
Abstract
Proper regulation of white and brown adipogenic differentiation is important for maintaining an organism’s metabolic profile in a homeostatic state. The recent observations showing that the p53 tumor suppressor plays a role in metabolism raise the question of whether it is involved in the regulation of white and brown adipocyte differentiation. By using several in vitro models, representing various stages of white adipocyte differentiation, we found that p53 exerts a suppressive effect on white adipocyte differentiation in both mouse and human cells. Moreover, our in vivo analysis indicated that p53 is implicated in protection against diet-induced obesity. In striking contrast, our data shows that p53 exerts a positive regulatory effect on brown adipocyte differentiation. Abrogation of p53 function in skeletal muscle committed cells reduced their capacity to differentiate into brown adipocytes and histological analysis of brown adipose tissue revealed an impaired morphology in both embryonic and adult p53-null mice. Thus, depending on the specific adipogenic differentiation program, p53 may exert a positive or a negative effect. This cell type dependent regulation reflects an additional modality of p53 in maintaining a homeostatic state, not only in the cell, but also in the organism at large.
Similar content being viewed by others


Main
The adipose tissue of mammals consists of two functional types: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT primarily contributes to energy storage and the regulation of energy balance at large.1, 2, 3 BAT’s principal function is to generate heat by fat burning.4 Disruption of normal metabolic homeostasis in obesity leads to the development of several pathological conditions such as insulin resistance, type 2 diabetes and increased risk of development of a variety of cancers.2, 5, 6 Results from several studies indicated that there is an inverse correlation between BAT activity and obesity.7, 8, 9 Thus, increasing brown adipose tissue mass or activity could be a useful approach to limit obesity and its associated disease states.
The two main factors that orchestrate the adipogenic network are Peroxisome Proliferator-Activated-Receptor-γ (PPARγ) and CCAAT-Enhancer-Binding-Protein-α (CEBPα). Being both necessary and sufficient for white and brown adipocyte formation, PPARγ is considered to be the master regulator of adipogenesis.1, 3 The brown adipocyte shares a common progenitor cell with the skeletal muscle lineage. The transcription factor Positive-Regulatory-Domain-Containing-16 (PRDM16) has been identified as the key molecular switch committing skeletal muscle progenitors into the development of brown adipocytes.10, 11 Adipocyte-Fatty-Acid-Binding Protein (AP-2) and Adiponectin (AdipoQ) are crucial for the differentiation of both WAT and BAT and serve as general terminal adipogenic differentiation markers. The formation of the mature brown adipocytes is achieved upon the expression of the specific markers for brown fat cells including Uncoupling-Protein-1 (UCP-1), PPARγ Coactivator-1α (PGC-1α), Elongation of Very Long chain fatty acids gene 3 (Elovl3) and Cell Death-Inducing DNA Fragmentation Factor-like Effector A (Cidea).12
p53, the well known tumor suppressor, that is central for the regulation of cell proliferation and apoptosis was also found to be involved in numerous other process.13, 14, 15, 16, 17 Notably, p53 has been demonstrated as an important regulator of development and differentiation programs, either via its control of cell proliferation or by regulating the expression of specific genes involved in these specific programs.18 This multitask protein may either induce or suppress key differentiation factors depending on the specific cellular fate.19
Although increasing evidence from in vitro and in vivo studies suggest that p53 exerts a negative effect on white adipogenic differentiation,19, 20, 21 its role in brown fat differentiation has not been determined.
The proposed inhibitory effect of p53 on white adipogenesis prompted us to further investigate the involvement of p53 along the adipocyte differentiation program and its effect on adipose tissues following diet-induced obesity. Additionally, we aimed to elucidate the putative role of p53 in brown adipogenic differentiation. Here, we report that p53 has an inhibitory role on white adipogenesis. This effect was observed both in mouse and human models representing various stages of white adipogenic differentiation. Moreover, the results of our in vivo analysis indicated that p53 protects against diet-induced obesity. Our data also present for the first time a role of p53 in the regulation of brown adipogenic differentiation program. In contrast to the suppressive effect of p53 on white fat differentiation, results from both in vitro and in vivo models revealed that p53 promotes proper BAT differentiation, during embryonic development and in adults.
These data position p53 as a new regulatory factor that maintains proper adipogenic differentiation, to prevent the development of obesity, its associated metabolic disorders and cancer formation.
Results
p53 inhibits the white adipogenic differentiation program
Our previous study demonstrated a negative regulatory role of p53 in white adipogenic differentiation of mouse embryonic fibroblasts (MEFs).19 To corroborate the negative effect that p53 exerts on white adipogenic differentiation, we used additional mouse and human cell lines that represent various stages of adipogenic differentiation.
