
Abstract
Side population (SP) cells within tumors are a small fraction of cancer cells with stem-like properties that can be identified by flow cytometry analysis based on their high ability to export certain compounds such as Hoechst 33342 and chemotherapeutic agents. The existence of stem-like SP cells in tumors is considered as a key factor contributing to drug resistance, and presents a major challenge in cancer treatment. Although it has been recognized for some time that tumor tissue niches may significantly affect cancer stem cells (CSCs), the role of key nutrients such as glucose in the microenvironment in affecting stem-like cancer cells and their metabolism largely remains elusive. Here we report that SP cells isolated from human cancer cells exhibit higher glycolytic activity compared to non-SP cells. Glucose in the culture environment exerts a profound effect on SP cells as evidenced by its ability to induce a significant increase in the percentage of SP cells in the overall cancer cell population, and glucose starvation causes a rapid depletion of SP cells. Mechanistically, glucose upregulates the SP fraction through ATP-mediated suppression of AMPK and activation of the Akt pathway, leading to elevated expression of the ATP-dependent efflux pump ABCG2. Importantly, inhibition of glycolysis by 3-BrOP significantly reduces SP cells in vitro and impairs their ability to form tumors in vivo. Our data suggest that glucose is an essential regulator of SP cells mediated by the Akt pathway, and targeting glycolysis may eliminate the drug-resistant SP cells with potentially significant benefits in cancer treatment.
Similar content being viewed by others


Main
Accumulating evidence suggests that tumors of various tissue origins, including the brain, breast, and lung, contain a small subpopulation of special cells with stem-like properties, often referred to as cancer stem cells (CSCs) or tumor-initiating cells.1, 2, 3, 4, 5, 6, 7 In addition to the ability of CSCs to self-renew and initiate tumor formation, one important biochemical feature of CSCs is their ability to export certain toxic compounds and resistance to many chemotherapeutic agents due in part to their high expression of ATP-dependent efflux pump ABCG2, their increased DNA repair capacity, and activation of survival pathways.8, 9, 10 The increase in expression of ABCG2 also confers CSCs the ability to effectively export the DNA-binding dye Hoechst 33342 out of the cells, leading to a low retention of the fluorescent signal in these cells, which appear at the low-left corner in flow cytometry analysis and thus are known as ‘side population’ or SP cells.2, 11 As SP cells can be readily identified by flow cytometry in a quantitative manner, the measurement of SP cells has been widely used as a quantitative assay for the relative number (%) of stem-like cancer cells in the bulk of the overall cancer cell population. Importantly, as this assay is functionally based on the ability of SP cells to export Hoechst 33342 and certain toxic compounds, it is also a quantitative analysis of the subpopulation of cancer cells that are resistant to conventional chemotherapeutic agents, including doxorubicin (Adriamycin, or ADM), cisplatin (CDDP), and vincristine. As such, quantitative analysis of SP cells and investigation of the factors that affect SP cells are of high clinical importance with potential therapeutic implications.
Although certain key molecules and signaling pathways that affect the stemness of CSCs and their differentiation have been identified,12, 13, 14, 15 the metabolic features of CSCs and the effect of nutrients and metabolites on CSC largely remain to be investigated. Recent studies suggest that CSCs may have special metabolic properties that distinguish them from the bulk of tumor cells, and that such biochemical properties may constitute a basis for developing new therapeutic strategies to eliminate CSCs.16 Our recent study showed that brain tumor stem-like cancer cells exhibited low mitochondrial respiratory activity and preferred hypoxic microenvironment to maintain their stemness.17 We further demonstrated that glioma stem cells were active in glycolysis and were intrinsically sensitive to glycolytic inhibition.18 Although cancer cells are in general active in glycolysis, CSCs seem to have even higher glycolytic activity. The findings that CSCs are highly dependent on glycolysis are consistent with the hypothesis of Warburg, who considered mitochondrial respiration injury and a switch from mitochondrial oxidative phosphorylation to cytosolic glycolysis as the origin of cancer.19, 20 Based on the observation that glucose may function as an essential nutrient for CSCs, we reasoned that glucose might have a significant effect on the CSC subpopulation in the overall cancer cell population. This led us to evaluate the impact of glucose in the microenvironment on SP cells, and to investigate the underlying mechanisms.
In this study, we used several cancer cell lines containing various percentages of SP cells as in vitro models to test the effect of glucose on the SP subpopulation. Flow cytometry sorting method was employed to separate SP cells from the non-SP cells, which were then compared for their metabolic properties and for the expression of relevant genes. We found that SP cells are more active in glycolysis when compared to the non-SP cells. Addition of glucose to the culture medium induced a significant increase in SP subpopulation in culture. We also revealed that several key genes involved in glucose metabolism were differentially expressed in SP and non-SP cells, and that the Akt pathway seemed to play a key role in mediating glucose-induced increase in SP cells. Finally, we investigated the potential therapeutic effect of glycolytic inhibition on the viability of SP cells in vitro and their ability to form tumor in vivo, and showed that the glycolytic inhibitor 3-BrOP potently compromised the viability of SP cells and impaired their tumorigenic capacity in animals.
Results
SP cells in human cancer cell lines and their expression of ABCG2
We first examined the presence of SP cells in several human cancer cell lines in order to identify proper cell lines for isolation of SP and non-SP cells for metabolic study. SP cells were detected based on their ability to exclude Hoechst 33342 dye. These cells appeared as a distinct dim ‘tail’ in the flow cytometry plots, which revealed that the percentage of SP cells varied significantly among different cancer cell lines (Figure 1a). The human non-small cell lung cancer (NSCLC) cell lines A549 (cultured in RPMI 1640 medium) and NCI-H460 (in RPMI 1640) contained 16.57% and 3.25% SP cells, respectively, while the colon cancer cell line LoVo (cultured in F12K medium) contained 1.73% SP cells. Other cancer cell lines tested (Raji, HCT 116, CaPan-2, HT 29, and PANC-1 cells) contained less than 1% of SP population (data not shown). The ABC transporter inhibitor Verapamil (Vera, 50 μM) effectively blocked the export of the Hoechst dye and thus led to the disappearance of the SP subpopulation (Figure 1a), confirming that the ABC transporter was responsible for the SP phenotype.

