
Abstract
Vasculogenesis, the establishment of the vascular plexus and angiogenesis, branching of new vessels from the preexisting vasculature, involves coordinated endothelial differentiation, proliferation and migration. Disturbances in these coordinated processes may accompany diseases such as cancer. We hypothesized that the p53 family member p73, which regulates cell differentiation in several contexts, may be important in vascular development. We demonstrate that p73 deficiency perturbed vascular development in the mouse retina, decreasing vascular branching, density and stability. Furthermore, p73 deficiency could affect non endothelial cells (ECs) resulting in reduced in vivo proangiogenic milieu. Moreover, p73 functional inhibition, as well as p73 deficiency, hindered vessel sprouting, tubulogenesis and the assembly of vascular structures in mouse embryonic stem cell and induced pluripotent stem cell cultures. Therefore, p73 is necessary for EC biology and vasculogenesis and, in particular, that DNp73 regulates EC migration and tube formation capacity by regulation of expression of pro-angiogenic factors such as transforming growth factor-β and vascular endothelial growth factors. DNp73 expression is upregulated in the tumor environment, resulting in enhanced angiogenic potential of B16-F10 melanoma cells. Our results demonstrate, by the first time, that differential p73-isoform regulation is necessary for physiological vasculogenesis and angiogenesis and DNp73 overexpression becomes a positive advantage for tumor progression due to its pro-angiogenic capacity.
Similar content being viewed by others

Main
Vascular system formation is one of the earliest events during organogenesis.1 The original vascular plexus is established by vasculogenesis, through differentiation and assembly of mesodermal precursors.2 The angiogenesis process allows the formation of new blood vessels from the existing vasculature and is perturbed in many diseases, including cancer.3 Although efforts have been made to identify factors that control vascular development, the understanding of the molecular networks remains incomplete.
The formation of new capillaries and the remodeling of preexisting blood vessels is linked by signal transduction pathways.4 The members of the p53 family (p53, p73 and p63) coordinate cell proliferation, migration and differentiation, and could act as regulators of vascular development. TP73 function in angiogenesis is quite controversial,5, 6, 7 and it has never been addressed using developmental models.
TP73 has a dual nature that resides in the existence of TA and DNp73 variants. TAp73 is capable of transactivating p53 targets8, 9, 10 whereas DNp73 can act as p53 and TAp73 repressor.11, 12, 13 TP73 final outcome will depend upon the differential expression of the TA/DNp73 isoforms in each cellular context, as they can execute synergic, as well as antagonist, functions.
TP73 role during development is emphasized by the p73-knockout mice (Trp73−/−, p73KO from now on) multiple growth defects.14 These mice, which lack all p73 isoforms, exhibit gastrointestinal and cranial hemorrhages,14 suggestive of vascular fragility. Furthermore, TAp73 directly regulates GATA-1,8 which is essential for endothelial and hematopoietic differentiation.15, 16 This compounded information led us to hypothesize that p73 could be implicated in the regulation of vasculogenesis/angiogenesis.
Regulation of these processes involves a broad range of signaling molecules essential for vascular growth and stability,17 such as vascular endothelial growth factor (VEGF)18 and transforming growth factor-β (TGF-β).19 TGF-β operates as a rheostat that controls endothelial cell (EC) differentiation, having an inhibitory effect on EC migration and proliferation by the TGF-β/TGFRI (ALK5)/Smad2/3 pathway, while the TβRII–ALK5/ALK1 complex activates Smad1/5/8, ID1 expression and a pro-angiogenic state.20, 21, 22
Regulation of the TGF-β and VEGF pathways by p53 family members has been documented.23, 24 However, p73's function in these pathways during development remains largely unexplored. In this work, we have used mouse embryonic stem cells (mESC) and induced pluripotent stem cells (iPSCs) as models that recapitulate early vascular morphogenesis.25, 26, 27 ESC and iPSC form multi-cellular aggregates (embryoid bodies, EBs) that, under appropriate conditions, generate functional EC.28 mESC and iPSC differentiation capacity into ECs has been fully addressed.29, 30 We have also performed retinal vascularization analysis to assess vascular processes in vivo.31, 32
We demonstrate that p73 deficiency perturbs density and stability of mouse retinal development by affecting VEGF and TGF-β signaling. Furthermore, p73 is necessary for the assembly of vascular structures under physiological conditions in mESC and iPSC. Moreover, DNp73 positively affects angiogenesis through regulation of the TGF-β pathway in human umbilical vein cells (HUVEC) and DNp73-overexpression results in enhanced angiogenic potential of B16-F10 melanoma cells.
Results
Defects in retinal vessel migration and vascular morphology in p73-deficient mice
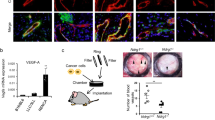
P5-p73KO and wild-type (WT) mice retinas were analyzed to ascertain p73 developmental function in vascular formation using isolectin B4 (IB4) staining. At the leading edge of the vascular plexus (sprouting zone), WT-tip cells and their extending filopodia were directed toward the periphery, guiding the direction of vascular growth (Figure 1a, arrows). In contrast, p73KO retinas had disoriented tip cells (arrows) with long and thin filopodia (Figures 1a, circles and 1b). The central plexus was also abnormal, appearing disorganized, less dense and branched than WT (Figure 1a, right panel). Morphometric analysis showed increased lacunarity and moderate, but highly significant, decrease in vessel covered area, vessel length and branching index, indicating a less dense p73-deficient vascular plexus (Figure 1b).

Absence of p73 perturbs development of the retinal vasculature. (a) Retinas from P5 WT and p73KO mice were stained with isolectin B4 (IB4) to analyze the morphology and orientation (white arrows, left panels) of filopodia at the leading edge of vascularization. Higher-magnification images for each genotype (medium panels) show abundant and disorientated filopodia (circles) and tissue macrophages (arrow heads) in p73KO retinas. Scale bars=50 μm. Right panels illustrate perturbed vascular branching and decreased vascular density in p73KO central retinal plexus compared with WT. (b) Quantification of vessel coverage (percentage of area covered by IB4+ endothelial cells), total vessel length, vascular branching index (branch points/unit area) and lacunarity (distribution of the gap sizes surrounding the object). Representative images were analyzed independently using the AngioTool software (https://ccrod.cancer.gov/confluence/display/ROB2/Home). (c) IB4 retinal flat-mount staining of P5 and P7 retinas. Radio was measured from the optic nerve to the sprouting zone. The spreading of the vasculature toward the periphery is highly significantly reduced in p73KO retinas. (d) Visualization of empty matrix sleeves by IB4 (green) and collagen IV (red) staining, with increased presence of collagen IV sleeve segments lacking endothelial cells (IB4-negative; white arrows) in p73KO retinas. (e) GFAP/IB4 double staining to visualize astrocytes (red) and vasculature (green), respectively. p73KO retinas display a disorganized astrocyte network underlying a chaotic vasculature; tufts are indicated by arrow heads. (f) VEGF-A immunostaining (red) in P5 retinas. Note that VEGF-A expression is markedly decreased in the absence of p73. Yellow arrow heads indicate IB4+/VEGF-A+ microglial cells commonly found at sites of prospective sprout anastomosis. All statistical analysis were performed with data from at least five animals. Bar graphs represent mean±S.D. Equal-variance Student's t-test was performed to evaluate statistical differences. **P<0.01; ***P<0.001
P5 and P7-p73KO vascular plexus had less spreading suggesting that the abnormal filopodia was affecting tip cell migration33 (Figure 1c). IB4 positive (IB4+) macrophages, which act as cellular chaperones for EC fusion,34 appeared as isolated cell clusters, rather than interacting with tip cell filopodia (Figure 1a, middle panel, arrow heads), supporting the idea of a defective guidance and migration in p73-deficient ECs.
