR signaling.
While we delineated the P2Rs agonist (ATP), the TLR agonist remained obscure. During pathogenic infections apart from protein PAMPs (lipoproteins/flagellin/viral proteins), cell wall-derived (LPS/lipoteichoic acid) or nucleotidic-PAMPs (viral/bacterial nucleic acids) also drive TLR signaling.26, 27 As the current setup exhibited features of pathogen response-like signaling, we wondered whether MTX-treated cells released analogous nucleotidic DAMPs.28 Analysis of cell-free CM from MTX-treated cells revealed significant enrichment of nucleic acids (Figure 6g) including double-stranded DNA (Figure 6h). The release of nucleic acids was a general consequence of cell death but their overall quantities were inducer-dependent (Supplementary Figures S9A and B). Therefore, we degraded the nucleic acids released by MTX-treated cells (via DNase/RNase enzymes) and observed remarkable reduction in neutrophil maturation (similar to Myd88−/−neutrophils) (Figure 6i). Nucleic acids-based danger signals tend to bind/signal through various cognate TLRs, for example, TLR3, TLR7/8 and TLR9.26, 27 Hence we blocked these through selective pharmacological inhibition (CU-CPT4a against TLR3) or specific oligodinucleotide (ODN) antagonists of TLR7/8 (ODN2087) or TLR9 (ODN2088).29 Inhibition of TLR7/8/9 (wherein TLR9>TLR7/8), but not TLR3, ablated neutrophil stimulation (Figure 6j). Interestingly, a GO bioinformatics analysis of TLR7/8/9-MyD88 axis significantly enumerated for GO terms relevant for pathogen response-like signaling (especially anti-viral response) (Supplementary Figure S9C).
These effects of ATP-P2Rs and nucleic acids–TLR7/8/9-MyD88 axes were not confined to neutrophil phenotypic maturation. Blockade of these signaling axes also drastically shifted the (extracellular) cytokines profile of neutrophils interacting with MTX-treated cells, from pro-inflammatory (IL6highIL1βhighIL10low) to anti-inflammatory (IL6lowIL1βlowIL10high) (Figure 6k). Here, IL1β was co-induced by both axes, whereas IL6 was induced (and IL10 was suppressed) only by nucleic acids–TLR7/8/9-MyD88 axes (Figure 6k). Blockers of FPR1/extracellular HMGB1/extracellular AnxA1/TLR3 failed to affect the cytokines patterns (Figure 6k).
Neutrophils stimulated by immunogenic apoptosis exert cytotoxicity against residual live cancer cells
PAMPs-activated neutrophils typically target/eliminate residual pathogens to facilitate resolution-of-inflammation.12 Hence, we determined whether the DAMPs-activated neutrophils exerted cytotoxicity against residual cancer cells that managed to survive MTX-induced cell death (Supplementary Figure S1B). Indeed, MTX-treated LLC cells (CD11b−Ly6G−) co-cultured with neutrophils (CD11b+Ly6G+) underwent significantly more cell death (~90%) than MTX-treated LLC cells alone (~70%) (Figure 6l). Remarkably, blockade of ATP-P2Rs and nucleic acids–TLR7/8/9-MyD88 axes reduced the neutrophil-dependent cell death of residual cells that survived the MTX insult (Figure 6m). Thus, neutrophils interacting with immunogenic apoptotic cells gain a pro-inflammatory profile, culminating into neutrophil-dependent cytotoxicity against residual cancer cells.
