
Abstract
Following acute-phase infection, activated T cells are terminated to achieve immune homeostasis, failure of which results in lymphoproliferative and autoimmune diseases. We report that sterile α- and heat armadillo-motif-containing protein (SARM), the most conserved Toll-like receptors adaptor, is proapoptotic during T-cell immune response. SARM expression is significantly reduced in natural killer (NK)/T lymphoma patients compared with healthy individuals, suggesting that decreased SARM supports NK/T-cell proliferation. T cells knocked down of SARM survived and proliferated more significantly compared with wild-type T cells following influenza infection in vivo. During activation of cytotoxic T cells, the SARM level fell before rising, correlating inversely with cell proliferation and subsequent T-cell clearance. SARM knockdown rescued T cells from both activation- and neglect-induced cell deaths. The mitochondria-localized SARM triggers intrinsic apoptosis by generating reactive oxygen species and depolarizing the mitochondrial potential. The proapoptotic function is attributable to the C-terminal sterile alpha motif and Toll/interleukin-1 receptor domains. Mechanistically, SARM mediates intrinsic apoptosis via B cell lymphoma-2 (Bcl-2) family members. SARM suppresses B cell lymphoma-extra large (Bcl-xL) and downregulates extracellular signal-regulated kinase phosphorylation, which are cell survival effectors. Overexpression of Bcl-xL and double knockout of Bcl-2 associated X protein and Bcl-2 homologous antagonist killer substantially reduced SARM-induced apoptosis. Collectively, we have shown how T-cell death following infection is mediated by SARM-induced intrinsic apoptosis, which is crucial for T-cell homeostasis.
Similar content being viewed by others


Main
The immune system clears pathogens via a coordinated network of cells, tissues and organs. The activated immune system must be downregulated in a timely manner to restore homeostasis. Although abundant information is available on immune activation,1 less is known on its restoration to basal level.
Apoptosis is a crucial mechanism of immune defense. It prevents excessive inflammation, limits the spread of microbes and initiates adaptive immune response.2 Apoptosis is vital for T-cell development and maintenance. Following infection, an antigen-specific T-cell clonal population expands to kill the pathogen. Subsequently, two distinct forms of cell death clear the expanded T cells: (i) mitochondria-dependent neglect-induced cell death (NID), elicited by the lack of survival cytokines like interleukin-2 (IL-2); (ii) mitochondria-independent activation-induced cell death (AICD), attributable to T-cell receptor (TCR) restimulation or death receptor activation.3, 4, 5 Failure in apoptosis impairs lymphocyte homeostasis, leading to autoimmunity and lymphoproliferative diseases.6
Extranodal nasal natural killer (NK)/T-cell lymphoma is a clinically aggressive tumor caused by proliferation of either cytotoxic T/NK cells. Malignant lymphocytes proliferate uncontrollably and amass in lymph nodes, spleen and other sites in the body. Pathogenesis involves either mutation or altered gene expression of tumor suppressor genes and oncogenes.7, 8 Although failure in cellular apoptosis is the generally accepted reason, the molecular mechanisms mediating T-cell apoptosis has eluded us for decades. The understanding of lymphocyte homeostasis is vital towards clinical intervention of T-cell lymphoma. Here we present findings on a novel factor in T-cell apoptosis.
Sterile α- and heat Armadillo-motif-containing protein (SARM), a purported Toll-like receptor (TLR) adaptor, is highly conserved. Unlike other TLR adaptors, SARM is prominently expressed in brain and T lymphocytes rather than macrophages.9 The C. elegans SARM homolog, Tir-1, acts independent of TLR signaling to protect the worm from infections.10, 11 Overexpression of SARM homologs downregulates TLR signaling.12, 13, 14, 15 However, SARM−/− mice macrophages respond normally to TLR agonists,9, 16 suggesting that SARM probably functions differently from other TLR adaptors.
Neurons from SARM−/− mice are protected from death by glucose and oxygen deprivation.9 Similarly, the C. elegans Tir-1 regulates anoxic death.17 The mouse and Drosophila SARM homologs are involved in axonal death.18 Global gene expression profiling of human NK/T-cell lymphoma indicated that SARM is downregulated in lymphoma.7 Our preliminary study also suggested that SARM is attenuated upon T-cell proliferation (Supplementary Figure S1a). Hence, we explored the mechanism of action of SARM on the survival/death of activated T cells.