We sought to analyze whether p73-loss affected vessel architecture and stability. The vascular basement membrane, marked by type IV-collagen staining, is closely associated with the EC abluminal surface.35 Thus, empty basement membrane sleeves (collagen-IV+/IB4−) identify vessel regression.36 In WT central plexus, most vessels showed collagen-IV+/IB4+ staining with only a few empty collagen-IV sleeves (Figure 1d, arrows). However, in p73KO, empty sleeves were frequent, suggesting that p73 deficiency results in vessel regression.
Next, we examined the resident astrocytic network, which provides guidance cues for ECs by establishing a VEGF-A gradient. Although in WT retinas, astrocytes and ECs closely followed one another, in p73-deficient retinas astrocytes displayed a chaotic reticulation with astrocytes clumps (Figure 1e, white arrows) surrounded by IB4+cells (arrow heads). p73KO retinas had significantly reduced VEGF-A expression and distribution (Figures 1f, yellow arrows and 2a), confirming a hindered pro-angiogenic signaling in the absence of p73. WT retinas showed IB4+/VEGF-A+-microglial cells commonly found where two tip-cells were contacting each other, at sites of prospective sprout anastomosis (Figure 1f, arrow heads). In p73KO, microglia was isolated and not fully interacting with the ECs. Thus, lack of p73 affects the retinal microglia, hindering the VEGF-A gradient for EC migration.
TGF-β signaling is essential for EC organized migration and determines EC function during development.20, 37 We detected a significant decrease in TGF-β1, TGF-βR1 and ALK1 levels in p73KO retinas (Figures 2b–d), and significantly lower ID1 levels (Figure 2e), suggesting an attenuated pro-angiogenic state. Taken together, our data demonstrate that p73 is necessary for retinal vasculature organization and suggest that p73 could be a positive regulator of EC migration, vessel sprouting and tubulogenesis in vivo regulating the VEGF and TGF-β pathways.

Lack of p73 affects TGF-β signaling in vivo. Quantification of VEGF-A expression and analysis of the TGF-β/ALK1/ID1 signaling pathway in P5 retinas from WT and p73KO mice. qRT-PCR analysis demonstrated a significant decrease in VEGF-A expression (a), TGF-β 1 expression (b), TGF-βR1 expression (c), ALK1 expression (d) and ID1 expression levels (e) in the absence of p73. Analysis was performed with data from three independent experiments. Mean±S.D. are represented; equal-variance Student's t-test was performed to evaluate statistical differences. *P<0.05, **P<0.01, ***P<0.001
p73 deficiency results in impaired endothelial differentiation and reduced angiogenic sprouting in mESCs
We used mESC and iPSC to address p73 function in EC differentiation and vasculogenesis. Both TA and DNp73 isoforms were expressed in mESCs at low but consistent levels, with DNp73 being the most abundant isoform (Figure 3a). Stable cell lines with constitutive p73-dominant-negative mutant (DDp73) expression were generated38 (Supplementary Figure 1a). The clones exhibited the typical ES morphology, alkaline phosphatase activity as well as Nanog expression (Supplementary Figure 1b), and were able to differentiate into derivatives of the three germ layers (Supplementary Figure 1c). Although DDp73 cells expressed the mesodermal/endothelial marker CD31, they failed to form vascular networks like the WT cells (Supplementary Figures 1c and d), supporting a possible role for p73 in endothelial morphogenesis.

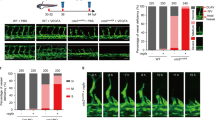
Lack of p73 impairs the formation of vascular structures and endothelial sprouting of mESC and iPSC in the EB endothelial differentiation model. (a) Quantitative analysis of p73 isoform expression (TA and DN) by qRT-PCR in proliferating E14TG2a mESC. Ct-TAp73 : 38.22 and ΔCt-TAp73: 0.099; Ct-DNp73: 33.78 and ΔCt-DNp73: 2.15 (b–c) EBs size, morphology and formation of vascular structures in WT and DDp73 mESC EBs under 2D differentiation culture conditions. (b) EBs were stained for CD31 expression and EB average diameter for individual EBs (n≥10) was calculated after 7 days in vitro (7 DIV). (c) VE-cadherin immunostaining (green) shows that DDp73-EBs did not assemble into vascular networks. (d–f) Angiogenic sprouting in WT and DDp73 mESC EBs under 3D differentiation conditions. (d) Phase contrast images correspond to 7 DIV and 10 DIV EBs. Graph shows percentage of sprouting EBs at day 18. (e) CD31 expression (red) and (f) VE-cadherin expression (green) demonstrate that DDp73 mESC EBs failed to form a branched vascular network. Scale bars=500 μm (e, left panel), 100 μm (e, right panel) and 250 μm (f). (g,h) Angiogenic sprouting in WT and p73KO iPSC-EBs under 3D differentiation conditions. (g) Phase contrast images of 12 DIV iPSC-EBs illustrate sprouts in WT EBs, but not in the p73KO-iPSC-EBs. Graph represents percentage of sprouting EBs at day 18. (h) CD31 expression (red) highlighted that p73-deficient iPSC cannot form vascular structures. Data represent mean values±S.D.; equal-variance Student's t-test was performed to evaluate statistical differences. *P<0.05, **P<0.01
Next, we differentiated mESCs under two-dimensional (2D) and three-dimensional (3D) endothelial-specific conditions (Supplementary Figure 1e). The 3D model recapitulates the developmental steps of vasculogenesis and sprouting angiogenesis, whereas the 2D EBs recapitulate the formation of the primitive vascular network in vivo.25 EBs were immunostained for CD31 and vascular endothelial (VE)-cadherin and EB size and morphology were analyzed (Figures 3b and c). Control EBs had CD31+ cells forming a branching network from the center of the EB to the outer rim (Figure 3b, upper panel), whereas DDp73-EBs had a smaller diameter and their ECs (CD31+ or VE-cadherin+) did not assemble into a vascular network (Figures 3b and c).
In 3D conditions, control EBs formed a vascular network after 7 days in vitro (7 DIV) whereas DDp73-EBs failed to sprout (Figure 3d). After 10 DIV, more than 75% of control EBs showed branched CD31+ and VE-cadherin+ sprouts (Figures 3e and f). However, DDp73-EBs had fewer and shorter sprouts that failed to form a branched network. In DDp73-EBs only some VE-cadherin− cells migrated away from the EB core (Figure 3f, arrows), supporting the notion that p73 deficiency affects EC migration and tube formation.