First, we used the multipotent C3H10T1/2 cell line.22 These cells represent a model for investigation of early events in adipogenesis such as commitment of stem cells to adipocyte lineage.23 To investigate the effect of p53 on adipogenic differentiation of C3H10T1/2 cells, we knocked down p53 in these cells (C3H10T1/2-sh-p53) and compared their differentiation capacity with control cells (C3H10T1/2-sh-con). p53 expression was downregulated and followed by reduction of its target gene p21 in the C3H10T1/2-sh-p53 cells (Figure 1a). To test whether p53 knockdown affected adipogenic differentiation capacity of C3H10T1/2 cells, we analyzed the expression levels of adipogenic differentiation markers at several time points throughout the differentiation process. C3H10T1/2-sh-p53 exhibited a substantially higher expression of adipogenic markers than in their control counterparts. (Figure 1b). The Oil Red O staining indicated extensive adipocyte differentiation with numerous large lipid droplets of the induced sh-p53 cells compared with the sh-con cells (Figure 1c). These data further validate the notion that p53 exerts an inhibitory effect on white adipogenic in MEFs as suggested before.19

p53 knockdown enhances white adipogenic differentiation of C3H10T1/2 cells. (a) C3H10T1/2 cells were infected with retroviruses encoding for either p53-directed sh-RNA (sh-p53) or control sh-RNA (sh-con). Relative expression levels of p53 (left panel) and p21 (middle panel) were determined by QRT-PCR or by western blot analysis (right panel). GAPDH serves as a loading control. (b and c) Confluent cultures of C3H10T1/2 sh-con and C3H10T1/2 sh-p53 cells were subjected to induction of adipogenic differentiation. Total RNA was isolated before the induction of differentiation (0d), as well as 2 and 7 days later. Relative expression of PPARγ, CEBPα, Ap2 and AdipoQ were determined by QRT-PCR analysis. The results indicate the mean±S.D. of two duplicate runs. (b) Adipogenic differentiation was assessed using Oil Red O staining for lipid droplets (c)
The 3T3-L1 pre-adipocyte cell line serves as a genuine model for differentiation of committed white pre-adipocytes into adipocytes.23 Indeed these pre-adipocytes that carry wild-type (wt) p53 have been extensively used to explore the molecular regulation of terminal adipogenic differentiation.1 Thus, to investigate the role of p53 at advanced steps of adipogenic differentiation, we downregulated p53 expression in this cell line as well (Figure 2a). In agreement with others,1 our 3T3-L1-sh-con cells displayed extensive adipogenic differentiation following induction. As judged by Oil Red O staining, knockdown of p53 in the 3T3-L1 cells did not result in significant enhancement of their differentiation capacity (Figure 2b). This raised a possibility that p53 that is usually kept at very low levels in cells needs to be activated in order to affect adipogenic differentiation in these cells. Therefore, to further characterize the role of p53 in the adipogenic differentiation of the 3T3-L1, we treated the cells with Nutlin-3 that induces stabilization and activation of p53.24 As expected, treatment with Nutlin-3 led to an increase in p53 and p21 protein levels (Figure 2c). Accordingly, we speculated that if activated p53 would inhibit adipogenesis, it should be reflected by reduced levels of PPARγ, which is critical for adipogenesis and is required for maintenance of the differentiation state. Indeed, Nutlin-3 treatment resulted in a considerable reduction in PPARγ basal levels in a p53-dependent manner (Figure 2d). Moreover, elevation of p53 activity by Nutlin-3 prior to differentiation induction, led to repression of terminal adipogenic differentiation of the 3T3-L1-sh-con cells, as manifested by a marked reduction in the formation of cytoplasmic lipid droplets, and lower expression of terminal adipogenic differentiation markers compared with the sh-p53 cells (Figures 2e and f). Hence, although basal p53 activity did not restrict the differentiation potential of the 3T3-L1 cells, p53-dependent suppressive effect could be observed upon elevation of p53 activity.

Activation of p53 in the 3T3-L1 cells leads to attenuation of their differentiation capacity. (a) 3T3-L1 cells were infected with retroviruses encoding for either p53-directed sh-RNA (sh-p53) or control sh-RNA (sh-con). The subsequent downregulation of p53 expression levels was estimated by western blot analysis. GAPDH serves as a loading control. (b) 3T3-L1-sh-con and 3T3-L1-sh-p53 cells were subjected to the induction of adipogenic differentiation that was assessed using Oil Red O staining for lipid droplets. (c and d) 3T3-L1-sh-con and 3T3-L1-sh-p53 cells were either treated with Nutlin-3 at a final concentration of 25 μM, for 48 h (Nutlin) or maintained in control medium (Control). Western blot analysis was performed for p53 and p21. GAPDH serves as a loading control (c). Relative expression of PPARγ was determined by QRT-PCR. Normalized expression levels in control samples were set to 100%. The results are presented as the mean±S.D. of two duplicate runs (d). (e and f) 3T3-L1-sh-con and 3T3-L1-sh-p53 cells were pretreated with Nutlin-3 at a final concentration of 25 μM for 48 h and then subjected to induction of adipogenic differentiation. Adipogenic differentiation was assessed using Oil Red O staining for lipid droplets (e). Total RNA was isolated before the induction of differentiation (Control) as well as 7 days later (Induced). Relative expression of Ap2 and AdipoQ were determined by QRT-PCR analysis. The results indicate the mean±S.D. of two duplicate runs (f)
Next, we examined whether p53-dependent repression of white adipogenesis is also applicable in cells from human origin. Human adipose-derived stem cells (hASCs) were knocked down for p53 and exhibited significant reduction in p53 and p21 mRNA and p53 protein levels (Figure 3a). sh-p53 hASCs displayed enhanced lipid droplet formation and elevated expression of adipogenic markers both at basal conditions and following differentiation induction (Figures 3b and c). These data suggest that the negative effect of p53 on white adipogenic differentiation is a general phenomenon that is present both in mouse and human cell lines.