Neutrophils-mediated cytotoxicity against residual live cells is driven by respiratory burst
Normally neutrophils exert cytotoxic effects through extrinsic molecular mechanisms (e.g., FasL/perforins-based, against eukaryotic cells) or respiratory burst (e.g., H2O2-/NO-based, typically against pathogens).12, 26, 30, 31 Neutrophils interacting with MTX-induced dying cells did not appreciably upregulate FasL/perforins (Supplementary Figures S10A and B); however, they significantly released H2O2 and NO (Figure 6n). The ATP-P2Rs and nucleic acids–TLR7/8/9-MyD88 axes also tightly regulated neutrophil-driven respiratory burst (NO production was co-induced by both, whereas H2O2 production was driven by ATP-P2Rs axis) (Figure 6n). Next, we blocked the respiratory burst through various agents,30, 31 for example, apocynin (blocks NADPH oxidase complex-based H2O2 production), catalase (degrades H2O2) and L-NMMA (blocks NOS-based NO production) (Supplementary Figures S11A–C). Curiously, blockade of either H2O2 or NO production inhibited neutrophil-dependent residual cell killing (Figure 6o) suggesting combinatorial effect of these two oxidative species. Furthermore, the pan-caspase inhibitor, zVAD, significantly reduced neutrophil-dependent residual cell killing (Figure 6o) thereby indicating that H2O2 and/or NO induce apoptosis in residual cancer cells.
Discussion
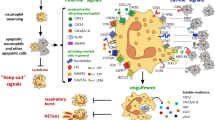
Collectively, the current study demonstrates elicitation of pathogen response-like innate immune signaling or 'altered-self mimicry' during sterile immunogenic apoptosis on multiple levels (Figure 7), that is, (i) dying cell autonomous co-release of CXCL1/CCL2/CXCL10 constituting a PARC signature, observed against bacteria/bacterial products and viruses (bacteria>viruses) rather than protozoan parasites, (ii) neutrophils as first innate immune responders to these dying cells in vivo, (iii) cross-species evolutionary conservation of PARC signature-based neutrophil recruitment (mice versus zebrafish), (iv) regulation of neutrophil activation and neutrophil-dependent cytotoxicity through ATP-P2Rs and nucleic acids–TLR7/8/9-MyD88 axes and (v) a pathogen response-like elimination of residual cells via (H2O2-/NO-driven) respiratory burst. Our results are partially supported by another study where doxorubicin-treated immunogenic dying cells exhibited autonomous ‘dsRNA virus mimicry’, that is, self-dsRNA:TLR3 interaction causing production of type I interferons.25 However, our study reveals a much broader 'altered-self mimicry' phenotype consisting of not only a dying cell autonomous 'mimicry', that is, the PARC signature that at least partially resembles responses to both bacteria/bacterial products and viruses, but also neutrophil-level 'mimicry' involving pathogen response-like recruitment/activation and residual cell-targeting activity. In future it would be interesting to uncover the cell autonomous pathways governing the co-release of CCL2, CXCL1 and CXCL10 from immunogenic dying cells.

Schematic representation of ‘altered-self mimicry’ by immunogenic apoptotic cancer cells. This 7-step model of ‘altered-self mimicry’ is initiated with treatment of a heterogeneous cancer cell population with a specific cell death stimuli like MTX (step 1) that induces immunogenic apoptosis in cancer cells susceptible to its cell death and molecular signaling-inducing effects (step 2). At the outset, these immunogenic apoptotic cells orchestrate a PARC signature consisting of concurrent release of CXCL1, CCL2 and CXCL10 in the current setup (step 3). This PARC signature in turn preferentially attracts neutrophils as first innate immune responders (step 4). Thereafter, while on one hand the neutrophils engage in phagocytic interactions with some of these dying cells, predominantly guided by the ‘eat me’ signal, surface CRT (step 5); yet in parallel, some dying cells release danger signals or DAMPs-like extracellular ATP and nucleic acids, which together pave way for a pathogen response-like activation of neutrophils via purinergic receptors (P2Rs) and TLR7/8/9-MyD88 axes, respectively (step 6). The ATP-P2Rs and nucleic acids–TLR7/8/9-MyD88 axes together regulate the neutrophil phenotypic maturation (CD86high/MHC-IIhigh) and its pro-inflammatory profile (IL1βhighIL6high). This ultimately paves way for activation of a respiratory burst consisting of NADPH oxidase complex and NO synthase-based extracellular production of H2O2 and NO, respectively. These reactive oxygen intermediates (ROIs) or nitric oxide intermediates (NOIs) thereafter exert cytotoxicity against residual cancer cells that managed to survive the initial drug-based cell death stimulus (step 7)