Analysis of SP cells in cancer cell lines and their expression of ABCG2. (a) Comparison of the % of SP cells in lung cancer cell lines (A549 and NCI-H460) and colon cancer LoVo cell line. Approximately 1 × 106 cancer cells were stained with Hoechst33342 dye in the presence or absence of 50 μM verapamil (an efflux pump inhibitor) for 90 min and analyzed by flow cytometry. SP cells appeared as a dim ‘tail’ with low red and low blue Hoechst signals due to active efflux of the dye. Inhibition of the efflux pump function by verapamil (Vera) led to accumulation of Hoechst33342 in the cells and the SP subpopulation disappeared. (b) Expression of ABC transporters MRP2, ABCG2, and p-glycoprotein in A549 cells, measured by flow cytometry analysis. The expression of ABCG2 in SP and non-SP cells sorted by flow cytometry from A549 cells was further compared
To determine the specific type of ABC transporters expressed in SP cells, we measured the expression of MRP2, P-glycoprotein, and ABCG2 in A549 cells, which contained relatively high SP cells. Flow cytometry analysis showed that A549 cells mainly expressed ABCG2, while MRP2 and P-glycoprotein were undetectable (Figure 1b). To further compare the expression of ABCG2 in SP cells and non-SP cells, we used flow cytometric sorting to separate the two subpopulations and then measured ABCG2 expression. As shown in Figure 1b, expression of ABCG2 was mainly found in the SP cells, while very little expression of ABCG2 was seen in the non-SP cells. These data together suggest that ABCG2 might be the key efflux molecule in SP cells.
Active glycolysis and high expression of PDK-1 in SP cells
The active utilization of glucose by tumor cells constitutes the basis for 18FDG-PET imaging for cancer diagnosis, and positive FDG-PET signals in the tumor sites after treatment predict poor prognosis.21, 22, 23, 24 Because CSCs are resistant to conventional chemotherapeutic agents, it is possible that the tumor cells in the residual lesions after chemotherapy might represent CSCs with elevated glycolytic activity. This prompted us to compare glucose metabolic activity in SP and non-SP cells. SP and non-SP cells were isolated from A549 cells by flow cytometry sorting, and glucose consumption and lactate production were measured. As shown in Figure 2a, glucose uptake and lactate production were significantly increased in SP cells compared to non-SP cells. Analysis of cellular ATP showed that SP cells had higher ATP content (Figure 2b). Interestingly, we also noted that the lung cancer cells (A549) were more glycolytic than the non-tumorigenic bronchus epithelial cells (BEAS-2B), as indicated by higher glucose uptake, higher lactate production, and lower oxygen consumption in A549 cells (Supplementary Figure S1). Thus, the lung cancer A549 cells are more glycolytic than their normal counterparts, while the SP cells sorted from A549 seem to have the highest glycolytic activity.

Comparison of glycolytic activity and expression of the relevant genes in SP and non-SP cells. (a) SP and non-SP cells were separated by flow cytometry sorting from A549 cells, seeded in 12-well plates at a density of 3.5 × 105 cells/well for 8 h. The medium was replaced with fresh one, and glucose and lactate in the culture medium were measured at 12 and 24 h. (b) Comparison of intracellular ATP levels in SP and non-SP cells. Cellular ATP levels were measured after SP and non-SP cells were sorted from A549 cells and cultured in fresh medium for 2 h. (c) Expression of HK-1, HK-2, PDK-1, MCT1, MCT4, GluT1, c-Myc, and GAPDH in SP and non-SP cells sorted from A549 cell line determined by western blot. (d) Real-time RT-PCR analysis of mRNA expression of HK-1, HK-2, PDK1, MCT1, MCT4, GluT1, HIF-1α, and GAPDH in SP and non-SP cells. (e) Comparison of hexokinase activity between SP and non-SP cells. Cell lysates were prepared from SP and non-SP cells immediately after sorting, and then used fresh for analysis of hexokinase activity. The values in a, b, d, and e present the mean±S.D. of three independent experiments. *P<0.05
To investigate the molecular events associated with high glycolytic activity in SP cells, we compared the expression of several key molecules involved in glucose metabolism, including hexokinase-1 (HK-1), hexokinase-2 (HK-2), pyruvate dehydrogenase kinase 1 (PDK-1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and monocarboxylate transporter 1 (MCT1), in SP and non-SP cells. As shown in Figure 2c, the expression of PDK-1 was substantially higher in SP cells. Because PDK-1 is responsible for the phosphorylation of pyruvate dehydrogenase (PDH) and suppresses its ability to convert pyruvate to acetyl-CoA for further metabolism in the mitochondria via the tricarboxylic acid (TCA) cycle,25 the high expression of PDK-1 in SP cells would suppress the metabolic flow of pyruvate into the mitochondria and thus promote the conversion of pyruvate to lactate in the cytosol. Unexpectedly, the expression of HK-2, an enzyme that catalyzes the first step of the glycolytic reaction (glucose to glucose-6-phosphate), was significantly downregulated in SP cells. However, the expression of HK-1, another enzyme that catalyzes the conversion of glucose to glucose-6-phosphate, remained highly expressed in SP cells (Figures 2c and d). These data, together with the high glycolytic activity observed in SP cells (Figure 2a), suggest a possibility that SP cells might use HK-1 to maintain their high glycolysis. In fact, enzyme activity analysis showed that the overall hexokinase activity (including HK1 and HK2) was higher in the SP cells (Figure 2e). The high expression of PDK-1, low expression of HK-2, and high expression of HK-1 in SP cells were further confirmed by qRT-PCR analysis (Figure 2d). Interestingly, the expression of HIF1α (known to affect HK-2 and PDK-1 expression) and c-Myc (known to affect HK-2 expression) appeared similar in SP and non-SP cells (Figures 2c and d), suggesting that the high expression of PDK1 and low expression of HK2 in SP cells are unlikely due to differential expression of HIF-1α or c-Myc in SP and non-SP cells.