p53 knockdown enhances adipogenic differentiation of human ASCs. (a) ASCs were infected with retroviruses encoding for either p53 directed sh-RNA (sh-p53) or control sh-RNA (sh-con). Relative expression levels of p53 (left panel) and p21 (right panel) were determined by QRT-PCR, or by western blot analysis (middle panel). GAPDH serves as a loading control. (b and c) Confluent cultures of ASCs-sh-con and ASCs-sh-p53 cells were maintained in either adipogenic differentiation medium (Induced) or in control medium (Control) for 3 weeks. Adipogenic differentiation was assessed using Oil Red O staining for lipid droplets (b). Relative expression of PPARγ2, Ap2 and AdipoQ were determined by QRT-PCR analysis. The results indicate a mean±S.D. of two duplicate runs (c)
p53 protects against diet-induced obesity
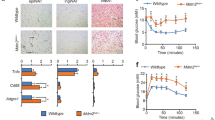
Our in vitro studies have prompted us to examine whether the suppressive effect of p53 is retained in WAT development, in vivo. First, we collected epididymal fat from wt and p53 knockout (KO) mice and measured the basal levels of PPARγ mRNA expression in it. Figure 4a shows that p53 KO mice exhibited a statistically significant elevation of this key adipogenic transcription factor, compared with the wt mice.

p53 KO mice exhibit a higher increase in body fat percentage. (a) Total RNA was isolated from epididymal fat from wt and p53 KO mice. The relative expression level of PPARγ mRNA was determined with QRT-PCR. Each rhombus represents the expression level of one mouse from the indicated group. (b and c) p53 wt and KO male mice, 7 weeks of age, were fed for 4 weeks with either a high-fat high sucrose diet (HFD) or a normal diet (ND). The p53 KO and wt groups contained 11 mice that were kept on the HFD and 5 mice that were kept on a ND. Total body weight and body fat percentage were measured weekly. All data is presented as mean±S.D. (b) Curves represent the gain of body fat percentage. Asterisks indicate a P-value<0.05, generated from t-test analysis. (c) Curves represent the gain of weight percentage of the mice along the experiment
In order to elucidate the physiological role of p53 in adipogenesis in vivo, we decided to compare wt and p53 KO mice in the context of diet-induced obesity. To this end, wt and p53 KO mice were maintained, either on a normal chow diet (ND) or fed with high-fat high sucrose diet (HFD) for 4 weeks during which their body weight and fat mass was measured. Both wt and p53 KO mice kept on a ND maintained their initial proportion of body fat throughout the experiment (Figure 4b),demonstrating that at normal conditions, there is no difference between the fat mass of p53 KO and wt mice. As for the results obtained following a HFD, the increase of body fat percentage was higher in the p53 KO mice compared with the wt mice throughout the experiment. Comparing the gain of body weight of the mice, there was no statistically significant difference between p53 wt and KO mice kept on either of the two diets. However, p53 KO HFD mice exhibited a trend of higher weight gain, suggesting a tendency of these mice to be more prone to obesity (Figure 4c). This suggests that these mice are more prone to obesity, implying the regulatory role of p53 as an inhibitor in the process of white fat accumulation in vivo. Obesity may be induced by increased energy intake or/and decreased energy expenditure. To examine these possibilities, we monitored daily food intake and locomotor activity in wt and p53 KO mice. We found that both these parameters were comparable in wt and p53 KO mice (Supplementary Figure S1).
Collectively, our results demonstrate that the negative effect of p53 on white adipogenesis observed in our in vitro experiments, is retained in vivo, strongly suggesting that these observations represent an authentic inhibitory role of p53 on white adipogenic differentiation.
p53 is essential for brown adipocyte differentiation in vitro
The role of p53 in brown fat differentiation is still unknown. To investigate whether p53 might be involved in brown fat differentiation program, we adopted the skeletal muscle myogenic cell-line, C2, as a model system, as the brown adipocyte shares a common progenitor cell with the skeletal muscle lineage. The induction of these cells into the brown fat lineage requires the expression of the transcription factor PRDM16, which, upon stable expression, commits these cells towards the differentiation into brown adipocytes.25 To this end, C2 cells were infected with either a retroviral vector encoding for PRDM16 or a control vector (Figures 5a and b). Upon induction of differentiation, the PRDM16 expressing C2 cells underwent efficient brown fat differentiation (Figure 5c). To evaluate a possible role of p53 in brown adipocyte differentiation, we altered the p53 status in the PRDM16 expressing C2 cells, by either knockdown with sh-RNA, or overexpression of a dominant negative mutant form of p53, p53R172H (mutp53). mutp53 acts as a dominant negative through heterooligomerization, with wt-p53 preventing its activity,26 and thus mirrors wt-p53 knockdown. The impaired wt p53 function was reflected by the low protein levels of the p53 target p21 (Figure 5d). Gene-expression analysis showed that wt p53 abrogation, by knockdown or mutation, substantially diminished the induced expression of both the general adipocyte differentiation marker Ap-2 and the selective genes for brown adipocytes, such as Cidea and Elovl3 (Figure 5e). Consistently, the Oil Red O staining displayed that the p53 abrogated cultures exhibited decreased capacity of lipid droplets formation, reflecting impaired differentiation (Figure 5f).