We demonstrate how SARM mediates intrinsic T-cell apoptosis by modulating B cell lymphoma-extra large (Bcl-xL) and pERK. The proapoptotic function of SARM is mapped to the C-terminal sterile alpha motif (SAM) and Toll/IL-1 receptor (TIR) domains. During in vitro T-cell activation, the SARM level initially fell before rising, correlating inversely with the progression of T-cell proliferation and subsequent death. A similar drastic decrease in SARM was observed during in vivo activation of T cells. SARM-specific RNAi prolonged T-cell survival and rescued them from NID and AICD, supporting that SARM contributes to T-cell termination, failure of which affects T-cell homeostasis. Importantly, during influenza infection in vivo, SARM-knocked down T cells proliferated more than wild-type T cells. Consistently, the proapoptotic SARM is downregulated in NK/T-cell lymphoma patient tissues compared with healthy cells.
Results
The evolutionary conservation of SARM
SARM possesses two N-terminal Armadillo motif (ARM), two SAM and a C-terminal TIR domains (Supplementary Figure S1b). SARM is evolutionarily conserved for millions of years, suggesting its functional significance (Supplementary Figure S1c). The human and mouse SARM share 93% homology. Two mouse isoforms of 724 and 764 amino acids are reported. The shorter isoform is the splice variant of the longer isoform. RT-PCR indicated that mouse T cells express only the shorter isoform (Supplementary Figures S1b and d). Henceforth, we studied the role of SARM 724 in T cells using transgenic OTI+RAG−/− mouse strain, which contains homogenous populations of cytotoxic T cells (Supplementary Figure S1e) and a B6.PL-Thy1a/CyJ mouse strain, which expresses the congenic Thy1.1 marker. We employed HEK 293T cells to demonstrate SARM activity. To enable our study, we generated a series of constructs that expressed various domains of SARM (Figure 1a).

Induction of cell death by SARM in (b–g) HEK 293T and (h–i) Primary CD8 T cells. (a) A schematic representation of all the truncated constructs of SARM used in this study. HEK cells in 12-well plate were transfected with 800 ng of the indicated GFP fusion construct. (b) Cell death was quantified by dead cell stain, 7-AAD. (c) Cell lysates from 24 h post-transfection were immunoblotted with anti-GFP. The membrane was stripped and reprobed with anti β-actin. (d) White arrow indicates that SAM–TIR-transfected cells at 24 h post-transfection show distinctive morphological rounding up compared with GFP-transfected controls. (e) Nuclear condensation of SAM–TIR-transfected cells was visualized by DAPI staining. SAM–TIR–GFP-positive cells in 20 random microscopic fields were enumerated and the percentage was calculated based on GFP-positive transfected cells. (f) At 24 h post-transfection, the fragmented nuclei were quantified by labeling the nicked ends of the DNA by TUNEL assay. DNase I-treated cells were used as positive control. (g) SAM–TIR-transfected cells at 12 h were double-stained with Hoechst and propidium iodide to study cell membrane integrity and condensation of the nuclei. (h) Activated primary CD8 T cells were nucleofected with 4 μg of indicated truncated SARM constructs. Cell death was quantified 24 h post-nucleofection by 7-AAD staining. (i) Kinetic profile of primary CD8 T-cell death following nucleofection with SAM–TIR compared with GFP alone cells. Panels b, e and h are repeated for three independent times. Data in b, e and h represent means±S.D. of at least two independent experiments. *P<0.005; **P<0.0005
Expression of SARM induces primary T-cell death
To study the function of SARM, we expressed SARM-FL (full length) and various deletion mutants of SARM (ARM–SAM–TIR, SAM–TIR, SAM and TIR) in HEK 293T cells (Figure 1b). The expression competency of these truncated constructs was confirmed by immunodetection (Figure 1c). Cell death analysis indicated progressive death in SARM-FL-, ARM–SAM–TIR- and SAM–TIR-expressing cells (Figure 1b). SAM–TIR induced cell death similar to that of SARM-FL, suggesting that the SAM and TIR are the death domains. Consistently, ARM–SAM and ARM–TIR constructs (Figure 1a) were less potent compared with SARM-FL and SAM–TIR, highlighting the requirement of both SAM and TIR for cell death induction (Supplementary Figure S1f). Notably, overexpression of SAM–TIR led to rounding up of cells, (Figure 1d) nuclear condensation (Figure 1e) and nuclear fragmentation (Figure 1f). Hoechst and propidium iodide double staining of SAM–TIR-transfected cells showed chromatin condensation before the cell membrane was compromised (Figure 1g). To exclude possibility of cell death due to green fluorescent protein (GFP) fusion, we cotransfected deletion constructs of SARM and GFP, and showed that the SAM–TIR:GFP-coexpressing cells exhibited dose-dependent decrease in GFP mean fluorescence intensity and greater cell death compared with the control, suggesting specificity of SARM-mediated cell death (Supplementary Figure S2a–e).