Glucose induces a reversible increase of SP cells in the cancer cell population
Based on the observation that SP cells were highly glycolytic (Figure 2a), we postulated that glucose in the tissue environment might have a significant impact on SP cells. To test this possibility, we first cultured A549 cells in medium containing various concentrations of glucose and analyzed the percent of SP cells. As shown in Figure 3a, A549 cells in their routine culture medium (F12K) with 1260 mg/l glucose contained 5.04% SP cells. When the cells were switched to a medium containing a higher level of glucose (2000 mg/l, RPMI1640), there was a time-dependent increase in SP cells, which reached 26.48% at 72 h. In contrast, when the cells were switched to glucose-free RPMI1640 medium, the SP population dramatically decreased to 0.86% in 24 h and to less than 0.1% in 72 h (Figure 3a). Interestingly, A549 cells continued to proliferate during the first 24 h in the glucose-free medium, while the % of SP cells decreased substantially during this time period (Supplementary Figure S2). Cell proliferation stopped when the cells were cultured in the absence of glucose for a prolonged period of time (48–72 h, Supplementary Figure S2).

Effect of glucose on SP cell fraction in lung cancer and colon cancer cell lines. (a) The lung cancer A549 cells were maintained in standard F12K medium containing 1260 mg/l glucose. A portion of the cells was switched to RPMI 1640 medium containing higher glucose (2000 mg/l) and another portion of the cells was switched to glucose-free RPMI 1640 medium. The cells cultured under three different conditions were analyzed for % of SP cells at 24, 48, and 72 h. (b) LoVo cells (human colon cancer) were maintained in F12K medium containing 1260 mg/l glucose, switched to glucose-free RPMI 1640 medium for 24 h, and then replaced with fresh medium containing 2000 mg/l glucose for 24 and 48 h. The cells cultured under each of these conditions were collected for SP analysis. (c) Lung cancer cells (NCI-H460) were maintained in RPMI 1640 medium containing 2000 mg/l glucose, switched to glucose-free RPMI 1640 medium for 24 h, and then replaced with fresh medium containing 2000 mg/l glucose for 24 and 48 h. The cells cultured under each of these conditions were collected for SP analysis. Each experiment was repeated at least three times. Representative data are shown
The impact of glucose on SP cells was consistently observed using two other cell lines. As shown in Figure 3b, the human colon cancer cell line (LoVo) contained 1.73% SP cells when maintained in F12K medium (1260 mg/l glucose). The percentage of SP cells decreased to 0.86% in 24 h after switching to glucose-free RPMI1640 medium. Addition of glucose (2000 mg/l) back to the cultured medium caused a time-dependent increase in SP fraction. A similar phenomenon was also observed in lung cancer H460 cell line (Figure 3c). These data suggest that glucose has a major effect in inducing SP cells in multiple cancer cells.
To further test the ability of glucose to induce the conversion of non-SP cells to SP cells, we used flow cytometry sorting to obtain purified non-SP cells, which were then incubated in medium containing various concentrations of glucose. As shown in Supplementary Figure S3, the sorted non-SP cells were highly purified and contained 0.0% SP cells as measured by flow cytometry after sorting. Incubation of the sorted non-SP in medium containing 0, 1260, and 2000 mg/l of glucose resulted in a concentration-dependent induction of SP cells. These data indicate that non-SP cells could be induced to become SP cells by glucose.
Role of Akt in mediating glucose-induced increase in SP cells
To investigate the molecular events associated with glucose-induced upregulation of SP cells, we examined the effect of glucose addition and deprivation on the expression of molecules involved in regulation of CSCs. A549 cells were cultured in RPMI 1640 with or without glucose for 72 h, and RNA was isolated for qRT-PCR analysis. As shown in Figure 4a, the expression of ABCG2 and SOX2 was significantly higher in the sorted SP cells than in non-SP cells. Importantly, the expression of SOX2, ALDH1, and Notch1 was significantly induced by glucose (Figure 4b). The mRNA expression of ABCG2, HK2, and PDK1 was also increased in the presence of glucose (Figure 4b).

Impact of glucose on the Akt signaling pathway and the effect of LY294002 on SP cells. (a) Comparison of expression of ABCG2 and SOX2 in sorted SP and non-SP cells. (b) Effect of glucose on expression of genes involved in stem cell regulation and glycolysis. A549 cells were cultured in RPMI 1640 without or with glucose (2000 mg/l) for 72 h, and RNA was isolated for real-time RT-PCR for analysis of expression of SOX2, ALDH1, ABCG2, Notch1, HK2, and PDK1. β-Actin was used as an internal control. (c) A549 cells were cultured in RPMI 1640 medium without or with glucose (2000 mg/l) for 72 h, and cell lysates were subjected to western blotting to measure the expression of p-AMPK, total AMPK, p-Akt(ser473), total Akt, ABCG2, HK2, SOX2, PDK1, and β-actin. (d) Comparison of p-AKT and total AKT in sorted SP and non-SP cells measured by western blotting. (e) A549 cells maintained in F12K medium were incubated with 10 μM LY294002 for 6 h, and cell extracts were analyzed by western blot for p-Akt(ser473) and total Akt protein. (f) A549 cells maintained in F12K medium were incubated with 10 μM LY294002, and SP cells were quantified by flow cytometry analysis. *P<0.05
Because Akt seems to affect ABCG2 expression and localization,26 we therefore investigated the potential role of Akt in mediating glucose upregulation of SP cells. Western blot analysis showed that glucose significantly upregulated Akt activity, as evidenced by a striking increase in phosphorylated Akt and an increase in total Akt protein (Figure 4c). The presence of glucose also caused a significant suppression of AMPK activity, as indicated by a substantial decrease in phosphorylated-AMPK, while the total AMPK protein remained unchanged. These data together suggest that glucose enhanced Akt activity, possibly through suppression of AMPK, which is known to negatively regulate Akt.27 Associated with the suppression of AMPK and activation of Akt by glucose, there was a dramatic increase in ABCG2 expression (Figure 4c). Consistent results were observed when A549 were incubated with various concentrations of glucose, which inhibited AMPK phosphorylation, activated Akt, and promoted ABCG2 expression in a concentration-dependent manner (Supplementary Figure S4). The activation of Akt in SP cells was confirmed in flow cytometry-sorted cells (Figure 4d).