DDp73 effect on vascular morphogenesis may involve a crossed effect on other p53 family members.38 To rule out this possibility, iPSC reprogrammed from p73KO or WT mouse embryonic fibroblasts were used (Martin-Lopez et al., manuscript under preparation). WT-iPSC-EBs in 3D culture exhibited abundant sprouts that constituted an intricate network (Figures 3g and h). In contrast, only few p73KO-iPSC-EBs had sprouts that were short and unable to form networks (Figure 3g). CD31-immunostaining highlighted the lack of sprouts of p73KO-iPSC-EBs (Figure 3h). To rule out the possibility that reprogramming had affected the genetically modified iPSCs capacity to form 3D vascular sprouts, we also differentiated p53KO and p73KO/p53KO iPSC. p53KO-iPSC EBs generated a profuse vascular network, demonstrating that reprogramming did not affect the capacity of KO cells to differentiate into ECs and form vascular sprouts. Thus, p53 was not required for this process, but lack of p73, even in the context of p53-deficiency, resulted in lack of vascular sprouts (Supplementary Figure 2a), confirming p73 requirement and that the defect in sprout formation was not due to enhanced p53-dependent cell death.
To address whether p73 deficiency could have a non-cell autonomous effect, we performed a co-culture experiment mixing GFP+-mESC with either WT-iPSC or p73KO-iPSC, and differentiating them under 3D endothelial-specific conditions (Supplementary Figure 2b). Although GFP+-mESC/WT-iPS EBs were able to differentiate into ECs that form vascular sprouts, only few GFP+-cells from the GFP+-mESC/p73KO-iPS EBs were capable to migrate and form sprouts. These results supported p73 critical role in vascular morphogenesis, but also indicated that p73 deficiency has a non-cell autonomous effect over neighboring WT-ECs.
To study the mechanism of p73 regulation on EC commitment, endothelial CD31+ cells and differentiated (but non-endothelial) CD31− cells from 12 DIV-EBs were isolated. The number of CD31+ cells from DDp73-EBs was significantly lower than from control-EBs (Figure 4a), indicating that p73 function is required for efficient endothelial differentiation. To determine the contribution of p73 isoforms in this process, we compared TA and DNp73 expression in undifferentiated mESC (gray bars) with that in differentiated CD31+ ECs (black bars). Both isoforms were significantly upregulated in the CD31+ population compared with the undifferentiated mESC, with DNp73 being predominant in ECs (Figure 4b). The non-endothelial differentiated CD31− cells (white bars) showed the opposite pattern of isoform expression (TA>DN). Analysis of EC markers revealed that CD31 and VE-cadherin levels were significantly lower in DDp73-CD31+ cells (Figure 4c), confirming that p73 functional inhibition impairs EC differentiation.

p73 functional inhibition hinders mESC endothelial differentiation, decreases endothelial marker expression and blunts the pro-angiogenic state of the obtained endothelial cells. (a) CD31 positive cells were isolated with magnetic beads and the obtained cell number was quantified. (b) qRT-PCR analysis of p73 isoform expression in proliferating undifferentiated E14Tg2a (square bars), differentiated non-endothelial CD31− negative cells (CD31−, white bars) and differentiated CD31 positive endothelial cells (CD31+, black bars) demonstrated that TA and DNp73 are differentially regulated during mESC endothelial differentiation. (c–e) Quantitative qRT-PCR analysis of endothelial cell marker expression (c), VEGF signaling (d) and TGF-β/ID1 signaling (e), in CD31− (white bars) and CD31+ cells (black bars) isolated from either control or p73DD-EBs. Bars represent mean values±S.D.; experiments were repeated twice; equal-variance Student's t-test was performed to evaluate statistical differences. *P<0.05, **P<0.01, ***P<0.001
So far our results suggest that p73 could be regulating some of the molecular pathways that control endothelial differentiation, migration and/or assembly of endothelial precursors. To address this, we analyzed VEGF-A and TGF-β signaling, both altered in p73KO retinas. In agreement with previous publications,6, 7 DDp73-CD31+ cells expressed lower levels of VEGF-A (Figure 4d). The expression of VEGFR1, a decoy of angiogenic signaling, was significantly higher in DDp73-CD31+, while VEGFR2 expression, the VEGF signaling transducer, was lower (Figure 4d). The VEGFR2/VEGFR1 ratio, which may be considered as a pro-angiogenic indicator,39 was about five times lower in DDp73-CD31+. Furthermore, we detected a significant reduction in TGF-β expression, with a concomitant attenuation in ID1 levels, in DDp73-CD31+ cells (Figure 4e), all consistent with a decrease in the number of cells committed to endothelial lineage.40
Taken together, our data indicate that the lack of p73 in mESC attenuates endothelial differentiation, diminishes the angiogenic potential of the differentiated cells and hinders angiogenic sprouting of 3D-EBs, therefore demonstrating that p73 is a positive regulator of in vitro vasculogenesis/angiogenesis, at least in part, by modulating pro-angiogenic signaling through the VEGF and TGF-β pathways.
DNp73 silencing interferes with HUVECs to undergo migration and tubulogenesis
To show that p73 has a specific role in EC biology, independently of its possible effect in other cell types within the retina or in the EBs, we used HUVECs. p73 isoforms were differentially expressed in HUVECs, with DNp73 expression being about 2.7 times higher than TAp73 (2.7±0.0002; Ct-TAp73: 36.70 and ΔCt-TAp73: 0.00019; Ct-DNp73: 35.60 and ΔCt-DNp73: 0.00045). We suppressed p73 function with DDp73 or by an RNA interference knockdown (p73KD) using previously validated small interfering RNA (siRNA) oligos: p73i.4 for total-p73KD, TAp73i for specific TAp73 or DNp73i for DNp73-KD.8 The maximal interference using these siRNAs occurs 72 h after transfection, lasting more than 48 h.8 All angiogenic assays were carried out during this period. It is important to point out that p73KD did not affect HUVEC proliferation kinetics within this time frame (Supplementary figure 3).
HUVECs were transfected with either DDp73 or siRNAi-p73i.4 and tube formation was monitored and quantitatively analyzed. p73-deficient HUVEC showed reduced tube formation capacity and networks with reduced area and fewer branching points and loops (Figures 5a and b). Total-p73KD and DNp73 in particular, but not TAp73 alone, provoked a significant reduction in all the analyzed parameters (Figure 5c). These parameters revealed a defective capacity of the interfered ECs to migrate and assemble into tubular structures, indicating that DNp73 deficiency results in an impaired endothelial morphogenesis. On the contrary, under normoxic conditions, there was no significant effect on any of the analyzed parameters after TAp73 KD.

p73 function, and DNp73 in particular, is required for tube morphogenesis in HUVEC. Tube formation assays on Matrigel in HUVEC upon (a) p73 functional inhibition – DDp73, (b) knockdown of total p73 – p73i.4 or (c) specific knockdown of TA or DNp73 isoforms – Tap73i and DNp73i, respectively. Tube formation was monitored microscopically and images were analyzed using WimTube software (Wimasis GmbH). Covered area, total tubes, total tube length, total branching points and total number of loops are shown as representative parameters. Experimental data were normalized to the control (a) or to the scrambled (Scr.; b,c). Data represent mean values±S.D.; n≥3, experiments were repeated at least three times; equal-variance Student's t-test was performed to evaluate statistical differences: *P<0.05, **P<0.01, ***P<0.001
Next, p73 role in EC migration was addressed by wound healing or ‘scratch’ assays.41 The percentage of wound closure was significantly reduced upon functional inhibition or total p73KD (Figure 6a). DNp73KD, but not TAp73KD alone, attenuated wound closure (Figure 6c), indicating that DNp73 is a positive regulator of EC migration. We examined TA and DN expression kinetics after the wounding. As shown in Figure 6b, both isoforms were upregulated to a similar extent, indicating that the EC-characteristic DNp73>TAp73 ratio was maintained. To conclusively demonstrate that DNp73 function was necessary for EC migration, we performed a phenotype recovery experiment. DNp73 overexpression in siRNA-p73i4 cells restored the migratory capacity (Figure 6d), confirming DNp73 as a positive regulator of EC migration.