The limited accountability for neutrophil modulation by dying cells perhaps stems from the particular emphasis on mechanisms of antigen recovery and presentation from cell corpses,5 a process better executed by DCs and macrophages.3, 24 Thus, chemokines or 'find me’ signals, ‘eat me’ signals and DAMPs operating at the neutrophil-dying cell interface and influencing neutrophil-dependent cytotoxicity are poorly understood. Here we provide several intriguing insights into how neutrophils sense dying/dead cells by uncovering hitherto unclear mechanisms collectively coordinated through chemokines, ecto-CRT, nucleic acids and ATP. Also, previously most studies have focused on neutrophil-dependent cytotoxicity against a subset of untreated cancer cells,30, 31 whereas mechanisms of targeting residual cancer cells that have survived a drug-induced cytotoxic insult were seldom described. Henceforth, co-ordination of ATP-P2Rs and nucleic acids–TLR7/8/9-MyD88 axes underlying neutrophil-dependent cytotoxicity against residual cells that survived MTX insult is an interesting finding. Similarly, positioning of ecto-CRT as neutrophils’ preferred ‘eat me’ signal is captivating. The discovery of (cross-species) evolutionarily conserved ability of murine CCL2/CXCL10 to chemoattract zebrafish neutrophils is also intriguing. Although existence of CCL2-like23 and CXCL10-like32 chemokinetic activity in zebrafish has been reported previously, the ability of these chemokines to attract neutrophils was seldom described. Thus our results provide interesting insights into the versatility of zebrafish chemokine biology and redundancy among vertebrate chemokine activity. Moreover, the dominant neutrophil-attracting effect of CXCL10 (but not CXCL1) across both mice and zebrafish is noteworthy.
The existence of PARC signature in different cell types (and induction by various immunogenic apoptotic stimuli), indicates its broad relevance as neutrophil recruitment signal. However, it is not clear whether PARC signature is restricted to immunogenic sub-form of only apoptosis or also of other regulated cell death pathways. Moreover, considerable redundancy in chemokines' functions12, 21 means that we cannot exclude the possibility of chemokines within the PARC signature being replaced or co-supported by other (homologous) chemokines with overlapping neutrophil-recruiting potential in a context-dependent fashion (Figure 4). Last but not the least, the tissue milieu where these dying cells were injected may also be an influencing factor in the final outcome of the innate immune responses.11
As such, the results presented here are not exhaustively identical to pathogen responses, which tend to be highly multi-factorial and complex, but merely resembling them through ‘mimicry’ on certain specific levels (i.e., dying cells and neutrophils).11 Moreover, chemokine-based recruitment and TLRs/P2Rs-driven activation of neutrophil-dependent cytotoxicity is not the only distinguishing characteristic of innate immune response to pathogens. There are several other pathogen response pathways (e.g., complement pathway, epithelial antimicrobial mechanisms)11 whose exact relationship with cell death immunology remains enigmatic.
In a physiological sense, these data have potential implications for how dying cells may modulate their immediate extracellular microenvironment. Similarly, the intricate details of the neutrophil-dying cell interface uncovered here may have implications for various diseased scenarios, for example, cancer (owing to MTX, immunogenic apoptosis, DAMPs and residual cells-targeted cytotoxicity), transplantation biology (concerning self/non-self model), autoimmune or inflammatory diseases (owing to nucleic acids and heightened neutrophils' activity), and even neurodegeneration (as MTX is also a multiple sclerosis therapeutic). In future it would be crucial to uncover the overall immunological- and disease-related implications of the cell death-associated ‘altered-self mimicry’.