We then used two strategies to further test the role of the Akt signaling pathway in mediating glucose-induced ABCG2 expression and increase in SP subpopulation. First, we examined the effect of a PI3K inhibitor LY294002 on Akt and % of SP cells. As shown in Figure 4e, treatment of A549 cells with 10 μM LY294002 for 6 h caused a substantial inhibition of Akt phosphorylation, and this led to a rapid decrease in SP cell population (Figure 4f). Secondly, we used siRNA strategy to knock down Akt expression in A549 cells and tested its effect on glucose-induced SP cells. Cells treated with scrambled RNA were used as control. After siRNA transfection, the cells were first incubated in glucose-free medium for 24 h, and then were cultured in glucose-replenished medium for another 24 and 48 h. Each sample was analyzed for Akt expression and for SP cells. As shown in Figure 5a, siRNA was effective in knocking down Akt expression 24 and 48 h after transfection, whereas the scrambled RNA did not cause a change in Akt expression. Flow cytometry analysis showed that removal of glucose caused a rapid decrease in SP cells from 13.56 to 0.26% in 24 h (Figure 5b). Replenishment of glucose led to a time-dependent reappearance of SP cells in the control samples treated with scrambled RNA, but not in the samples transfected with siRNA against Akt (Figure 5b), indicating that the knockdown of Akt effectively prevented the induction of SP cells by glucose. These data together with the LY294002 experiment suggest that Akt may play an important role in glucose-induced upregulation of SP cells. Consistently, treatment of A549 cells with AICAR (5-Aminoimidazole-4-carboxamide 1-β-D-ribofuranoside) caused an activation of AMPK (a negative regulator of Akt), leading to a lower ABCG2 expression and a decrease in SP cells (Supplementary Figure S5).

The role of Akt in mediating glucose-induced increase in SP cells. A549 cells were transfected with siRNA against Akt, scrambled RNA (s.c.r.m), or no transfection (n.t.). The transfected cells were cultured in glucose-free medium for 24 h, and then shifted to fresh medium containing glucose (2000 mg/l) for 24 or 48 h as indicated. The cells were collected for (a) western blot analysis of Akt expression, and (b) for flow cytometry analysis of SP cells. Cells without transfection cultured in RPMI 1640 medium with glucose (Glu +) or without glucose (Glu −) were used as an additional control
SP cells are resistant to conventional chemotherapy and sensitive to glycolytic inhibition
CSCs are thought to be intrinsically resistant to conventional anticancer agents, and new strategies are needed to eliminate these cells. Based on the observation that SP cells were highly glycolytic, we speculated that this subpopulation might be sensitive to glycolytic inhibition. We first tested if 3-BrOP, an effective glycolytic inhibitor,18, 28 could kill SP cells by disrupting their energy metabolism. As shown in Figure 6a, incubation of A549 cells with 3-BrOP caused a significant decrease in glucose uptake and lactate production, indicating an inhibition of glycolysis. This led to a time-dependent depletion of cellular ATP (Figure 6b). Importantly, when A549 cells were treated with equal toxic concentrations of 3-BrOP and several conventional chemotherapeutic agents, there was a striking difference in their impact on SP cells. As shown in Figures 6c and d, 3-BrOP at its IC50 concentration (50 μM) significantly reduced SP cells from 16 to 6.7% in 72 h, while the equal toxic concentrations of cisplatin (CDDP, IC50=3.57 μM) or (ADM, IC50=159 nM) caused a substantial increase of SP cells, from 16.03 to 30.01% and 26.47%, respectively, indicating that CDDP and ADM mainly kill non-SP cells and thus enrich SP cells. Incubation with the IC50 concentration of Paclitaxel (PTX, 20.3 nM) did not alter the percent of SP cells. Consistent with the ability of 3-BrOP to decrease SP cells, western blot analysis showed that treatment of A549 cells with 3-BrOP caused a significant decrease in phosphorylated Akt and a decrease in expression of ABCG2 and SOX-2, whereas AMPK was activated in the same samples, as evidenced by a substantial increase in p-AMPK (Figure 6e). The overall inhibitory effect of 3-BrOP, CDDP, ADM, and PTX on A549 cells with the corresponding IC50 values determined by MTT assay is shown in Supplementary Figure S6. The cytotoxic effect of these compounds, measured by annexin-V/PI double staining, is shown in Supplementary Figure S7.

Effect of the glycolytic inhibitor 3-BrOP on SP cells and their Akt signaling pathway. (a) Inhibition of glucose uptake and lactate production by 3-BrOP in A549 cells. A549 cells were seeded in 6-well plates (5 × 105 cells/well) overnight. The culture medium was replaced with 1 ml fresh medium/well in the presence or absence of 3-BrOP (80 μM, 6 h). Glucose uptake and lactate production were measured. (b) A549 cells were cultured with 80 μM 3-BrOP, and cellular ATP was measured at different time points as indicated. (c) Effects of various drugs on SP fractions at their respective IC50 concentrations. A549 cells were seeded in a culture plate (5 × 105 cells/well) for 24 h, then replaced with fresh medium containing each compound at their respective IC50 concentrations as determined by MTT assay (Supplementary Figure 6). The cells were harvested and stained with Hoechst 33342 in the presence and absence of Verapamil to determine SP cells. (d) Quantitation of SP cells under the experimental conditions described in (c). Data are means±S.D. of 3–5 independent experiments. (e) A549 cells were treated with 3-BrOP (100 μM for 12 h). Cell lysates were subjected to western blotting for detection of p-AMPK, total AMPK, p-Akt(ser473), total Akt, ABCG2, HK2, SOX2, and β-actin. *P<0.05
Inhibition of tumor formation in vivo by pretreatment of cancer cell with glycolytic inhibitor in vitro
Based on the evidence that the glycolytic inhibitor 3-BrOP could reduce SP cells while the conventional chemotherapeutic agents CDDP and ADM increased the SP subpopulation, we compared the effect of 3-BrOP and CDDP on the ability of A549 cells to form tumor in vivo. As shown in Figure 7a, A549 cells were first incubated with 3-BrOP or CDDP at their respective IC50 concentrations for 24 h, and then cultured in drug-free fresh medium for another 24 h (3-BrOP) or 48 h (DDP) to allow time for the cell death to occur (24 h for 3-BrOP and 48 h for CDDP due to different mechanisms of action) and for the viable cells to recover. The detached dead cells were removed, and equal numbers of viable cells from the control and drug-treated samples were inoculated subcutaneously into the flanks of athymic mice. The mice were then observed for tumor formation without further drug treatment. As shown in Figure 7b, when mice were inoculated with 1.5 × 105 viable cells per injection site, almost all the mice in the control group and the CDDP-treated group developed tumors, while 3-BrOP pre-treatment reduced tumor incidence to 45%. When the inoculated cell number was 1 × 105 cells, 27% mice in the control group developed tumors and 55% mice in the CDDP group developed tumors. The tumor incidence in the 3-BrOP group was dramatically reduced to 9%. Since A549 cells were treated CDDP or 3-BrOP at equal cytotoxic concentrations (IC50), the significant decrease in tumor formation observed in the 3-BrOP-treated group demonstrated that this compound effectively killed the SP cells. Consistently, tumor growth was retarded substantially by pre-treatment with 3-BrOP (Figure 7c). In contrast, as CDDP mainly kill non-SP cells, the remaining viable cells were enriched in SP cells and thus had higher tumor incidence when equal number of viable cells were inoculated.