Next, we examined whether SARM induces death in primary cytotoxic T cells. We observed that expression of ARM–SAM–TIR or SAM–TIR in CD8 T cells induced >80% cell death, higher than that induced by SARM-FL (Figure 1h). The cytotoxic effect of ARM–SAM–TIR and SAM–TIR was much more pronounced in CD8 T cells than in HEK 293T. SAM–TIR-expressing cells started to die as early as 3 h post-nucleofection, and by 24 h ∼90% were non-viable (Figure 1i). To substantiate the proapoptotic role of SARM, we assessed other cell types like neuronal (Neuro-2a), NK (NK-YS) and B (Raji B) cells, which express SARM. We found that both SARM-FL and SAM–TIR induced significant cell death in all these cell types (Supplementary Figures S3a–e). Taken together, our findings demonstrate that overexpression of SARM exhibited hallmark apoptotic features. The proapoptotic function was delineated to the SAM and TIR domains, and the role of SARM is conserved in various cell types.
SARM executes apoptosis via the initiator caspase-9
To confirm that SARM induces apoptosis, we investigated caspase activities. The proportion of SARM-FL- and SAM–TIR-transfected HEK 293T cells displaying caspase-3 activity increased significantly over 12–24 h (Figure 2a). Around 35% of SARM-FL-expressing T cells showed increased caspase-9 activity (Figure 2b). Furthermore, within 3 h post-nucleofection, a higher proportion of SAM–TIR-expressing T cells were active for caspase-9 relative to caspase-3 and -8. At 24 h, fewer cells exhibited caspase-8 activity than caspase-3 and caspase-9 (Figure 2c and Supplementary Figures S4a–c). Hence, the order of activation was caspases-9>3>8, supporting that SARM induces intrinsic apoptosis. Corroborating our observation, caspases-9 and -3 have been reported to activate caspase-8 during the amplification of apoptosis.19, 20 It is likely that during T-cell clearance, these caspases collaborate to execute apoptosis efficiently.

SARM induces MPTP, generates ROS and inhibits ERK phosphorylation. (a) HEK 293T cells were transfected with 800 ng of SARM-FL/SAM–TIR/GFP plasmids. At 12, 18 and 24 h post-transfection, cells were loaded with fluorogenic caspase-3 substrate and the proportion of caspase-3 active cells was quantified. (b) Activated CD8 T cells expressing SARM was assessed for caspase-9 activities at 12 and 24 h post-nucleofection. (c) Activated primary CD8 T cells were nucleofected with SAM–TIR GFP/GFP alone and assayed for caspases-9, -3 and -8 activities. Representative flow cytometry data depict the overlay of caspase activity histogram of SAM–TIR- and GFP-transfected cells at 3 h (first 3 panels). The line graph (fourth panel) indicates the proportion of caspases-3, -9 and -8 active cells during the indicated time points of 3, 6, 12 and 24 h post-nucleofection. (d) HEK 293T cells transfected with SARM-FL/ARM–SAM–TIR/SAM–TIR/GFP alone were loaded with 20 nM of TMRE dye at 12, 18 and 24 h post-transfection. Decrease in mitochondrial transmembrane potential (ΔΨm) was computed based on TMRE fluorescence for each of the constructs. (e) Activated primary CD8 T cells were nucleofected with SAM–TIR/GFP and ΔΨm was quantified as above. (f) Intracellular ROS was detected in HEK 293T cells transfected with SARM-FL/SAM–TIR, using CM-H2DCFDA, at 12, 18 and 24 h post-transfection. The proportion of cells with increased intracellular ROS were calculated and plotted. (g) Cell lysates were prepared from SARM-FL-, SAM–TIR-, GFP-transfected and control untransfected (UT) HEK 293T cells, and immunoblotted with anti-pERK. ERK was used as the loading control. UT and GFP transfection did not affect ERK phosphorylation. Data in a–f represent means±S.D. of at least three independent experiments. *P<0.0005; **P<0.008