p73, and DNp73 in particular, acts as a positive regulator of HUVEC migration. Wound healing assays upon (a) p73 functional inhibition (DDp73, upper panel) and total p73 knockdown (p73i.4, lower panel) or (c) specific knockdown of TA or DNp73 (TAp73i and DNp73i, respectively). Endothelial cell migration into the ‘wound’ denuded area was monitored microscopically and the percentage of wound closure after 10 h was calculated relative to the control. (b) Expression kinetics of p73 isoforms during wound-healing assays were analyzed by qRT-PCR. (d) HUVEC were cotransfected with p73i4 siRNA (silencing of total p73) or scrambled oligos (Scr.) together with a DNp73 expressing plasmid, and a wound healing assay was performed. Data represent mean values±S.D.; n=3; experiments were repeated at least three times; equal-variance Student's t-test was performed to evaluate statistical differences. *P<0.05, **P<0.01, ***P<0.001
To elucidate the molecular mechanisms underlying the DNp73 regulation, we analyzed the VEGF and TGF-β pathways. We examined phosphorylated ERK1/2 as a downstream indicator of VEGF signaling42 and, as published,43 both p73 isoforms regulated this pathway (Figure 7a). VEGF-A levels were strongly downregulated upon total p73 and DNp73 silencing, (Figures 7a and b), and barely affected after TAp73KD (Figure 7b), pointing to DNp73 as the predominant regulator on HUVEC normoxic cultures. However, transcriptional analysis indicated that ectopic DNp73 expression did not induce VEGF-A promoter activity (Supplementary Figure 4a), suggesting the existence of other downstream posttranscriptional regulatory mechanisms.

Total p73 and specific DNp73 knockdown in HUVEC led to a decrease in the downstream angiogenic signaling. The indicated knockdown experiment was performed in HUVEC (p73i.4: total p73 knockdown; TAp73i: TAp73 specific knockdown; DNp73i: DNp73 specific knockdown). (a) Expression of P-ERK1/2, ERK, VEGF-A, P-Smad1/5 and Smad1 were analyzed by western blot and protein levels were quantified using Quantity One software (Bio-Rad). (b) Quantitative analysis of VEGF-A mRNA levels was done by qRT-PCR. (c, d) Quantitative analysis of TGF-β1, ALK1 and ID1 mRNA levels was done by qRT-PCR. Data represent mean values±S.D.; experiments were repeated twice; equal-variance Student's t-test was performed to evaluate statistical differences. *P<0.05, **P<0.01, ***P<0.001
On the other hand, analysis of TGF-β1/ALK1/ID1 pathways showed that only DNp73 is upregulated by TGF-β1 treatment in a dose-dependent manner (Figure 7c), linking p73 differential regulation to the angiogenic TGF-β1 signaling axis. Indeed, DNp73 silencing resulted in significantly reduced p-SMAD1/5 and total SMAD1 proteins (Figure 7a), and TGF-β1, ALK1 and ID1 levels (Figure 7d), unequivocally demonstrating that DNp73 is required for TGF-β1/ALK1/ID1 signaling in ECs.
We have demonstrated p73 requirement for EC biology and vasculogenesis and, in particular, that DNp73 regulates EC migration and tube formation. However, our in vivo data also indicated that p73 deficiency could affect non-EC resulting in reduced pro-angiogenic milieu. We sought to address whether DNp73 could affect tumor angiogenesis, using the syngeneic transplantation model of B16-F10 melanoma.44 Conditioned media (CM) was prepared from B16-F10 melanoma cells with either silenced (Supplementary figure 4b) or transiently overexpressed DNp73 (Supplementary figure 4c), and its angiogenic effect on HUVEC was tested in scratch assays. DNp73-deficient CM retard, but do not suppress, HUVEC wound healing, while DNp73-overexpressed CM failed to accelerate the healing rate. These data could be interpreted as that DNp73 does not alter B16-F10 cells angiogenic potential. However, we hypothesized that DNp73 positively regulates angiogenesis by repressing an anti-angiogenic factor. In this scenario, the previous assay might not be sensitive enough. Thus, constitutively expressing DNp73 clones (F10-DNp73), as well as vector controls, were generated. Cells were injected subcutaneously into C57BL/6 mice and palpable tumors were collected 2 or 3 weeks post injection. DNp73 protein levels in B16-F10 cells under normoxic culture conditions were compared with those from 2 or 3 weeks tumor extracts. As observed in Figure 8a, cultured cells had very low DNp73 levels, but these were sensibly increased in tumors, indicating that DNp73 expression was favored and upregulated in the tumor environment by transcriptional independent mechanisms. Two weeks post injection B16-DNp73 tumors had higher vessel density, quantified by the number of CD31+ structures (blue arrows), and higher mitotic index and VEGF-A expression than controls (Figures 8b–d). As microvessel density reflects the intercapillary distance determined by the net balance between the pro- and anti-angiogenic factors that stimulate vessel growth,45 we can conclude that DNp73 expression enhances the angiogenic potential of B16-F10 cells. Therefore, our data demonstrate TP73 requirement for endothelial differentiation, angiogenesis and vasculogenesis under physiological conditions, and the role of DNp73 as a pro-angiogenic factor in EC and non-ECs.

Constitutive DNp73 expression in the B16-F10 mouse melanoma cell line results in enhanced tumor vascularization. (a) DNp73 expression in B16-F10 cell extracts and tumor extracts were derived from a syngenic transplantation experiment. B16-F10 parental cells, or stable clones transfected with either DNp73 (B16-F10 DN) or vector control (B16-F10 C) were injected in C57BL/6 mice and tumors were collected after the indicated week number (2 and 3). (b) Histology and immunohistochemistry analysis of paraffin-embedded tissue sections of melanomas. Haematoxylin and eosin (HE) staining, as well as CD31 staining, revealed an increase in vessel density (blue arrows) in B16-F10 DN sections. Ki-67 proliferation antigen expression is also shown. Scale bar=500 μm (HE and CD31 staining); 100 μm (Ki-67). (c) Quantification of vessel number (10 high-power fields per section; 5 sections per genotype) and Ki-67+ cells (10 high-power fields; objective x40; 1000 cells/slide) from images in b. (d) VEGF-A expression in paraffin sections from B16-F10 and B16-F10 DN melanomas
Discussion
p73 potential role in vascular biology has been addressed before, but not using developmental models. Here, we revealed TP73 as a regulator of VEGF and TGF-β signaling in vasculogenesis/angiogenesis under physiological conditions. Our in vivo data demonstrate that p73 is necessary for proper development of the retinal vasculature and to establish the appropriate pro-angiogenic signaling. The data presented here strongly supports the role of p73 as positive regulator of EC migration, vessel sprouting and tubulogenesis in vivo. However, our in vivo data indicated that p73 deficiency also affects non-ECs, hindering the guidance cues secreted by them, resulting altogether in reduced pro-angiogenic milieu.