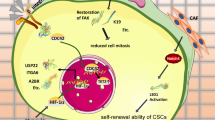
Suppression of tumor formation by glycolytic inhibition. (a) Experimental scheme. A549 cells were pretreated with the IC50 concentrations of 3-BrOP (50 μM) and DDP (3.5 μM) for 24 h, washed, and cultured in fresh medium without drug for 1 day (3-BrOP group) or 2 days (CDDP group) to allow time for the cell death to occur (24 h for 3-BrOP and 48 h for CDDP due to different mechanisms of action). The detached dead cells were removed, and equal numbers of viable cells from the control and drug-treated samples were inoculated subcutaneously into the flanks of athymic mice at the cell number of 1.5 × 105/injection site (group 1) or 1 × 105 cells (group 2). The mice were monitored for tumor incidence (b) and tumor growth (c). (d) A proposed model of glucose-induced SP cells through activation of the Akt pathway
Discussion
It has been recognized for decades that cancer cells actively use glycolytic metabolism even in the presence of oxygen, a phenomenon known as the Warburg effect.19, 20 A high rate of glycolysis may be beneficial to cancer cells by providing ATP and metabolic intermediates,29 and constitutes the biochemical basis of 18F-fluoro-2-deoxy-glucose position emission tomography (18FDG-PET) for clinical diagnosis of cancer.30 Because elevated glycolysis seems prevalent in cancer cells and is of clinically relevance, such metabolic alteration has recently been considered as a hallmark of cancers.31 Recent studies from our laboratory and other groups suggest that CSCs may have even more active glycolytic activity compared to the bulk of the general cancer cells.17, 18, 32 Consistently, the current study showed that SP cells were more glycolytic compared to the non-SP cells. These observations underscore the importance of glucose as an essential nutrient for cancer cells, especially for CSCs, and suggest a possibility that the levels of glucose in the tumor microenvironment might significantly affect CSCs.
Although the important role of tumor tissue niches in affecting CSCs has been recognized in recent years, the impact of key nutrients in the tumor microenvironment on CSCs remains largely unknown. In this study, we used flow cytometry analysis of SP cells as a quantitative end point to evaluate the effect of glucose on CSCs in the overall cancer cell population, and used qRT-PCR and western blot analyses to investigate the underlying mechanism. Our results showed that glucose plays a major role in promoting CSC phenotype through the AMPK→Akt→ABCG2 axis (Figure 7d). This conclusion is supported by the following evidences: (1) glucose in the culture medium significantly increased the % of SP cells in the overall cancer cell population, whereas removal of glucose from the medium caused a depletion of SP cells in multiple cell lines; (2) the presence of glucose suppressed AMPK activity and activated Akt, leading to high expression of ABCG2, while removal of glucose led to activation of AMPK and suppression of Akt and ABCG2 expression; (3) activation of AMPK by AICAR lead to a decrease in ABCG2 expression and a lower SP cells; (4) siRNA knockdown of Akt expression prevented the glucose-induced increase of SP cells; (5) inhibition of glycolysis by 3-BrOP caused ATP depletion, activation of AMPK, and suppression of Akt, leading to suppression of ABCG2 expression and a decrease in SP cells.
The induction of SP cells by glucose appears to be a reversible phenotype. As shown in Figure 3a, switch of cancer cells from low-glucose medium to high-glucose medium led to a time-dependent increase in SP cells for up to 72 h. Consistently, glucose deprivation caused a rapid depletion of SP cells, which re-appeared when glucose was replenished. This rapid inducible phenotype suggests that induction of SP cells by glucose is likely by promoting activation of Akt and expression of ABCG2 in a subpopulation of pre-existing cancer cells, and not by inducing proliferation of SP cells, since it is unlikely that SP cells could proliferate 40 folds (from 0.26 to 10.23%) in a 24-h period. The ability of the purified non-SP cells (with 0% SP by flow cytometry sorting) to produce 5.7% SP cells when cultured in glucose-containing medium further supports the conclusion that SP cells are inducible (Supplementary Figure S3). The fact that only 10–20% cells were induced by glucose to become SP cells suggests a possibility that only a fraction of the A549 cells could be induced to exhibit the SP phenotype. This interesting phenomenon and the underlying regulatory mechanisms need to be further investigated.
Activation of the serine/threonine kinase Akt is frequently observed in cancer cells and is associated with drug resistance.33 This molecule seems to have pro-oncogenic property and has been implicated in enhancing glucose transport and aerobic glycolysis in cancer cells. Our study suggests that Akt activation plays a key role in mediating glucose-induced increase in SP cells, likely by promoting the expression of ABCG2, an ATP-dependent pump capable of exporting certain chemotherapeutic agents out of the cells. This is consistent with the finding that Akt could modulate the expression of ABCG2.14, 15, 26, 34, 35 Interestingly, a recent study showed that inhibition of Akt by LY294002 induces a loss of SP fraction accompanied by a rapid translocation of ABCG2 from the cell membranes to the intracellular compartment, and that forced expression of Akt led to an increase in SP fraction but has no effect on SP fractions in cells from ABCG2-deficient mice,26 supporting the notion that ABCG2 may be an effector of Akt in promoting cancer stem cell phenotype.