SARM induces mitochondria permeability transition pore, generates ROS and inhibits ERK phosphorylation
Mitochondrial depolarization occurs during intrinsic apoptosis.21, 22 Thus, we examined the decrease in mitochondrial potential (ΔΨm) due to mitochondria permeability transition pore (MPTP) formation. By 24 h post-transfection, >45% of SARM-FL-, ARM–SAM–TIR- and SAM–TIR-expressing cells showed decreased ΔΨm (Figure 2d and Supplementary Figure S4d). Around 70% of T cells expressing SAM–TIR showed decreased ΔΨm (Figure 2e). We also observed increased intracellular reactive oxygen species (ROS) in SARM-expressing HEK 293T cells (Figure 2f and Supplementary Figure S4e). Consistently, the antioxidant, N-acetyl cysteine (NAC), dose dependently protected these cells from SARM-induced apoptosis (Supplementary Figure S5a). Interestingly, NAC-treatment reduced SARM-induced MPTP formation, highlighting that ROS production precedes loss of ΔΨm (Supplementary Figure S5b). However, antioxidant treatment did not completely prevent the drop in ΔΨm, suggesting that other parallel pathways are also operational.
Extracellular signal-regulated kinase (ERK) phosphorylation is important for cell survival, proliferation and differentiation,23 and is upregulated in cancer cells.24, 25 SARM-FL expression inhibited ERK phosphorylation (Figure 2g). Treatment with ERK inhibitors further enhanced SARM-induced apoptosis (Supplementary Figure S5c). Moreover, SARM-induced ROS was significantly increased during ERK inhibitor treatment, suggesting that downregulation of ERK phosphorylation by SARM might contribute to ROS production (Supplementary Figure S5d). However, antioxidant treatment did not affect SARM-mediated inhibition of ERK phosphorylation (Supplementary Figure S5e). These data clearly demonstrate that SARM mediates apoptosis by downregulating ERK phosphorylation, generating ROS and depolarizing mitochondria.
SARM mediates apoptosis via Bcl-2 family members
B cell lymphoma-2 (Bcl-2) family members guard mitochondrial integrity.26 We observed that expression of SARM-FL or SAM–TIR in T cells decreased the level of Bcl-xL (Figure 3a) without affecting Bcl-2. NAC and ERK inhibitor pretreatment indicated that downregulation of Bcl-xL is independent of both SARM-induced ROS production and downregulation of pERK (Supplementary Figure S5f and g). Reciprocally, overexpression of Bcl-xL caused 50% drop in SARM-FL-induced cell death (Figure 3b). Thus, SARM probably induces apoptosis by downregulating the anti-apoptotic Bcl-xL. To substantiate our findings, we transfected SARM-FL or SAM–TIR into HEK Bcl-xL stably expressing cell line, which showed reduced cell death by three-fold (Figure 3c and d). Consistently, using BAX−/− BAK−/− mouse embryo fibroblast (MEF) cells, we observed that Bcl-2 associated X protein (BAX) and Bcl-2 homologous antagonist killer (BAK)-mediated SARM-induced MPTP (Figure 3e). These data strongly corroborate that SARM mediates intrinsic apoptosis via the Bcl-2 family.