wt p53 is required for brown adipocyte differentiation of C2 cells. (a and b) For overexpression of PRDM16, C2 cells were infected with either flag-tagged PRDM16-vector (PRDM16) or empty control vector (vector). The overexpression was validated with both QRT-PCR analysis of PRDM16 relative mRNA expression. The results of QRT-PCR indicate the mean±S.D. of two duplicate runs (a). Western blot analysis using antibodies against flag-tag. β-tubulin serves as a loading control (b). (c) C2 cells overexpressing PRDM16 or control vector were treated with brown adipocyte induction medium. Adipogenic differentiation was estimated Oil Red O staining for lipid droplets. (d) C2-PRDM16 cells were infected with retroviruses encoding for either p53-directed sh-RNA (PRDM16-sh-p53), control sh-RNA (PRDM16) or mutant p53 form R172H (PRDM16-mutp53). Protein expression levels of p53 and p21 were determined by western blot analysis. β-tubulin serves as a loading control. (e and f) The cells were induced to undergo brown fat differentiation. Relative mRNA induction of differentiation markers was analyzed by QRT-PCR. The results are presented as a mean±S.D. of two duplicate runs (e). Adipogenic differentiation was estimated by Oil Red O staining for lipid droplets (f)
Taken together, these results suggest that in contrast to the negative effect that p53 exerts on white adipocytes differentiation, p53 is required for proper differentiation of brown adipocytes.
p53 deficiency leads to abnormalities in BAT development
The above results demonstrated that p53 is essential for brown adipocyte differentiation in vitro. In order to investigate whether p53 has a role during BAT development, we measured PRDM16 mRNA levels in p53 KO and p53 knockdown MEFs compared with wt controls. Figure 6a demonstrates that PRDM16 expression was markedly reduced in MEFs with abrogated p53 status suggesting that p53 might be involved in the development of BAT. Additionally, we examined the effect of p53 reduction in primary brown adipocytes. In concordance with the observation in MEFs, p53 knockdown in primary BAT cells also led to downregulation of PRDM16 expression (Supplementary Figures S2a and b). The induction of key brown fat thermogenic genes, such as Cidea and PGC-1α following cAMP treatment was abrogated by the p53 knockdown (Supplementary Figure S2c). While in mice, BAT emerges during fetal development, reaching maximum size close to birth, WAT development starts only after birth.2 Therefore, in consistent with our data obtained from primary cells, QRT-PCR analysis of BAT from 18.5 dpc wt and p53 KO sibling embryos indicated that the expression of PRDM16 was significantly lower in the p53 KO embryos compared with their wt counterparts (Figure 6b). In order to test whether the p53-mediated upregulation of PRDM16 mRNA results from binding of p53 to its promoter, we performed chromatin immunoprecipitaion (ChIP) analysis in BAT obtained from 18.5 dpc wt embryos. Chromatin was precipitated using antibodies against p53 or a non-specific antibody. The DNA region containing the PRDM16 promoter was significantly enriched in p53-precipitated samples compared with control-precipitated sample (Figure 6c). Histological analysis of brown fat sections obtained from wt and p53-null embryos showed that wt embryos exhibited a more condensed brown fat tissue than the corresponding p53 KO embryos (Figure 6d). Another interesting marker exclusively expressed in BAT is the proton transporter UCP1 that uncouples electron transport from ATP production, allowing the energy to dissipate as heat.27 Examination of UCP1 protein expression by immunohistochemistry and western blot demonstrated lower levels in BAT of p53 KO embryos compared with the wt embryos (Figures 6e–g). Thus, these in vivo data imply that p53 is required for proper brown fat development and function.

Abnormalities in BAT isolated from p53 KO mice. (a) Total RNA was isolated from MEFs-sh-con, MEFs-sh-p53 and p53 KO MEFs. The relative expression level of PRDM16 mRNA was determined by QRT-PCR. The results indicate the mean±S.D. of two duplicate runs. (b) Total RNA was isolated from brown fat tissues of 8wt and 8 p53 KO 18.5 dpc embryos and mRNA levels were determined using QRT-PCR. Each rhombus represents an individual embryo. The statistical significance was determined by Student’s t-test, P=0.012. (c) Protein–DNA complexes from BAT of wt 18.5 dpc embryos were immunoprecipitated with antibodies against p53 (monoclonal anti-p53 PAb 421 and anti-p53 polyclonal PAb) or a nonspecific antibody (anti-large T antigen). The amount of precipitated DNA was measured by QRT-PCR with specific primers directed against PRDM16 promoter and normalized to the 1% input. The results indicate the mean±S.D. of two duplicate runs. (d) Representative transverse sections obtained from 18.5 dpc sibling mouse embryos stained with H&E. High magnification images are indicated to the right of each section. (e) Representative transverse sections of similar anatomical levels obtained from 18.5 dpc sibling mouse embryos stained with α-UCP1 Ab (Red, 20 × magnification). (f) Western blot analysis of brown fat tissue, obtained from 18.5 dpc embryos, using anti-UCP1 antibody. β-tubulin was used as a loading control. (g) Quantification of the immunoblot from (f) using imageJ. Each dot represents an individual embryo; asterisks indicate a P-value<0.05, generated from t-test analysis. (h) Total RNA was isolated from interscapular BAT obtained from 5 wt and 5 p53 KO mice. The relative expression level of genes expressed selectively in brown adipocytes (PRDM16, PGC-1α and CEBPβ) was determined by QRT-PCR. Each rhombus represents the expression level of one mouse from the indicated group. Asterisks indicate a P-value <0.05, generated from t-test analysis. (i) Representative images of H&E stained sections of interscapular BAT from p53 wt and p53 KO mice. High magnification images are indicated to the right of each section
To evaluate the effect of p53 on BAT of adult mice, we performed gene expression analysis of BATs isolated from the interscapular region of wt and p53 KO adult mice. Owing to their central role in the maintenance of a brown adipocyte phenotype, PRDM16, PGC-1α and C/EBPβ can advantageously be used as selective markers of brown fat cells.25, 28 QRT-PCR analysis showed that expression levels of all the three markers were significantly reduced in the KO mice compared with their wt counterpart (Figure 6h). Histological analysis of these BATs revealed that p53 KO mice exhibited an atrophied BAT with smaller cells and vacuoles compared with the normal BAT characteristics of their wt counterparts (Figure 6i).