To address p73 function we used mESC and iPSC models. The p73KO-EBs had smaller size and lower expression of endothelial markers; this could reflect a defect in cellular fate46 but also, supports the idea that p73 function is required to effectively fulfill the endothelial differentiation process. In 3D EBs, p73 functional inhibition affected both the differentiation capacity and the ability to migrate and assemble into vascular networks. Similar altered vascular patterns have been described in EBs upon disruption of genes like VEGF-A,47, 48 its cellular receptor VEGFR2,49, 50 or the adhesion molecule VE-cadherin,51 among others. Consistently, p73 deficiency resulted in a significant decreased expression of VEGF-A and, more importantly, a lower VEGFR2/VEGFR1 ratio, which could be considered as a pro-angiogenic indicator. Moreover, in HUVEC, analysis of the morphogenic potential and migration capacity demonstrated that p73 function, DNp73 in particular, is necessary for the full angiogenic potential and the pro-angiogenic signaling through the VEGF pathway of these cells. These results are in agreement with the recently proposed VEGF regulation by the p53 family members.52 In this regard, p53 seems to have both a repressive and a required activating function on VEGF promoter activity.23 Similarly, p73 role in VEGF-A regulation is complex and dependent upon the environmental conditions. Although our data concerning DNp73 function seem to be clear, TAp73 data appears controversial. On one hand, endogenous attenuation of TAp73 expression in HUVEC did not decrease tube formation or migration, but rather seem to have the opposite effect, hinting to a TAp73 anti-angiogenic effect. On the other hand, TAp73KD resulted in reduced VEGF-A mRNA levels, and TAp73 overexpression induced VEGF promoter activity in a luciferase reporter assay (data not shown), implying a TAp73 pro-angiogenic function. This scenario might reflect a context-dependent regulation of TAp73 and of the nature of its angiogenic role. Nevertheless, the molecular mechanism of this regulation remains elusive.
The TGF-β pathway, inducer of EC fate in ESC, is another important regulator of angiogenic signaling and vascular homeostasis.40 TGF-β signaling operates as a rheostat that controls EC differentiation having both inhibitory and stimulating effects.53 EC sprouting has been explained by a model in which TGF-β synergizes with VEGF and Notch to drive migration, partly through induction of ID1, required for EC migration.54 The link between the p53 family members and TGF-β signaling in somatic cells has been previously established and it has been suggested, but never addressed, that the p53 homologs might interact with TGF-β in development.24 In this work we show that the absence of p73 results in a significant decrease in TGF-β1 and ID1 levels in all the models analyzed. Moreover, in HUVEC, specific DNp73KD has a strong and significant effect, downregulating EC migration and the TGF-β1/ALK1/ID1 signaling axis. These data demonstrate, for the first time, that DNp73 is a positive modulator of EC migration and angiogenic potential under physiological conditions through the regulation of the TGF-β pathway. These DNp73 functions are in agreement with recent data indicating that DNp73 drives migration and invasion of non-metastatic melanoma cells facilitating the epithelial–mesenchymal transition phenotype of these cells.55
It has become increasingly clear that tumor vessel abnormalities contribute to create a microenvironment that facilitates tumor progression. Therefore, it is highly relevant to identify novel molecules involved in angiogenesis and to integrate them in the molecular networks that orchestrate vascular morphogenesis. The described DNp73 role is in accordance with the proposed TP73 function in oncogenesis, which seems to arise from the preferential expression of the DNp73 isoforms. Indeed, our data demonstrate that DNp73 protein levels are upregulated in the tumor environment by a transcriptional independent process. DNp73 upregulation is a frequent abnormality in solid cancers but, unlike p53, p73 is rarely mutated.56 In this regard, DNp73 capacity to enhance the angiogenic potential of tumor cells will provide a positive advantage for tumor progression.
These results suggest that in the analyzed cellular milieu (standard normoxic cell culture conditions), DNp73, but not TAp73, is the positive regulator in EC migration and morphogenesis. Taken together, our data reveal, for the first time, that p73 is a positive regulator of EC migration, vessel sprouting and tubulogenesis under physiological in vivo and in vitro conditions of angiogenesis, by modulating TGF-β and VEGF signaling pathways. Moreover, the differential p73 isoform regulation, as well as the context-dependent p73 function, is necessary for the physiological control of the angiogenesis and vasculogenesis processes, and its deregulation might be an important event in tumor angiogenesis.
Materials and Methods
Mice husbandry and animal breeding
Animal experiments were conducted in agreement with European (Council Directive 2010/63/UE) and Spanish regulations (RD 53/2013) on the protection of experimental animals with the appropriate institutional committee approval. Mice heterozygous for Trp73 on a mixed background C57BL/6_129/svJae14 were backcrossed to C57BL/6, at least five times, to enrich for C57BL/6 background. Mice were genotyped as described before.14, 57
Retina dissection and flat-mount preparation
Postnatal mice on day 5 and 7 were decapitated, and eyes were enucleated and fixed in 4% paraformaldehyde (PFA) in PBS for 40 min. Retinas were dissected and washed with PBS for further processing. For whole-mount preparations, retinas were blocked in PBLEC buffer (0.1 mM CaCl2, 0.1 mM MnCl2, 0.1 mM MgCl2, 1% TX-100 in PBS, pH 6.8) for 1 h at room temperature (RT), followed by incubation with 2 μg/ml biotinylated isolectin B4 (Sigma, St. Louis, MO, USA) in PBLEC, at 4 °C overnight. Retinas were washed five times in PBS and incubated with 4 μg/ml Streptavidin-Alexa488 (Invitrogen, Carlsbad, CA, USA) in 5% fetal bovine serum (FBS), 3% BSA, 0.1% TX-100 in PBS, for 2 h at RT. After the washes, retinas were blocked with 3% BSA for 2 h at RT and incubated with primary antibody anti GFAP 1 : 200 (Thermo Scientific, Waltham, MA, USA), anti Colagen IV 1 : 100 (Serotec, Kidlington, Oxford, UK) or anti VEGFA 1 : 100 (Abcam, Cambridge, UK), in PBS, 3% BSA, 5% FBS, at 4 °C overnight. On the next morning, retinas were washed five times with PBS at RT (each washing for 30 min) and incubated thereafter with secondary antibody (donkey anti-rabbit Alexa594, Molecular Probes, Carlsbad, CA, USA) in PBS, 3% BSA, 5% FBS. After PBS washing, retinas were mounted with Fluoromont G (Electron Microscopy Sciences, Hatfield, PA, USA). Representative images were analyzed independently using the AngioTool software (https://ccrod.cancer.gov/confluence/display/ROB2/Home). At least five animals from each individual genotype were used for the statistical analysis by Student's t-test.
mESC and iPSCs culture, EB formation and endothelial 2D and 3D differentiation
iPSC have been generated from early passage fibroblast from various Trpp73−/− E13.5 embryos or WT littermates (C57BL/6 background) by reprogramming using the piggyBac transposon system (kindly provided by Dr. Hämäläinen, Biomedicum Stem Cell Centre, Finland).58 Eight independent WT-iPSC clones and five p73KO iPSC clones were generated and cultured as previously described (Martin-Lopez et al., manuscript under preparation).