SP cells are defined by their ability to effectively export Hoechst 33342 due to high expression of ATP-dependent transporters such as ABCG2.36 The high expression of these efflux pumps enables the CSCs to export chemotherapeutic agents and confer drug resistance. This is thought to contribute to the persistence of residual disease after chemotherapy and is a major reason for cancer recurrence. In the clinic, a positive 18FDG-PET signal at the tumor sites after chemotherapy often predicts a poor treatment outcome due to persistence of cancer cells with high glycolytic activity. Consistently, our observation that in vitro treatment of A549 cells with CDDP or ADM resulted in an increase in SP cells in the surviving cell population (Figure 6c) supports the notion that CSCs are resistant to standard drug treatment, and underscores the urgent need to develop new strategies and novel agents to kill CSCs. Our study showed that SP cells are more active in glycolysis compared with non-SP cells. Furthermore, the high expression of PDK1 in SP cells would inhibit pyruvate dehydrogenase and thus suppress pyruvate entry into the TCA cycle. Under this circumstance, pyruvate would be converted into lactate. This may explain why SP cells have higher glucose consumption and higher lactate production. This metabolic property also suggests that CSCs may be more dependent on glycolysis and thus more sensitive to glycolytic inhibition. Indeed, we showed that incubation of A549 cells with glycolytic inhibitor 3-BrOP led to a substantial decrease in SP cells. This is in a sharp contrast to CDDP and ADM. As such, inhibition of glycolysis may be a promising strategy to eliminate CSCs. This hypothesis was further supported by the findings that pre-treatment of A549 cells with 3-BrOP in vitro significantly reduced tumorigenesis in vivo (Figure 7). The inhibitory effect of 3-bromopyruvate and its analog 3-BrOP on glycolysis has been attributed to their ability to suppress the enzyme activity of the glycolytic enzymes hexokinase (HK) and GAPDH.18, 37, 38 It appears that GAPDH is a preferred target of 3-BrOP, as this compound inhibits GAPDH at low concentrations (0.3–3 μM), while much higher concentrations (over 300 μM) were required to inhibit hexokinase.28 Interestingly, a recent study by Sabatini and coworkers39 suggested that MCT1 is a transporter of 3-bromopyruvate, and that a high expression of MCT1 may render cells more sensitive to this compound due to a higher drug uptake.
It is of interest to note that the high expression of the active Akt in SP cells may confer survival advantage to this subpopulation of cells and render them resistant to anticancer agents. This may in part explain why the SP cells were less sensitive to conventional chemotherapeutic agents such as CDDP and ADM as shown in Figures 6c and d. Paradoxically, the high expression of active Akt may also render SP cells highly glycolytic, resulting in high sensitivity to glycolytic inhibition. As inhibition of glycolysis by 3-BrOP is effective in killing SP cells, it seems logical to combine standard chemotherapeutic agents with glycolytic inhibitors such as 3-BrOP to simultaneously eliminate the bulk of cancer cells as well as SP cells. This new therapeutic strategy may potentially improve the outcomes of cancer treatment, and merits further test in pre-clinical and clinical settings.
Materials and Methods
Chemicals and reagents
Hoechst 33342, verapamil, ADM, paclitaxel, 3-bromopyruvate, and 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were obtained from Sigma (St Louis, MO, USA). 3-Bromo-2-oxopropionate-1-propyl ester (3-BrOP) was synthesized as described previously.37 AICA-Riboside (AICAr) was supplied by Calbiochem (San Diego, CA, USA). PE-labeled mouse anti-human P-glycoprotein, APC-labeled mouse anti-human ABCG2, and their respective control antibodies were purchased from BD Bioscience (San Jose, CA, USA). Mouse monoclonal anti-MRP2 and FITC-labeled anti-mouse IgG were supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit monoclonal anti-hexokinase I and anti-hexokinase II, rabbit monoclonal anti-PDK1 (C47H1), rabbit monoclonal anti-GAPDH, rabbit polyclonal anti-AMPK-alpha, rabbit monoclonal anti-phospho-AMPK-alpha (Thr172) (40H9), rabbit monoclonal anti-Akt, rabbit monoclonal anti-phospho-Akt (Ser473) (D9E), rabbit monoclonal anti-Sox2, rabbit polyclonal anti-ABCG2, rabbit monoclonal anti-MCT1, and mouse monoclonal anti-β-actin antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Small interfering RNA (siRNA) that targets Akt1 and Akt2 (Akt siRNA) and scramble siRNA were obtained from Cell Signaling Technology. Rabbit polyclonal anti-MCT4, rabbit polyclonal anti-GluT1, and rabbit polyclonal anti-c-Myc antibodies were purchased from Santa Cruz Biotechnology. Lipofectamine-2000 and Opti-MEM were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). The PI3K inhibitor LY294002 was purchased from Cell Signaling Technology. Chemiluminescence kit was purchased from Keygen Biotech. Co., Ltd (Nanjing, China).
Cell culture and proliferation assays
Human non-small cell lung carcinoma (NSCLC) A549 and NCI-H460 cell lines and colon cancer LoVo cell line were obtained from the American Type Culture Collection (ATCC) (Rockville, MD, USA) and routinely maintained in culture medium as recommended by ATCC. All media were supplemented with 10% fetal bovine serum (FBS; Invitrogen Life Technologies). All cell lines were incubated in a humidified incubator at 37 °C supplied with 5% carbon dioxide. Cell growth inhibition by ADM, CDDP, paclitaxel (PTX), and 3-BrOP was determined by MTT assay. Briefly, 2.5 × 103 cells/well were seeded in triplicate in 96-well plates. After 24 h of incubation, the cells were exposed to various concentrations of ADM, DDP, 3-BrOP, and PTX for 3 days. At the end of the incubation, 20 μl of MTT reagent (5 mg/ml) was added to each well and incubated at 37 °C for 4 h. After removal of the culture medium, 200 μl DMSO was added to each well to dissolve the formazan precipitates. The optical density (OD) was measured at wavelengths of 570 and 630 nm with a Multiskan MK3 microplate-reader (Thermo Labsystem, Franklin, MA, USA). To directly quantify cell proliferation, cells were trypsinized and counted using a cell counter (Cellometer Auto T4, Nexcelom Bioscience, Lawrence, MA, USA).
Glucose and lactate assay
Cells were cultured in complete medium with 10% FBS, and replaced with fresh medium when cells in exponential growth phase for 6–24 h. Media from the samples were collected and analyzed for glucose and lactate concentration using colorimetric kits according to the manufacturer’s instructions (Biovision, Mountain View, CA, USA). The absorbance was measured at 450 nm using a Multiskan MK3 microplate-reader (Thermo Labsystem).
Hexokinase enzymatic activity assay
Hexokinase activity was measured using a hexokinase assay kit (ScienCell Research Laboratories, Carlsbad, CA, USA) according to the manufacturer’s instructions. In brief, cells were lysed in the assay buffer, and then incubated with the assay mix for 1 h in room temperature in the dark. The optical absorbance was measured at OD490nm to quantify the color production from the hexokinase reaction.