These findings suggest that p53 KO mice display an impaired phenotype of BAT both at embryonic and adult stage, further supporting the notion that intact p53 function is required for maintaining normal characteristics of brown adipocytes and the integrity of BAT morphology.
Discussion
Balancing between white and brown adipogenic differentiation has an important role in maintaining an organism’s normal metabolic homeostasis. A deeper understanding of the pathways and intermediates between the mesenchymal stem cells and the two types of mature adipocytes may yield therapeutic interventions to prevent obesity and its associated disorders. In this study, we set out to examine the involvement of p53 in differentiation of both white and brown adipocytes. By using several mouse and human in vitro models, we demonstrate that p53 inhibits white adipogenesis, and suggest for the first time that p53 may antagonize diet induced obesity. In contrast to the suppressive effect p53 seem to exert on white adipocyte differentiation, we report that p53 is required for proper brown adipocyte differentiation program both in vitro and in vivo.
As most studies use cell lines that have a restrictive potential to differentiate into other cell types, the molecular mechanism regulating terminal adipogenic differentiation stages was extensively studied and thus is well characterized compared with the early determination events.1 Here, we provide evidence that p53-dependent repression of white adipogenesis is a general phenomenon that is present in cells representing various differentiation stages from both mouse and human origin. It should be noted that although the anti-adipogenic effect of p53 observed in 3T3-L1 cell line is at least partially due to its ability to block cell division,29 the mechanism for suppression of adipogenesis in the C3H10T1/2 multipotent stem cells and hASCs that do not undergo mitotic clonal expansion is probably different and independent of p53 primarily function as an inducer of growth arrest.
Although previous examination of p53 expression levels in proliferating ASCs and terminally differentiated adipocytes did not reveal any differences,30 we show that knockdown of p53 in hASCs resulted in a significant increase in their adipogenic differentiation capacity, supporting the restrictive effect of p53 on white adipogenic differentiation as demonstrated in other cellular systems. Recent reports show that ASCs may represent a cell population linking obesity and cancer.31, 32 However, the biological mechanisms that link obesity to cancer and the role of p53 in this pathway still need further extensive research.
In normal physiology, it is conceivable that the increased accumulation of intracellular lipids that occurs both during differentiation and upon excess energy intake will induce a stress signal to large adipocytes, leading to p53 activation. In this situation, p53 might be required for the maintenance of a homeostatic condition in the hypertrophied adipocytes. This potential of p53 might be mediated through its repression of the key adipogenic transcription factor PPARγ. Our in vitro study demonstrated that p53 serves as a potent transcriptional repressor of PPARγ (Figure 2d). Our finding that the basal level of PPARγ expression was increased in p53 KO mice (Figure 4a), further supports this hypothesis as a mechanistic basis for how p53 exerts its inhibitory effect on adipogenesis.
Following these observations, we examined the role of p53 in adipogenesis in the context of diet-induced obesity. The results showed a significant increase in the gain of body fat percentage in p53 KO mice fed with HFD compared with wt mice fed with the same diet. In agreement with our findings regarding body fat percentage, we observed that p53 KO mice have a tendency to gain more body weight compared with wt mice, suggesting that these mice are more prone to obesity.
Results from several studies have implicated a role of BAT in the protection against a surplus energy intake.33 Thus, increasing BAT mass or activity could be a useful approach to limit obesity and its associated disease states. In contrast to white adipogenic differentiation, factors and mechanisms governing brown adipogenic differentiation and BAT development have not been extensively investigated. Here, we show for the first time that p53 is essential brown fat differentiation program and is implicated in proper BAT development and function.
Our in vitro data demonstrates that abrogation of p53 function results in impaired differentiation and reduction in mRNA expression levels of BAT-specific markers in C2 cells (Figure 5). These findings imply a tight p53-dependent regulation of this cellular program. Hence, although p53 suppresses white adipogenic differentiation, it seems to be required for brown adipogenic differentiation.
In consistency with the in vitro results, our in vivo data demonstrated that p53 is implicated in the integrity of BAT both during embryonic development and in adults. The significant reduction in the levels of the brown adipocyte markers observed in p53 KO mice, illustrates this broad impact. ChIP analysis of BAT indicated that PRDM16 promoter is bound by wild-type p53 in vivo (Figure 6c). These data provide a mechanistic link between p53 function in brown adipogenic differentiation and PRDM16 transcriptional induction.These results imply that brown fat cells in p53 KO mice are incapable of expressing a complete brown-adipocyte phenotype.
Interestingly, it was recently reported that p53 was identified during proteomic analysis of immunopurified differentiation competent PRDM16 complexes.28 This analysis was performed in immortalized brown fat cells in which PRDM16 was introduced via overexpression. This suggests a possible protein–protein interaction between p53 and PRDM16. Together with our data, the data provided in the manuscript by Kajimura S et al.28 may indicate that p53 affects brown adipogenesis by both the transcriptional induction of PRDM16 and protein–protein interaction with PRDM16.
As for the histological analysis of p53 null mice, we observed that BAT from p53 KO mice displayed abnormal morphology. The above described reduction in expression levels of key genes for brown adipocyte function, is likely to account for a substantial portion of this altered morphology. This effect, suggests that p53 might be involved not only in brown fat differentiation and development, but also in maintaining the full integrity of a brown fat phenotype. Together, these findings suggest the requirement of p53 function in sustaining a brown adipocyte phenotype.