Murine ESC (E14TG2a cells, DDp73-E14TG2a and GFP+mESC) and iPSC were cultured on mouse fibroblast feeder cells in ESC medium (Dulbecco’s modified Eagle’s medium/Glutamax; Gibco, Carlsbad, CA, USA, 25 mM HEPES, 1.2 mM sodium pyruvate, 19 mM monothioglycerol; Sigma, 15% FBS, and 1000 units/ml leukemia inhibitory factor (LIF)).
Generation of EBs and endothelial differentiation of mESC and iPSC
The differentiation protocols were identical for ESC and for iPSC. For differentiation assays, EBs were formed by the hanging drop method as previously described.59 Briefly, feeder cells were removed from either ESC or iPSC cultures by tripsinizing the cells and plating the obtained suspension on 1% gelatin-coated dishes in ESC medium for 30 min at 37 °C, 5% CO2. The supernatant was collected and cells were counted. To form the EBs by hanging drops, a cell suspension of 60 000 cell/ml was prepared in EB medium (ESC medium without LIF) and 20 μl drops were placed into a Petri dish lid. The drops were incubated at 37 °C, 5% CO2, for 4 days. Then, EBs were flushed down from the lid with EBs medium and transferred to either gelatin-coated surfaces for 2D culture or into a collagen I matrix for 3D differentiation. For 2D, EBs were placed in eight-well glass culture slides (BD Falcon, Becton Dickinson, Franklin Lakes, NJ, USA) in EB differentiation media (EB medium with 50 ng/ml VEGF165; Peprotech, Princeton, NJ, USA). For 3D differentiation, EBs were embedded into a collagen I suspension composed of Ham’s F12 medium (Promocell, Heidelberg, Germany), 6.26 mM NaOH, 20 mM HEPES, 0.117% NaHCO3, 1% glutamax I (Gibco) and 1.5 mg/ml collagen I (Advanced Biomatrix, San Diego, CA, USA) supplemented with 100 ng/ml VEGF165. In both protocols EB differentiation media was changed every 2 days. After 8–12 days for 2D differentiation, or 12–20 days for the 3D protocol, the differentiated EBs were processed and analyzed.
Immunostaining of EBs
EBs on glass slides (days 8–12) or EBs in collagen gels (days 12–20) were fixed with 3.7% PFA and processed for whole-mount immunohistochemical staining as previously described.59 The samples were analyzed in a LSM 510 META confocal microscope (Carl Zeiss, Jena, Germany). The following dilutions and primary antibodies were used: rat anti CD31 1 : 1000 (Becton Dickinson), goat anti VE-Cadherin 1 : 500 (R&D Biosystems, Minneapolis, MN, USA), rabbit anti GFP 1 : 100 (Santa Cruz Biotechnology, Dallas, TX, USA), mouse anti Tuj1 1 : 1000 (Covance, Princeton, NJ, USA), mouse anti AFP 1 : 1000 (Inmunostep, Salamanca, Spain), secondary antibodies were: Alexa568 donkey anti-rat IgG, Alexa488 donkey anti-goat IgG (Molecular Probes), FITC anti-IgG rabbit (Jackson Immunoresearch, West Grove, PA, USA) and horseradish peroxidase-conjugated streptavidin (Vector, Burlingame, CA, USA).
Magnetic sorting to enrich for CD31 positive cells
CD31 positive cells were isolated at day 12 from differentiating EBs using DYNAL beads (Invitrogen) coated with rat anti-mouse CD31 antibody (Becton Dickinson), as previously described.60 CD31 positive cells were collected using a magnetic particle concentrator (Dynal MPC, Carlsbad, CA, USA, 120-20) following the manufacturer’s instructions. The supernatants, which contain the CD31 negative cells, were used as control.
HUVEC culture, transfection and siRNA silencing
HUVEC were kindly provided from Biomar Microbial Technologies (Leon, Spain). HUVEC were cultured on gelatin-coated dishes in endothelial basal medium, EBM (Lonza, Basel, Switzerland), with full supplements (Lonza) and they were used from passage 2 to passage 8.
Transient transfections of pcDNA3·DDp73 or pcDNA3 expression vectors were performed on 60–80% confluent HUVEC monolayers using Lipofectamine2000 Transfection Reagent (Invitrogen) following the manufacturer’s instructions. For gene silencing experiments, 100 nM of the indicated RNA interference oligos (Supplementary Table I) were transfected with Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions. Medium was changed 5–24 h later and maximal silencing was detected after 72 h of siRNA transfection.
HUVEC treatment with TGF-β
HUVEC were cultured for 12 h on gelatin-coated well-plates (15 800 cells/cm2) in EBM with full supplements (10% FBS). Cells were serum starved in EBM with full supplements and 0.2% FBS for 24 h. Then, cultures were treated with 0.5–50 ng/ml human TGF-β (Peprotech) and RNA was isolated after 24 h for qRT-PCR analysis.
In vitro migration assay
Wound healing or scratch assays were performed on HUVEC confluent monolayers either 48 h after plasmid transfection or 72 h after RNA interference. The monolayers were wounded with a micropipette tip making a straight scratch. After the scraping, cells were washed with PBS and fresh medium was added. Cell cultures were imaged at 0 and 10 h after the scratch, using a phase contrast microscope (Leica DMI 3000B, Wetzlar, Germany) and a digital camera (Leica DFC 310FX, Wetzlar, Germany). Wound width was measured using Image J software (NIH). Wound closure rate was calculated as follows: (wound width T0h–wound width T10h) × 100/wound width T10h.
In vitro tube formation assay
A 96-well plate was coated with 100 μl Matrigel matrix (Becton Dickinson) per well, and incubated at 37 °C for 30 min to allow gelling. HUVEC were seeded at a density of 125 000 cell/ml in 200 μl of EBM medium. After 6 h, images were acquired with a phase contrast microscope (Leica) and analyzed using WimTube software (Wimasis GmbH, Munich, Germany).
RNA isolation and real-time RT-PCR analysis
Total RNA from mice retinas or cultured cells was extracted with TRI reagent solution (Ambion, Austin, TX, USA) according to the manufacturer’s instructions. Up to 2 μg of total RNA was used to synthesize cDNA using High Capacity RNA-to-cDNA kit (Applied Biosystems, Carlsbad, CA, USA). Gene expression was detected by qRT-PCR in a StepOnePlus Real-Time PCR System (Applied Biosystems) using FastStart Universal SYBR Green Master (ROX; Roche, Basel, Switzerland). All protocols were performed according to the manufacturer’s instructions. Primers sequences and conditions were described before61, 62 and are indicated in the Supplementary Table II. mRNA expression levels were expressed as 2ΔCt and normalized to 18S.