Cellular oxygen consumption assay
To determine cellular oxygen consumption, equal numbers of cells were suspended in 1 ml fresh medium at 37 °C pre-equilibrated with 21% oxygen and transferred to a sealed respiration chamber equipped with a Clark oxygen sensor, a thermostat controller, and a micro-stirring device (Oxytherm, Hansatech Instrument, Norfolk, UK). Measurements were made at 37 °C with constant stirring. Oxygen consumption rate was expressed as nanomoles of O2 consumed per min per 5 million cells.
Measurement of intracellular ATP
Cellular ATP levels were determined using the ATP-based CellTiter-Glo luminescent Cell Viability kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions with the following modifications. Cells were plated in triplicate in 96-well plates to allow for attachment overnight. And then equal volume of the single-step reagent provided by the kit was added to each well and rocked for 2 min and kept at room temperature for 10 min to complete the reaction. Luminescence levels were measured using a luminescent plate reader (Thermo Fisher Varioskan Flash, Waltham, MA, USA). To test the inhibition effect of glycolytic inhibitor on intracellular ATP, 3-BrOP (80 μM) was added to the A549 cells and ATP was measured at various time points.
Flow cytometry
The expression of ATP-binding cassette transporters on the surface of SP and non-SP cells was analyzed using flow cytometry. Briefly, 1 × 106 cells were incubated with PE-labeled mouse monoclonal anti-P-glycoprotein and APC-labeled mouse monoclonal anti-ABCG2 antibodies for 15 min in darkness at 4 °C, or with mouse monoclonal anti-MRP2 antibody for 30 min at 4 °C, followed by FITC-labeled anti-mouse IgG antibody for 30 min in darkness at 4 °C. The cells were then washed and resuspended for analysis in cold PBS using a Cytomix FC500 cytometer (Beckman Coulter, Fullerton, CA, USA). For apoptosis assay, 1 × 106 cells were plated in 4 culture dishes overnight, and then treated with the indicated compounds for 72 h. The cells were then collected and stained with annexin-V/PI for analysis of cell viability using flow cytometer.Hoechst 33342 staining and flow cytometry sorting of SP and non-SP cells.
Cells were washed with PBS, detached by trypsinization, washed, and resuspended in pre-warmed F12K or RPMI 1640 medium containing 2% FBS with or without glucose at a density of 1 × 106 cells/ml. Cell staining was performed using the method described by Goodell et al40 with the following modifications. The cells were incubated with Hoechst 33342 (5 μg/ml) in the presence or absence of the ABC transporter inhibitor verapamil (50 μM) for 90 min at 37 °C in darkness with intermittent shaking. The cells were then washed with cold PBS, resuspended in PBS, and kept at 4 °C for flow cytometry analysis and sorting. Before sorting, the cells were filtered through a 70 μm cell strainer to obtain single cell suspension. Cell analysis and sorting were performed on a MoFlo XDP Cell Sorter (Beckman Coulter).
RNA extraction and quantitative real-time PCR analysis
Total RNA was extracted from sorted SP and non-SP cells or total A549 cells cultured in RPMI 1640 with or without glucose using TRIZOL reagent (Ambion, Austin, TX, USA). Equal amount of total RNA from each sample was subjected to reverse transcription using PrimeScript RT reagent kit with DNA Eraser (Takara Biotechnology, Dalian, Liaoning, China). Real-time PCR was performed using SYBR Premix Ex Taq II (Tli RNase H Plus) kit (Takara Biotechnology, Dalian, Liaoning, China) using the CFX96 real-time system (Bio-Rad Laboratories, Hercules, CA, USA). The RT-PCR amplification reaction program consisted of one cycle of 95 °C/30S and 40 cycles of 95 °C/5→60 °C/30S. β-actin was used as an internal control for normalization. The primers used for the amplification of the indicated genes were listed in Supplementary Table 1.
Protein extraction and western blot analysis
Cells were washed twice with ice-cold PBS and lysed in 50–150 μl lysis buffer for 10 min. Cell debris was removed by centrifugation at 12 000 × g for 20 min at 4 °C. Protein lysates (50 μg) from each experimental condition were subjected to electrophoresis in denaturing 10% SDS-polyacrylamide gel, and then transferred to a membrane, which was probed for HK2, PDK1, HK1, GAPDH, MCT1, MCT4, GluT1, c-Myc, AMPK-α, p-AMPK-α, Akt, p-Akt, ABCG2, Sox2, β-actin, and α-tubulin using the respective antibodies.
RNA interference assay
Small RNA interference (siRNA) for knockdown of Akt expression in A549 cells was performed using Lipofetamine RNAiMAX Reagent. Briefly, 3 × 105 A549 cells were plated in six-well plates. After overnight incubation, the culture medium was discarded and replaced with 900 μl/well transfection complex (containing Opti-MEM, Akt siRNA or scrambled RNA, and Lipofectamine 2000). After incubation for 6 h, the transfection complex was replaced with 2 ml fresh medium and cultured for 24–72 h before the transiently transfected cells were collected for western blot assay or SP analysis.
Evaluation of in vivo tumorigenicity
To test the effect of CDDP and 3-BrOP on tumor-initiating capacity, A549 cells were treated with each drug at their respective IC50 concentration for 24 h in vitro, washed, and cultured in fresh medium (no drug) for another 24–48 h. The cells were then harvested and injected subcutaneously into the flanks of athymic nude mice at 1.5 × 105 or 1.0 × 105 cells per injection site, 10 mice per group. The presence or absence of a visible or palpable tumor was evaluated and tumor growth was monitored every 2–3 days. The mice were killed at day 60 or when the tumors reached a maximum size of 1000 mm3. Tumor volume was calculated by the formula 0.52 × length × width2. All animal experiments were conducted in accordance with the institutional guidelines and approved by the Animal Care and Use Committee of Sun Yat-sen University Cancer Center.
Statistical analysis
Data are presented as mean±S.D. Comparisons between groups were performed using the Student’s t test on SPSS software (version 16.0). A P value less than 0.05 was considered statistically significant.