In summary, we found that although p53 inhibits WAT accumulation, which is associated with obesity and several pathological conditions, it is implicated in proper BAT differentiation, which seemingly is protective against obesity. It is plausible that this direct regulation of cell differentiation constitutes an additional modality, in which p53 operates to maintain a homeostatic state, not only in single cells, but in the organism at large. Epidemiological studies link obesity with several types of cancers.6, 34 However, the biological mechanisms underlying the link between obesity and cancer and the possible role of p53 in this pathway are still poorly understood. Abrogation of p53 function, by its deficiency or mutation, apparently results in aberrant differentiation, which may give rise to transformed cells. Additional oncogenic events, can result in their malignant transformation and tumor formation. Our findings demonstrate that p53 deficiency results in altered differentiation, as well as increased WAT accumulation and may provide a link between obesity, impaired metabolism and cancer.
Materials and Methods
Cell culture and differentiation induction
The multipotent C3H10T1/2 cells, pre-adipocyte cell line 3T3-L1 and the amphotropic and ecotropic Phoenix retrovirus-producing cells were obtained from the American Type Culture Collection (ATCC) (Rockville, MD, USA). MEFs were derived from p53 wt, p53 KO 13.5 dpc embryos. All were maintained with DMEM supplemented with 10% FCS and antibiotics. The skeletal muscle-committed cell-line C2 cells were cultured in DMEM supplemented with 20% FCS and antibiotics. ASCs were isolated as described in Folgiero et al.30 Cells were maintained in DMEM F12 with 10% FCS and antibiotics. Primary brown adipocytes were derived and from newborn wt mice and maintained as described in Klein et al.35 For cAMP treatment, cells were incubated with 10 μM forskolin (Sigma, St Louis, MO, USA) for 4 h.
For white adipogenic differentiation, induction confluent C3H10T1/2 cells were treated with growth medium supplemented with 20 nM insulin, 0.5 mM IBMX, 5 mM dexamethasone, 1 nM T3 and 0.125 mM indomethacin (Sigma). Following 48 h, cells were maintained in medium with 20 nM insulin and 1 nM T3 for an additional period of 5–7 days, with medium replacement every other day. 3T3-L1 cells were plated at a high density to reach confluence. At 2 days post confluence, cells were induced to differentiate with growth medium supplemented with 5 μg/ml insulin, 0.5 mM IBMX, 1 μM dexamethasone, (all from Sigma). After 48 h, the media were replaced with DMEM supplemented with 10% FCS and 5 μg/ml insulin for additional period of 5–7 days with medium renewal every other day. For Nutlin-3 treatment, cell cultures were treated with Nutlin-3 (Alexis Corporation, San Diego, CA, USA) at a final concentration of 25 μM for 24 h. Stock solution was prepared as 10 mM in DMSO. Confluent ASCs were treated with 10 μg/ml insulin, 0.5 mM IBMX, 1 μM dexamethasone and 0.1 mM indomethacin for 14 days with medium renewal every other day. Adipogenesis was detected by Oil Red O staining.
Brown adipocyte differentiation in C2 cells was induced by treating confluent cells with DMEM containing 10% FCS, 850 nM insulin, 0.5 mM IBMX, 125 nM indomethacin, 1 mM dexamethasone, 1 nM T3 (all from Sigma) and 1 μM rosiglitazone (Enzo Life Sciences, Farmingdale, NY, USA). After 48 h, cells were switched to the maintenance medium containing 10% FCS, 850 nM insulin, 1 nM T3 and 1 μM rosiglitazone (all from Sigma) for an additional period of 5–7 days.
Mice
C57BL/6 p53-wt and p53-KO mice were maintained inside a barrier facility, and experiments were performed in accordance with Weizmann Institute of Science regulations for animal care and handling. For diet-induced obesity, 6–8 weeks-old male wt and p53 KO mice were fed either a normal chow diet or on a high-fat high sucrose diet (Research Diets, product # D12451, New Brunswick, NJ, USA) for 5 weeks. Food and water were available ad libitum. Body mass and body fat percentage were weekly measured using a regular scale and an EchoMRI-100 instrument (Echo Medical Systems LLC, Houston, TX, USA), respectively. For measurement of food intake and detection of locomotor activity, mice were single housed. Food intake was measured by subtracting uneaten food from the initially premeasured food following 24 h. General locomotor activity was assessed using automated system with infrared detector (TSE Inframot-Activity System, TSE, Bad Homburg, Germany).
For histological analysis, adipose tissues were fixed in 4% paraformaldehyde, and embedded in paraffin blocks. Sections were stained with H&E.
Retroviral constructs and infections
For mouse p53 knockdown, the p53 short hairpin RNA (shRNA) vector and its human Rb shRNA control vector were kindly provided by Dr. SW Lowe (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, USA). For mutant p53 overexpression, pBABE-R172Hp53 was obtained following site-directed mutagenesis on pBABE-wt p53 construct.