Western blot analysis
Western blotting was performed as previously described43 with the following primary antibodies: rabbit anti-pSmad1/5 (Ser463/465) 1 : 1000 (Cell Signaling, Danvers, MA, USA), rabbit anti Smad1 1 : 1000 (Cell Signaling), mouse anti-pERK 1 : 1000 (Santa Cruz Biotechnology), rabbit anti-ERK1 : 10 000 (Santa Cruz Biotechnology), rabbit anti-VEGF-A 1 : 1000 (Abcam) mouse anti DNp73 1 : 1000 (Imgenex, San Diego, CO, USA), and rabbit anti Nanog 1 : 1000 (Chemicon, Billerica, TX, USA) followed by the appropriate HRP-conjugated secondary antibodies (Pierce, Waltham, MA, USA). The enhanced chemiluminescence was detected with Super Signal West-Pico Chemiluminiscent Substrate (Pierce).
Syngeneic transplantation model, histology and immunohistochemistry
We used the syngeneic transplantation model of B16-F10 melanoma injected subcutaneously in C57BL/6 mice described before.44 The B16-F10 murine melanoma cell line was a gift from Instituto Biomar SA, Leon, Spain (original source: American Type Culture Collection, Barcelona, Spain) and were maintained in RPMI 1640 (Sigma) supplemented with 10% FBS (Sigma), 2 mM L-Glutamine (Gibco) and 100 IU/ml penicillin and 100 IU/ml streptomycin (Sigma). Cells in log-phase growth were harvested by brief trypsinization and were injected subcutaneously in the dorsal neck region of C57BL/6 mice (Charles River Laboratories, Saint Germain Nuelles, France) in 0.10 ml PBS (4 × 105 cells/0.2 ml PBS; two injection/mice; two mice per group). Mice were killed by cervical dislocation when the tumor size was at least 200 mm3, that is, 10–14 days post-inoculation. Some mice were killed 3 weeks after implantation of the cells.
Formalin-fixed tumor sections were stained with hematoxylin and eosin (Sigma) or immunostained with the following antibodies: anti-CD31 (DAKO, Glostrup, Denmark), anti-VEGF-A (Abcam) and Ki-67 (Merk, Darmstadt, Alemania). The mitotic index was determined by quantifying Ki-67+ cells 10 high-power fields (objective x40; 1000 cells/slide).
Samples and statistical analyses
The analyses were performed with data from three independent experiments, with at least five mice from each genotype in the case of in vivo studies. Statistical analyses were performed using the Student’s two-tailed t-test (*P<0.05, **P<0.01, ***P<0.001). The mean±S.D. of each value is represented as indicated in each figure legend.
Abbreviations
- EB:
-
embryoid body
- EC:
-
endothelial cells
- HUVEC:
-
human umbilical vein cells
- iPSC:
-
induced pluripotent stem cells
- mESC:
-
mouse embryonic stem cells
- p73KO:
-
p73−/−
- TGF-β:
-
transforming growth factor-β
- VEGF:
-
vascular endothelial growth factor
- WT:
-
wild type
References
Heinke J, Patterson C, Moser M . Life is a pattern: vascular assembly within the embryo. Front Biosci (Elite Ed) 2012; 4: 2269–2288.
Risau W, Flamme I . Vasculogenesis. Annu Rev Cell Dev Biol 1995; 11: 73–91.
Carmeliet P, Jain RK . Angiogenesis in cancer and other diseases. Nature 2000; 407: 249–257.
Blagosklonny MV . Apoptosis, proliferation, differentiation: in search of the order. Semin Cancer Biol 2003; 13: 97–105.
Salimath B, Marme D, Finkenzeller G . Expression of the vascular endothelial growth factor gene is inhibited by p73. Oncogene 2000; 19: 3470–3476.
Vikhanskaya F, Bani MR, Borsotti P, Ghilardi C, Ceruti R, Ghisleni G et al. p73 Overexpression increases VEGF and reduces thrombospondin-1 production: implications for tumor angiogenesis. Oncogene 2001; 20: 7293–7300.
Guan M, Peng HX, Yu B, Lu Y . p73 Overexpression and angiogenesis in human colorectal carcinoma. Jpn J Clin Oncol 2003; 33: 215–220.
Marques-Garcia F, Ferrandiz N, Fernandez-Alonso R, González-Cano L, Herreros-Villanueva M, Rosa-Garrido M et al. p73 plays a role in erythroid differentiation through GATA1 induction. J Biol Chem 2009; 284: 21139–21156.
Jost CA, Marin MC, Kaelin WG Jr . p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 1997; 389: 191–194.
Jung MS, Yun J, Chae HD, Kim JM, Kim SC, Choi TS et al. p53 and its homologues, p63 and p73, induce a replicative senescence through inactivation of NF-Y transcription factor. Oncogene 2001; 20: 5818–5825.
Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S . Possible oncogenic potential of DeltaNp73: a newly identified isoform of human p73. Cancer Res 2002; 62: 636–641.
Zaika AI, Slade N, Erster SH, Sansome C, Joseph TW, Pearl M et al. Np73, a dominant-negative inhibitor of wild-type p53 and TAp73, is up-regulated in human tumors. J Exp Med 2002; 196: 765–780.
Buhlmann S, Putzer BM . DNp73 a matter of cancer: mechanisms and clinical implications. Biochim Biophys Acta 2008; 1785: 207–216.
Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000; 404: 99–103.
Adamo L, Garcia-Cardena G . The vascular origin of hematopoietic cells. Dev Biol 2012; 362: 1–10.
Iacovino M, Chong D, Szatmari I, Hartweck L, Rux D, Caprioli A et al. HoxA3 is an apical regulator of haemogenic endothelium. Nat Cell Biol 2011; 13: 72–78.
Papetti M, Herman IM . Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol 2002; 282: C947–C970.
Ferrara N, Gerber HP, LeCouter J . The biology of VEGF and its receptors. Nat Med 2003; 9: 669–676.
Bertolino P, Deckers M, Lebrin F, ten Dijke P . Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest 2005; 128: 585s–590s.
Pardali E, Goumans MJ, ten Dijke P . Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol 2010; 20: 556–567.
Goumans MJ, Liu Z, ten Dijke P . TGF-beta signaling in vascular biology and dysfunction. Cell Res 2009; 19: 116–127.
Wang X, Abraham S, McKenzie JA, Jeffs N, Swire M, Tripathi VB et al. LRG1 promotes angiogenesis by modulating endothelial TGF-beta signalling. Nature 2013; 499: 306–311.
Farhang Ghahremani M, Goossens S, Haigh JJ . The p53 family and VEGF regulation: "It's complicated". Cell cycle 2013; 12: 1331–1332.
Cordenonsi M, Dupont S, Maretto S, Insinga A, Imbriano C, Piccolo S . Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell 2003; 113: 301–314.
Jakobsson L, Kreuger J, Claesson-Welsh L . Building blood vessels—stem cell models in vascular biology. J Cell Biol 2007; 177: 751–755.
Yoshida Y, Yamanaka S . Recent stem cell advances: induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation 2010; 122: 80–87.
Kohler EE, Wary KK, Li F, Chatterjee I, Urao N, Toth PT et al. Flk1+ and VE-cadherin+ endothelial cells derived from iPSCs recapitulates vascular development during differentiation and display similar angiogenic potential as ESC-derived cells. PLoS One 2013; 8: e85549.
Yurugi-Kobayashi T, Itoh H, Yamashita J, Yamahara K, Hirai H, Kobayashi T et al. Effective contribution of transplanted vascular progenitor cells derived from embryonic stem cells to adult neovascularization in proper differentiation stage. Blood 2003; 101: 2675–2678.