Materials and Methods
Cell culture and cell death induction or treatments
LLC Lewis lung carcinoma cells, CT2A glioma cells, ID8 metastatic ovarian carcinoma cells (received from Dr. An Coosemans, KU Leuven, Belgium), B16 melanoma cells and 4T1 breast carcinoma cells were cultured at 37 °C under 5% CO2 in DMEM containing 4.5 g/l glucose and 0.11 g/l sodium pyruvate, and supplemented with 2 mM glutamine, 100 units per ml penicillin, 100 μg/l streptomycin and 10% fetal calf serum (FCS). To induce accidental necrosis, the cells were grown until the time point where other treated or untreated cells were recovered, exposed to three cycles of freezing (−80 °C) and thawing (37 °C), and immediately utilized. Tolerogenic apoptosis was induced by TUN (TUN, 50 μg/ml; Enzo Lifesciences, Farmingdale, NY, USA or Sigma-Aldrich, St. Louis, MO, USA) or cisplatin (CDDP, 100 μM; Sigma-Aldrich). Immunogenic apoptosis was induced by MTX (MTX, 2 μM; Sigma, Bornem, Belgium), radiotherapy (120 Gy, performed as described previously)9, 33 or Hypericin-based PDT (Hyp-PDT; incubation with 200 nM Hypericin, for 2 h in serum-free media, followed by light irradiation of 2.70 J/cm2 performed as described previously).17 Hypericin was prepared, purified and stored as detailed elsewhere.17 In certain cases as applicable, the cells were treated with E scherichia coli LPS (LPS, 1000 ng/ml; Sigma), Salmonella typhimurium flagellin (1000 ng/ml; Sigma-Aldrich), reovirus (Type 3 Dearing), parvovirus (H-1) or NDV (NDV; Hitchner B1) (the viruses were produced as described previously).19, 34 Of note, the highest cytotoxic doses possible of reovirus, parvovirus and NDV were used for LLC cells treatments.
Mice experiments
For mice experiments, female or male C57BL/6J mice (8–10 weeks old) were purchased from Harlan (Netherlands) or internal stock of the KU Leuven, Belgium. The animals’ care was in accordance with the institutional guidelines of University of Helsinki and/or KU Leuven (for these and subsequent sets of mice experiments). Three million cells were killed as described above (recovered 24 h post treatment), or PBS (control/CNTR) was injected (20 μl volume) intra-dermally into the mice ear pinna. On day 1 and day 5 post injections, the mice were killed and their ear pinna was recovered. Thereafter, the pinna were minced, enzymatically ‘digested’ (1 mg/ml collagenase H (Roche, Vilvoorde, Belgium), 0.8 U/ml Dispase II (Roche) in DMEM, 2% FCS for 1 h at 37 °C), disintegrated through rigorous pippetting, sterile filtered (through 100 and 30 μm filters, BD Biosciences, Erembodegem, Belgium) and pelleted. The cells were thereafter washed, exposed to RBC lysis buffer, treated with Fc-receptor block and stained for subsequent immunophenotyping. Following antibodies were used for immunophenotyping: anti-CD45 conjugated with APC (BD Biosciences), anti-CD11c conjugated with Alexa-488 (eBioscience, San Diego, CA, USA), anti-B220 conjugated with FITC (eBioscience), anti-Ly6G conjugated with FITC (BD Biosciences/eBioscience; RB6-8C5), anti-CD3 conjugated with Alexa-647 (BD Biosciences), anti-F4/80 conjugated with FITC (eBioscience) and anti-CD11b conjugated with PE (eBioscience/BD Biosciences). For in vivo analysis of stimulated neutrophils we used following antibodies: Brilliant Violet 421-conjugated anti-CD11b (BD Biosciences), FITC-conjugated anti-Ly6G (BD Biosciences), PerCP Cy5.5-conjugated anti-I-A/I-E or MHC-II (BD Biosciences) and PE-conjugated anti-CD86 (eBioscience). Blocking antibodies (and their respective isotype controls) against CXCL1/CCL2/CXCL10 (used at 75 μg/ml each, 1:1:1 proportion) were purchased from R&D Systems (Abingdon, UK). Data acquisition was performed either on BD FACSAria (Helsinki) or LSRFortessa (KU Leuven) flow cytometers (BD Biosciences) and the FlowJo software (Tree Star, Ashland, OR, USA) was used for data analysis. Of note, a number of mice experiments was performed across two independent laboratories in two different countries (i.e., CDRT Lab, KU Leuven, Belgium and Salven Lab, University of Helsinki, Finland).