SARM mediates apoptosis via Bcl-2 family members. (a) Cell lysates were prepared from SARM-FL-, SAM–TIR-, GFP-transfected and control untransfected primary CD8 T cells, and immunoblotted and probed with anti-Bcl-xL and anti-Bcl-2. Actin was used as the loading control. (b) HEK 293T cells were cotransfected with Bcl-xL and SARM-FL/SAM–TIR in the ratio of 5 : 1. At 24 h post-transfection, the cells were stained with 7-AAD and assessed for cell death by flow cytometry. (c) Immunoblot of the lysates obtained from HEK 293T cells and HEK Bcl-xL cells probed with anti-Bcl-xL shows very strong expression of Bcl-xL compared with control HEK 293T cells. Actin was used as loading control. Molecular weight marker was run in between HEK 293T and HEK Bcl-xL cell lysates. (d) SARM-FL/SAM–TIR was expressed in both HEK 293T and HEK Bcl-xL cell line. At 24 h post-transfection, cell death was assessed by counting the 7-AAD-positive cells by flow cytometry. (e) SARM-FL/GFP was expressed in both wild-type and BAX−/− BAK−/− mouse embryonic fibroblast cell lines. At 24 h post-transfection, decrease in mitochondrial transmembrane potential (ΔΨm) was computed based on TMRE fluorescence. Data in b and d represent means±S.D. of at least three independent experiments. *P<0.00005;**P<0.0008; ***P<0.005
SARM expression causes mitochondrial clustering
Next, we determined the exact subcellular localization of SARM. T cells expressing SARM-FL–GFP showed that SARM colocalized with the mitochondria (Figure 4a). Costaining of transfected HEK 293T cells with mitotracker and endosomal marker confirmed specific SARM–mitochondrial association (Figure 4b). Interestingly, the expression of SARM drastically altered the mitochondrial subcellular distribution, causing them to cluster (Figure 4b). Endogenous SARM was specifically detected in the mitochondrial fraction of T cells (Figure 4c). Transmission electron microscopy using immunogold labeling showed that SARM specifically localized in the mitochondria (Figure 4d and Supplementary Figure S6a).

SARM localized in mitochondria caused mitochondrial clustering. (a) Activated primary CD8 T cells were nucleofected with SARM-FL. Six hours later, the cells were stained with mitotracker (Mito), fixed and observed under fluorescence microscope. (b) HEK 293T cells were transfected with SARM-FL–GFP fusion construct. At 12 h post-transfection, cells were co-stained with either mitotracker (Mito) or labeled transferrin. The cells are fixed and then viewed under confocal microscope. (c) Mitochondria were isolated from primary T cells by differential centrifugation. Mitochondrial (Mito) and cytoplasmic (Cyto) lysates were blotted and probed with anti-SARM. The membrane was probed with anti-voltage-dependent anion channel (VDAC), a mitochondria marker and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a cytosol marker. (d) HEK 293T cells expressing SARM-FL were embedded in LR white resin and cut into ultrathin sections. The sections were then stained with anti-GFP followed by incubation with 10 nm gold-conjugated anti-rabbit secondary antibody. Yellow arrows point to the gold particles. The sections were then observed under transmission electron microscope. Scale bars are 100 nm. (e) Fluorescence localization of ARM–SAM–TIR, SAM–TIR and TIR–GFP fusion constructs at 12 h post-transfection with mitotracker. Fluorescence localization of (f) N-ARM–SAM, (g) N-ARM and (h) N124–GFP fusion constructs at 12 h post-transfection with mitotracker. (i) HEK 293 T cells were transfected for 24 h with SARM-FL, N124, N-ARM and N-ARM–SAM. Cell death was assessed by flow cytometry. (j) HEK 293T cells were transfected with N-ARM–GFP, and the percentage of cells displaying mitochondrial aggregation/clustering phenotype was counted microscopically. Data in j represent means±S.D. of at least three independent experiments. *P<0.00005. Scale bars are 10 μm (a, b, e, f, g and h). (k) Summary of truncated constructs of SARM, which is localized in the mitochondria and/or cause mitochondrial clustering
Next, we found that SARM truncates lacking the N-terminus (ARM–SAM–TIR, SAM–TIR and TIR, Figure 1a) did not associate with the mitochondria (Figure 4e), whereas N-ARM–SAM, containing the N-terminus associated with mitochondria (Figure 4f). To clarify how the N-terminus of SARM is responsible for mitochondria-associating ability, we subcloned the N-ARM and N124 of SARM in fusion with GFP (Figure 1a and Supplementary Figure S6b). Both N-ARM (Figure 4g and Supplementary Figure S6c) and N124 (Figure 4h and Supplementary Figure S6d) were associated with the mitochondria in the HEK 293T and T cells. The N-terminal 27 amino acids of SARM was shown to be the mitochondria-targeting signal,27 which confirms the importance of the N-terminus to mitochondrial association. When comparing cell death induced by these N-terminal constructs (lacking both the SAM and TIR domains), we noticed that N-ARM, N124 and N-ARM–SAM each conferred a three-fold drop in apoptosis (Figure 4i). These data strongly corroborate the requirement of both SAM and TIR for the proapoptotic function of SARM. Supplementary Figure S6e summarizes the apoptotic potential of different SARM truncates.