Abbreviations
- SP:
-
side population
- CSC:
-
cancer stem cells
- 3-BrOP:
-
3-bromo-2-oxopropionate-1-propyl ester
- Vera:
-
verapamil
- ADM:
-
adriamycin
- PTX:
-
paclitaxel
- CDDP:
-
cisplatin
- MTT:
-
3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
References
Regenbrecht CR, Lehrach H, Adjaye J . Stemming cancer: functional genomics of cancer stem cells in solid tumors. Stem Cell Rev 2008; 4: 319–328.
Ho MM, Ng AV, Lam S, Hung JY . Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res 2007; 67: 4827–4833.
Dalerba P, Cho RW, Clarke MF . Cancer stem cells: models and concepts. Annu Rev Med 2007; 58: 267–284.
Wicha MS, Liu S, Dontu G . Cancer stem cells: an old idea—a paradigm shift. Cancer Res 2006; 66: 1883–1890 1895-1896.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al. Identification of human brain tumour initiating cells. Nature 2004; 432: 396–401.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF . Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–3988.
Pardal R, Clarke MF, Morrison SJ . Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 2003; 3: 895–902.
Hambardzumyan D, Becher OJ, Holland EC . Cancer stem cells and survival pathways. Cell Cycle 2008; 7: 1371–1378.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444: 756–760.
Dean M, Fojo T, Bates S . Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5: 275–284.
Wu C, Alman BA . Side population cells in human cancers. Cancer Lett 2008; 268: 1–9.
Zhang W, Tan W, Wu X, Poustovoitov M, Strasner A, Li W et al. A NIK-IKKalpha module expands ErbB2- induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 2013; 23: 647–659.
Schieber MS, Chandel NS . ROS links glucose metabolism to breast cancer stem cell and EMT phenotype. Cancer Cell 2013; 23: 265–267.
Li H, Gao Q, Guo L, Lu SH . The PTEN/PI3K/Akt pathway regulates stem-like cells in primary esophageal carcinoma cells. Cancer Biol Ther 2011; 11: 950–958.
Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci USA 2007; 104: 16158–16163.
Menendez JA, Joven J, Cufi S, Corominas-Faja B, Oliveras-Ferraros C, Cuyas E et al. The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle 2013; 12: 1166–1179.
Zhou Y, Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M et al. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem 2011; 286: 32843–32853.
Yuan S, Wang F, Chen G, Zhang H, Feng L, Wang L et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cells 2013; 31: 23–34.
Warburg O . On respiratory impairment in cancer cells. Science 1956; 124: 269–270.
Warburg O . On the origin of cancer cells. Science 1956; 123: 309–314.
Oh JR, Seo JH, Chong A, Min JJ, Song HC, Kim YC et al. Whole-body metabolic tumour volume of 18F-FDG PET/CT improves the prediction of prognosis in small cell lung cancer. Eur J Nucl Med Mol Imaging 2012; 39: 925–935.
Poulou LS, Thanos L, Ziakas PD . Unifying the predictive value of pretransplant FDG PET in patients with lymphoma: a review and meta-analysis of published trials. Eur J Nucl Med Mol Imaging 2010; 37: 156–162.
Goerres GW, Stupp R, Barghouth G, Hany TF, Pestalozzi B, Dizendorf E et al. The value of PET, CT and in-line PET/CT in patients with gastrointestinal stromal tumours: long-term outcome of treatment with imatinib mesylate. Eur J Nucl Med Mol Imaging 2005; 32: 153–162.
Kostakoglu L, Coleman M, Leonard JP, Kuji I, Zoe H, Goldsmith SJ . PET predicts prognosis after 1 cycle of chemotherapy in aggressive lymphoma and Hodgkin's disease. J Nucl Med 2002; 43: 1018–1027.
Holness MJ, Sugden MC . Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans 2003; 31: 1143–1151.
Mogi M, Yang J, Lambert JF, Colvin GA, Shiojima I, Skurk C et al. Akt signaling regulates side population cell phenotype via Bcrp1 translocation. J Biol Chem 2003; 278: 39068–39075.
King TD, Song L, Jope RS . AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem Pharmacol 2006; 71: 1637–1647.
Tang Z, Yuan S, Hu Y, Zhang H, Wu W, Zeng Z et al. Over-expression of GAPDH in human colorectal carcinoma as a preferred target of 3-bromopyruvate propyl ester. J Bioenerg Biomembr 2012; 44: 117–125.
Lunt SY, Vander HM . Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011; 27: 441–464.
Gambhir SS . Molecular imaging of cancer with positron emission tomography. Nat Rev Cancer 2002; 2: 683–693.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Nakano A, Tsuji D, Miki H, Cui Q, El SS, Ikegame A et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS One 2011; 6: e27222.
Tazzari PL, Cappellini A, Ricci F, Evangelisti C, Papa V, Grafone T et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia 2007; 21: 427–438.
Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol 2009; 7: e1000121.
Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci USA 2009; 106: 268–273.
Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP . Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci USA 2002; 99: 12339–12344.
Xu RH, Pelicano H, Zhang H, Giles FJ, Keating MJ, Huang P . Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 2005; 19: 2153–2158.
Ko YH, Pedersen PL, Geschwind JF . Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett 2001; 173: 83–91.
Birsoy K, Wang T, Possemato R, Yilmaz OH, Koch CE, Chen WW et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat Genet 2013; 45: 104–108.
Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC . Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 1996; 183: 1797–1806.
Acknowledgements
This work was supported in part by a grant from the major science and technology project of the National Basic Research Program (973 Program) of China 2012CB967004, and grants CA085563 and CA100428 from the National Cancer Institute, National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by N Chandel
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Liu, PP., Liao, J., Tang, ZJ. et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ 21, 124–135 (2014). https://doi.org/10.1038/cdd.2013.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.131
Keywords
This article is cited by
-
Regulation and signaling pathways in cancer stem cells: implications for targeted therapy for cancer
Molecular Cancer (2023)
-
Cancer stem cells: an insight into the development of metastatic tumors and therapy resistance
Stem Cell Reviews and Reports (2023)
-
Cancer stem cells: advances in the glucose, lipid and amino acid metabolism
Molecular and Cellular Biochemistry (2023)
-
Warburg effect in colorectal cancer: the emerging roles in tumor microenvironment and therapeutic implications
Journal of Hematology & Oncology (2022)
-
Autophagy regulates the cancer stem cell phenotype of head and neck squamous cell carcinoma through the noncanonical FOXO3/SOX2 axis
Oncogene (2022)