Zebrafish experiments
The zebrafish transgenic model expressing fms:nfsB.mCherry (mCherry-labeled macrophages) and mpx:GFP (GFP-labeled neutrophils) was received from Dr. Timothy Chico, University of Sheffield, UK.22 Adult zebrafish (Danio rerio) were reared under standard aquaculture conditions at 28 °C on a 14/10 h light/dark cycle. Fertilized eggs were collected through natural spawning methodology. Embryos and larvae were maintained in Danieau's solution in an incubator at 28 °C. Three days post fertilization larvae were microinjected in the yolk sac with ~10–14 nl of PBS alone or PBS admixed with dead/dying cells (~50–100 cells per nl), in order to reach at least 400–800 cells per yolk sac per larvae. The microinjections were carried out using Eppendorf FemtoJet microinjector (injection pressure of ~500 hPa). Following injections, at specific post-injection recovery time points the larvae were sedated with a 1x tricaine solution (80 μg/ml tricaine in 0.02% w/v sodium phosphate) and live larvae imaging was done using a Leica MZ10 F stereomicroscope (Wetzlar, Germany) equipped with a DFC310 FX digital camera controlled via Leica Application Suite software (v.3.6.0, Wetzlar, Germany). The resulting images were processed and quantified via ImageJ software (Bethesda, MD, USA). Zebrafish larvae showing no relevant fluorescence (due to possible congenital defect) or dead larvae were excluded from the analysis.
Statistical analysis
All statistical analyses were performed using either Prism software (GraphPad Software, San Diego, CA, USA) or GraphPad QuickCalcs online software (http://www.graphpad.com/quickcalcs/index.cfm). The statistical analysis used is elaborated in the figure legends (significance level set at P<0.05).
References
Poon IK, Lucas CD, Rossi AG, Ravichandran KS . Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 2014; 14: 166–180.
Zitvogel L, Kepp O, Kroemer G . Decoding cell death signals in inflammation and immunity. Cell 2010; 140: 798–804.
Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol 2015; 6: 588.
Medzhitov R, Janeway CA Jr . Decoding the patterns of self and nonself by the innate immune system. Science 2002; 296: 298–300.
Matzinger P . The danger model: a renewed sense of self. Science 2002; 296: 301–305.
Jozefowski S . The danger model: questioning an unconvincing theory. Immunol Cell Biol 2016; 94: 164–168.
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL . Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13: 851–856.
Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3: e955691.
Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 2016; 8: 328ra327.
Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep 2016; 15: 274–287.
Janeway C Immunobiology: the Immune System in Health and Disease, 6th edn. Garland Science: New York, USA, 2005..
Kolaczkowska E, Kubes P . Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013; 13: 159–175.
Moser B, Wolf M, Walz A, Loetscher P . Chemokines: multiple levels of leukocyte migration control. Trends Immunol 2004; 25: 75–84.
Miest TS, Cattaneo R . New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol 2014; 12: 23–34.
Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 2012; 31: 1062–1079.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007; 13: 54–61.
Garg AD, Elsen S, Krysko DV, Vandenabeele P, de Witte P, Agostinis P . Resistance to anticancer vaccination effect is controlled by a cancer cell-autonomous phenotype that disrupts immunogenic phagocytic removal. Oncotarget 2015; 6: 26841–26860.
Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 2005; 202: 1691–1701.
Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L et al. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer 2015; 136: E313–E325.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009; 461: 282–286.
DeVries ME, Kelvin AA, Xu L, Ran L, Robinson J, Kelvin DJ . Defining the origins and evolution of the chemokine/chemokine receptor system. J Immunol 2006; 176: 401–415.
Gray C, Loynes CA, Whyte MK, Crossman DC, Renshaw SA, Chico TJ . Simultaneous intravital imaging of macrophage and neutrophil behaviour during inflammation using a novel transgenic zebrafish. Thromb Haemost 2011; 105: 811–819.
Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 2014; 505: 218–222.
Garg AD, Romano E, Rufo N, Agostinis P . Immunogenic versus tolerogenic phagocytosis during anticancer therapy: mechanisms and clinical translation. Cell Death Differ 2016; 23: 938–951.
Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014; 20: 1301–1309.
de Oliveira S, Rosowski EE, Huttenlocher A . Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 2016; 16: 378–391.
Thompson AJ, Locarnini SA . Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol Cell Biol 2007; 85: 435–445.
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 2012; 13: 832–842.
Krysko DV, Kaczmarek A, Krysko O, Heyndrickx L, Woznicki J, Bogaert P et al. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ 2011; 18: 1316–1325.
Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R . Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011; 20: 300–314.
Finisguerra V, Di Conza G, Di Matteo M, Serneels J, Costa S, Thompson AA et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015; 522: 349–353.
Torraca V, Cui C, Boland R, Bebelman JP, van der Sar AM, Smit MJ et al. The CXCR3-CXCL11 signaling axis mediates macrophage recruitment and dissemination of mycobacterial infection. Dis Model Mech 2015; 8: 253–269.
Vandenberk L, Garg AD, Verschuere T, Koks C, Belmans J, Beullens M et al. Irradiation of necrotic cancer cells, employed for pulsing dendritic cells (DCs), potentiates DC vaccine-induced antitumor immunity against high-grade glioma. Oncoimmunology 2016; 5: e1083669.
Alkassar M, Gartner B, Roemer K, Graesser F, Rommelaere J, Kaestner L et al. The combined effects of oncolytic reovirus plus Newcastle disease virus and reovirus plus parvovirus on U87 and U373 cells in vitro and in vivo. J Neurooncol 2011; 104: 715–727.
Acknowledgements
We would like to acknowledge Dr. Michael Dewaele and Dr. Tom Verfaillie (KU Leuven) for technical help; Dr. Shinjini Mukherjee (KU Leuven) for help with NMDS analysis. ADG is a recipient of European Molecular Biology Organization (EMBO) Short-term Fellowship (2012–2013); and FWO Postdoctoral Fellowship (2013–2016/2016–2019) from FWO-Vlaanderen, Belgium. MVW is supported by SBO grant (IWT-Flanders); NV is supported by FWO PhD Fellowship. This work is supported by grants from Olivia Hendrickx Research Fund to SVG, Academy of Finland and Sigrid Juselius Foundation to PS, and FWO (G0584.12N, K202313N and GA01111N), KU Leuven (C16/15/073) and Belgian State (IAP7/32) to PA.
Author contributions
ADG, LV, SF, TF performed the main experiments. SVG, JM, MVW, CK, NV provided supplementary experimental/technical support. ADG, SF, PS, PA designed the project/experiments. NG, PDW, SVG, PS provided technical/instrumental support and critically reviewed the manuscript. ADG wrote the manuscript and made the figures. PA critically revised and corrected the manuscript. PA, PDW, PS provided senior supervision/guidance.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Piacentini
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Garg, A., Vandenberk, L., Fang, S. et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ 24, 832–843 (2017). https://doi.org/10.1038/cdd.2017.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2017.15
This article is cited by
-
Shifting the paradigm: engaging multicellular networks for cancer therapy
Journal of Translational Medicine (2024)
-
Targeting of focal adhesion kinase enhances the immunogenic cell death of PEGylated liposome doxorubicin to optimize therapeutic responses of immune checkpoint blockade
Journal of Experimental & Clinical Cancer Research (2024)
-
Clearance of apoptotic cells by neutrophils in inflammation and cancer
Cell Death Discovery (2024)
-
Targeting TNFAIP2 induces immunogenic cell death and sensitizes glioblastoma multiforme to anti-PD-1 therapy
Journal of Neuro-Oncology (2023)
-
Stress-induced inflammation evoked by immunogenic cell death is blunted by the IRE1α kinase inhibitor KIRA6 through HSP60 targeting
Cell Death & Differentiation (2022)