For human p53 knockdown, the p53 short hairpin RNA (shRNA) vector, pWZL-shp53-blast and its mouse NOXA shRNA control vector, pWZL-shNOXA-blast were obtained by subcloning the p53 and NOXA fragments from pBabe-shp53-puro and pBabe-shNOXA-puro, correspondingly, which were kindly provided by Dr. D Ginsburg (Bar-Ilan University, Ramat Gan, Israel). For PRDM16 overexpression, MSCV-PRDM16-flag-puro was purchased from Addgene (Cambridge, MA, USA) and subcloned into pWZL-blast construct. The MSCV-puro empty vector was used as control for the infection. Retroviral infection procedures were done using Phoenix-producing cells as previously described in Milyavsky et al.36
RNA preparation and quantative real time PCR
Total RNA was isolated from cultured cells using a NucleoSpin II kit (Macherey-Nagel, Bethlehem, PA, USA). Total RNA from animal tissues was isolated following mechanical homogenization using TRI reagent (Molecular Research Center). A 2-μg aliquot of the total RNA was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (MMLV-RT; Bio-RT) and random hexamer primers. Quantitative real-time PCR (QRT-PCR) was performed using SYBR green PCR Master Mix (Applied BioSystems, Carlsbad, CA, USA) on an ABI 7300 instrument. Human and mouse mRNA levels were normalized to the level of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and HPRT (hypoxanthine phosphoribosyltransferase), respectively. PCRs were performed in duplicates, and error bars in charts represent the corresponding standard deviations. Primers designed for QRT-PCR are listed in Supplementary Table S1.
ChIP analysis
ChIP was performed as described by MacGill et al.37 Briefly, sonicated chromatin prepared from interscapular BAT tissues of 18.5 dpc wt mouse embryos was immunoprecipitated using the anti-p53 PAb421, anti-p53 polyclonal Ab and anti-SV40 PAb419 antibody. The enrichment of specific DNA sequences was measured by QRT-PCR.
Immunohistochemistry of adipose sections
Paraffin-embedded sections of 18.5 dpc embryos were deparafinizied, rehydrated and subjected to Tris–EDTA antigen retrieval buffer. Permeabilization was done in 0.2% Triton X-100 in PBS for 5 min, followed by incubating the sections in a blocking solution of 0.1% Triton and 3% BSA in PBS for 1 h, at room temperature. For immunostaining, sections were incubated overnight at room temperature (maintained in a humid chamber) with goat polyclonal anti-UCP1 (1 : 500, SANTA CRUZ). For fluorescent detection of UCP1, sections were incubated with donkey anti-goat antibody labeled with Cy3 flour (Jackson Immunoresearch Laboratories, West Grove, PA, USA), 1 : 5000 for 10 min and mounted with Elvanol.
Western blot analysis
For western blotting, total cell extracts were prepared; 50 μg protein of each sample was fractionated by gel electrophoresis, and transferred to nitrocellulose membranes. The following primary antibodies were used: rabbit polyclonal anti-p53 (1 : 1000, produced in our laboratory), rabbit polyclonal anti-p21 (1 : 1000, C-19; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal anti-UCP1 (1 : 200, Millipore, Billerica, MA, USA), mouse monoclonal anti-FLAG M2 (1 : 500, Sigma), mouse monoclonal anti-β-tubulin (1 : 2000, Sigma) and mouse monoclonal anti-GAPDH (1 : 1000, MAB374; Chemicon International, Chandlers Ford, UK). Horseradish peroxidase anti-mouse and anti-rabbit (Sigma) were used as secondary antibodies. The signal was detected by the super-signal-enhanced chemiluminescence system (Pierce, Rockford, IL, USA).
Statistical analysis
Statistical significance was evaluated using a either by one-tailed Wilcoxon signed rank test or one-tailed unpaired Student’s t-test, and asterisks in figures represent P values <0.05.
Abbreviations
- AdipoQ:
-
Adiponectin
- AP-2:
-
Adipocyte-Fatty-Acid-Binding-Protein
- hASC:
-
human Adipose-derived stem cells
- ATCC:
-
American Type Culture Collection
- BAT:
-
brown adipose tissue
- CEBPa:
-
CCAAT-Enhancer-Binding-Protein-α
- ChIP:
-
chromatin immunoprecipitaion
- Cidea:
-
Cell Death-Inducing DNA Fragmentation Factor-like Effector A
- Elovl3:
-
Elongation of Very Long chain fatty acids gene 3
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- HFD:
-
high fat high sucrose diet
- HPRT:
-
hypoxanthine phosphoribosyltransferase
- KO:
-
knockout
- MEFs:
-
mouse embryonic fibroblasts
- mutp53:
-
p53R172H
- ND:
-
normal chow diet
- PGC-1α:
-
PPARγ Coactivator-1α
- PPARγ:
-
Peroxisome Proliferator-Activated-Receptor-γ
- PRDM16:
-
Positive-Regulatory-Domain-Containing-16
- shRNA:
-
short hairpin RNA
- UCP-1:
-
Uncoupling Protein-1
- WAT:
-
white adipose tissue
- wt:
-
wild type
References
Rosen ED, MacDougald OA . Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 2006; 7: 885–896.
Farmer SR . Molecular determinants of brown adipocyte formation and function. Genes Dev 2008; 22: 1269–1275.
Cristancho AG, Lazar MA . Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol 2011; 12: 722–734.
Lidell ME, Enerback S . Brown adipose tissue--a new role in humans? Nat Rev Endocrinol 2010; 6: 319–325.
Gesta S, Tseng YH, Kahn CR . Developmental origin of fat: tracking obesity to its source. Cell 2007; 131: 242–256.
Khandekar MJ, Cohen P, Spiegelman BM . Molecular mechanisms of cancer development in obesity. Nat Rev Cancer 2011; 11: 886–895.
Vegiopoulos A, Muller-Decker K, Strzoda D, Schmitt I, Chichelnitskiy E, Ostertag A et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 2010; 328: 1158–1161.
Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med 2009; 360: 1509–1517.
Lowell BB, V SS, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB et al. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 1993; 366: 740–742.
Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab 2007; 6: 38–54.
Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL et al. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev 2008; 22: 1397–1409.
Fruhbeck G, Sesma P, Burrell MA . PRDM16: the interconvertible adipo-myocyte switch. Trends Cell Biol 2009; 19: 141–146.
Vousden KH, Ryan KM . p53 and metabolism. Nat Rev Cancer 2009; 9: 691–700.
Vousden KH, Prives C . Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137: 413–431.
Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G . The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol 2011; 12: 259–265.
Tapia N, Scholer HR . p53 connects tumorigenesis and reprogramming to pluripotency. J Exp Med 2010; 207: 2045–2048.
Zhu Y, Prives C . p53 and Metabolism: The GAMT Connection. Mol Cell 2009; 36: 351–352.
Molchadsky A, Rivlin N, Brosh R, Rotter V, Sarig R . p53 is balancing development, differentiation and de-differentiation to assure cancer prevention. Carcinogenesis 2010; 31: 1501–1508.
Molchadsky A, Shats I, Goldfinger N, Pevsner-Fischer M, Olson M, Rinon A et al. p53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS One 2008; 3: e3707.
Armesilla-Diaz A, Elvira G, Silva A . p53 regulates the proliferation, differentiation and spontaneous transformation of mesenchymal stem cells. Exp Cell Res 2009; 315: 3598–3610.
Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y et al. p53 Activation in adipocytes of obese mice. J Biol Chem 2003; 278: 25395–25400.
Reznikoff CA, Brankow DW, Heidelberger C . Establishment and characterization of a cloned line of C3H mouse embryo cells sensitive to postconfluence inhibition of division. Cancer Res 1973; 33: 3231–3238.
Bowers RR, Lane MD . A role for bone morphogenetic protein-4 in adipocyte development. Cell Cycle 2007; 6: 385–389.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848.
Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008; 454: 961–967.
Cadwell C, Zambetti GP . The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of-function on cell growth. Gene 2001; 277: 15–30.
Cannon B, Nedergaard J . Brown adipose tissue: function and physiological significance. Physiol Rev 2004; 84: 277–359.
Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP et al. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 2009; 460: 1154–1158.
Hallenborg P, Feddersen S, Madsen L, Kristiansen K . The tumor suppressors pRB and p53 as regulators of adipocyte differentiation and function. Expert Opin Ther Targets 2009; 13: 235–246.
Folgiero V, Migliano E, Tedesco M, Iacovelli S, Bon G, Torre ML et al. Purification and characterization of adipose-derived stem cells from patients with lipoaspirate transplant. Cell Transplant 2010; 19: 1225–1235.
Zhang Y, Bellows CF, Kolonin MG . Adipose tissue-derived progenitor cells and cancer. World J Stem Cells 2010; 2: 103–113.
Martin-Padura I, Gregato G, Marighetti P, Mancuso P, Calleri A, Corsini C et al. The white adipose tissue used in lipotransfer procedures is a rich reservoir of CD34+ progenitors able to promote cancer progression. Cancer Res 2012; 72: 325–334.
Lidell ME, Enerback S . Brown adipose tissue--a new role in humans? In: Nat Rev Endocrinol 2010; vol. 6: England pp 319–325.
Calle EE, Kaaks R . Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 2004; 4: 579–591.
Klein J, Fasshauer M, Ito M, Lowell BB, Benito M, Kahn CR . beta(3)-adrenergic stimulation differentially inhibits insulin signaling and decreases insulin-induced glucose uptake in brown adipocytes. J Biol Chem 1999; 274: 34795–34802.
Milyavsky M, Shats I, Erez N, Tang X, Senderovich S, Meerson A et al. Prolonged culture of telomerase-immortalized human fibroblasts leads to a premalignant phenotype. Cancer Res 2003; 63: 7147–7157.
McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY . Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc Natl Acad Sci USA 2006; 103: 18267–18272.
Acknowledgements
This research was supported by a Center of Excellence grant from Flight Attendant Medical Research Institute (FAMRI), Yad Abraham Center for Cancer Diagnosis and Therapy and by EC FP7-INFLACARE number-223151. VR is the incumbent of the Norman and Helen Asher Professorial Chair Cancer Research at the Weizmann institute. ED is the incumbent of the Henry J Leir Professorial Chair. We are grateful to Dr. Ori Brenner for pathological analysis and to Dr. Inbal Biton and Dr. Michael Tzoory for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by K Vousden
Disclaimer
This publication reflects the authors’ views and not necessarily those of the European Community. The EC is not liable for any use that may be made of the information contained herein.
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Molchadsky, A., Ezra, O., Amendola, P. et al. p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ 20, 774–783 (2013). https://doi.org/10.1038/cdd.2013.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.9
Keywords
This article is cited by
-
Effect of metabolically divergent pig breeds and tissues on mesenchymal stem cell expression patterns during adipogenesis
BMC Genomics (2024)
-
Regulatory modules of human thermogenic adipocytes: functional genomics of large cohort and Meta-analysis derived marker-genes
BMC Genomics (2021)
-
NADPH levels affect cellular epigenetic state by inhibiting HDAC3–Ncor complex
Nature Metabolism (2021)
-
The multifunctional protein E4F1 links P53 to lipid metabolism in adipocytes
Nature Communications (2021)
-
Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue
Cell Death & Disease (2020)

{kind=link}
{kind=link}