Narazaki G, Uosaki H, Teranishi M, Okita K, Kim B, Matsuoka S et al. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation 2008; 118: 498–506.
Taura D, Sone M, Homma K, Oyamada N, Takahashi K, Tamura N et al. Induction and isolation of vascular cells from human induced pluripotent stem cells—brief report. Arterioscler Thromb Vasc Biol 2009; 29: 1100–1103.
Uemura A, Kusuhara S, Katsuta H, Nishikawa S . Angiogenesis in the mouse retina: a model system for experimental manipulation. Exp Cell Res 2006; 312: 676–683.
Poulaki V . Angiogenesis assays. Methods Mol Biol 2011; 731: 345–358.
Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 2003; 161: 1163–1177.
Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S et al. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010; 116: 829–840.
Figg W, Folkman J . Angiogenesis: An Integrative Approach from Science to Medicine. Springer: NY, USA, 2008.
Baffert F, Le T, Sennino B, Thurston G, Kuo CJ, Hu-Lowe D et al. Cellular changes in normal blood capillaries undergoing regression after inhibition of VEGF signaling. Am J Physiol Heart Circ Physiol 2006; 290: H547–H559.
Allinson KR, Lee HS, Fruttiger M, McCarty JH, Arthur HM . Endothelial expression of TGFbeta type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLoS One 2012; 7: e39336.
Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000; 407: 645–648.
Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L . Signal transduction by vascular endothelial growth factor receptors. Biochem J 2011; 437: 169–183.
Puceat M . TGFbeta in the differentiation of embryonic stem cells. Cardiovasc Res 2007; 74: 256–261.
Wong MK, Gotlieb AI . In vitro reendothelialization of a single-cell wound. Role of microfilament bundles in rapid lamellipodia-mediated wound closure. Lab Invest 1984; 51: 75–81.
Abedi H, Zachary I . Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J Biol Chem 1997; 272: 15442–15451.
Fernandez-Garcia B, Vaque JP, Herreros-Villanueva M, Marques-Garcia F, Castrillo F, Fernandez-Medarde A et al. p73 cooperates with Ras in the activation of MAP kinase signaling cascade. Cell Death Differ 2007; 14: 254–265.
Fidler IJ, Nicolson GL . Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst 1976; 57: 1199–1202.
Hlatky L, Hahnfeldt P, Folkman J, Li Y, Berthiaume F, Toner M . Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn't tell us. J Natl Cancer Inst 2002; 94: 883–893.
Park J, Cho CH, Parashurama N, Li Y, Berthiaume F, Toner M et al. Microfabrication-based modulation of embryonic stem cell differentiation. Lab Chip 2007; 7: 1018–1028.
Bautch VL, Redick SD, Scalia A, Harmaty M, Carmeliet P, Rapoport R . Characterization of the vasculogenic block in the absence of vascular endothelial growth factor-A. Blood 2000; 95: 1979–1987.
Ng YS, Ramsauer M, Loureiro RM, D'Amore PA . Identification of genes involved in VEGF-mediated vascular morphogenesis using embryonic stem cell-derived cystic embryoid bodies. Lab Invest 2004; 84: 1209–1218.
Hidaka M, Stanford WL, Bernstein A . Conditional requirement for the Flk-1 receptor in the in vitro generation of early hematopoietic cells. Proc Natl Acad Sci USA 1999; 96: 7370–7375.
Jakobsson L, Kreuger J, Holmborn K, Lundin L, Eriksson I, Kjellén L et al. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev Cell 2006; 10: 625–634.
Vittet D, Buchou T, Schweitzer A, Dejana E, Huber P . Targeted null-mutation in the vascular endothelial-cadherin gene impairs the organization of vascular-like structures in embryoid bodies. Proc Natl Acad Sci USA 1997; 94: 6273–6278.
Bid HK, Roberts RD, Cam M, Audino A, Kurmasheva RT, Lin J et al. DeltaNp63 promotes pediatric neuroblastoma and osteosarcoma by regulating tumor angiogenesis. Cancer Res 2014; 74: 320–329.
Pardali E, Ten Dijke P . TGFbeta signaling and cardiovascular diseases. Int J Biol Sci 2012; 8: 195–213.
Holderfield MT, Hughes CC . Crosstalk between vascular endothelial growth factor, notch, and transforming growth factor-beta in vascular morphogenesis. Circ Res 2008; 102: 637–652.
Steder M, Alla V, Meier C, Spitschak A, Pahnke J, Fürst K et al. DNp73 exerts function in metastasis initiation by disconnecting the inhibitory role of EPLIN on IGF1R-AKT/STAT3 signaling. Cancer Cell 2013; 24: 512–527.
Alexandrova EM, Moll UM . Role of p53 family members p73 and p63 in human hematological malignancies. Leukemia & lymphoma 2012; 53: 2116–2129.
Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005; 7: 363–373.
Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009; 458: 766–770.
Li X, Claesson‐Welsh L, Shibuya M . Chapter 13 VEGF receptor signal transduction. Methods Enzymol 2008; 443: 261–284.
Magnusson P, Rolny C, Jakobsson L, Wikner C, Wu Y, Hicklin DJ et al. Deregulation of Flk-1/vascular endothelial growth factor receptor-2 in fibroblast growth factor receptor-1-deficient vascular stem cell development. J Cell Sci 2004; 117: 1513–1523.
Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22: 2677–2691.
Kudryashova E, Wu J, Havton LA, Spencer MJ . Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet 2009; 18: 1353–1367.
Acknowledgements
This work was supported by Grants SAF2012-36143 from Spanish Ministerio de Economía y Competitividad and cofinanced by FEDER funds (to MCM), and Grant LE015A10-2 from the Junta de Castilla y Leon (to MCM). RF-A was holder of a predoctoral scholarship from the Spanish Ministry of Science and Education (AP2007-02022). MM-L is a holder of a predoctoral scholarship from Junta de Castilla y Leon (PIRTU 2010). Work in the LCW lab was supported by Grants from the Swedish Cancer Foundation and Swedish Science Council. We thank the Instituto Biomar SA (Leon, Spain) for kindly supplying HUVEC and B16-F10 cells. We also thank the Pathology Deparment at the Complejo Hospitalario de Leon (Leon, Spain) for technical help and assistance in tumor analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Candi
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article

Cite this article
Fernandez-Alonso, R., Martin-Lopez, M., Gonzalez-Cano, L. et al. p73 is required for endothelial cell differentiation, migration and the formation of vascular networks regulating VEGF and TGFβ signaling. Cell Death Differ 22, 1287–1299 (2015). https://doi.org/10.1038/cdd.2014.214
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.214
This article is cited by
-
p73 isoforms meet evolution of metastasis
Cancer and Metastasis Reviews (2022)
-
p73 is required for vessel integrity controlling endothelial junctional dynamics through Angiomotin
Cellular and Molecular Life Sciences (2022)
-
Transplanted embryonic retinal stem cells have the potential to repair the injured retina in mice
BMC Ophthalmology (2021)
-
The p53 family member p73 in the regulation of cell stress response
Biology Direct (2021)
-
The hypoxic tumour microenvironment
Oncogenesis (2018)