The expression of SARM-FL or N-ARM–SAM or N-ARM clusters the mitochondria to the perinuclear region (Figures 4b, f and g). Kim et al.9 also reported altered distribution of mitochondria in SARM-transfected cells. A significant number of cells expressing N-ARM exhibited mitochondrial clustering within 10 h, which increased with time (Figure 4j). Although N124 (lacking the ARM domain) was associated with the mitochondria, it did not cause mitochondrial clustering, indicating that the ARM domain is necessary for mitochondrial clustering (Figure 4k). Thus, SARM localized in the mitochondria alters the mitochondrial distribution in the cell.
SARM knockdown prolongs primary T-cell survival and rescues them from AICD and NID
To interrogate the biological relevance of SARM, we studied its expression profile following activation of primary T cells. Plate-bound anti-CD3 successfully activated T cells in vitro as indicated by upregulation of CD44 and CD69 (Supplementary Figures S7a and b). Endogenous SARM decreased rapidly post activation and recovered to above the basal level from days 4 to 8 (Figure 5a). Carboxy fluorescein succinimidyl ester labeling indicates that T cells underwent rapid proliferation immediately post activation (Supplementary Figure S7c). The SARM levels dropped in the proliferating activated T cells. Treatment with a proteasome inhibitor suggested that the decrease in SARM expression is due to protein degradation (Supplementary Figure S7d). The increase in SARM in the activated T cells was accompanied by a gradual rise in the expression of death receptor and death receptor ligand, indicating the increased sensitivity to death (Supplementary Figure S7e). On activation, the T cells expanded in size up to day 3, and rounded up by day 6 (Supplementary Figure S7f). These observations consistently implicate SARM in T-cell apoptosis, during which SARM accumulates sufficiently and becomes active during the T-cell clearance phase. Plausibly, the differential expression of SARM determines its proapoptotic function.

SARM knockdown prolonged the survival of primary T cells and rescued them from activation-induced and neglect-induced cell death. (a) Activated primary CD8 T cells were lysed on the indicated days post activation and immunoblotted with anti-SARM. The membrane was stripped and reprobed with anti-actin. (b) OTI+ RAG−/− mice were subcutaneously injected with 5 μg of SIINFEKL peptide in complete Freund’s adjuvant. Popliteal and inguinal lymph nodes were isolated on days 1 and 2 post injection. RNA was prepared using Trizol. cDNA, reverse-transcribed from RNA was used as template for quantitative real-time PCR, and data were normalized to mouse tubulin. Data represent means of four mice per group. (c) Activated primary CD8 T cells were transduced with retrovirus containing either SARM-specific or non-targeting control shRNA. The SARM knockdown cells were tracked for 9 days by counting GFP-positive cells. Similar results were obtained in two other independent experiments. (d) Retroviral-transduced cells were grown in plate bound with anti-CD3 to induce AICD. Cell death was measured 24 h post activation. (e) Twenty-four post-nucleofection of primary T cells with increasing concentration of SARM-specific or control siRNA, the cells were deprived of IL-2. Cell death was measured 24 h post deprivation. Cells cultured with IL-2 served as control. Data in d and e represent means±S.D. of three independent biological replicates. *P<0.01; **P<0.001; ***P<0.0005
Next, we examined the kinetics of SARM expression in T cells activated in vivo. We challenged OTI+RAG−/− mice with SIINFEKL peptide, which OTI TCR respond to. On days 1 and 2 post-footpad injection, the corresponding draining lymph nodes (popliteal and inguinal) were collected. Upregulation of CD69 indicated successful in vivo activation of T cells (Supplementary Figure S7g). Consistently, we observed drastic reduction in the levels of SARM post T-cell activation (Figure 5b), and a significant increase in the lymph node cell number, denoting activation-induced T-cell proliferation (Supplementary Figure S7h). Consistent with in vitro data, the in vivo activation of T cells also suggests that expression of SARM is reciprocal to cell proliferation. To further confirm the proapoptotic role of SARM, we suppressed endogenous SARM using SARM-specific shRNAs (Supplementary Figures S7i and k) and siRNA (Supplementary Figures S7j and l). We tracked stable SARM knockdown and the non-targeting control T cells for up to 9 days post transduction and found significant knockdown of SARM by day 3 (Supplementary Figure S7m). We observed ∼50% increased survival of SARM knockdown CD8 T cells compared with the control (Figure 5c), suggesting that SARM knockdown prolongs T-cell survival.
Lymphocytes undergo AICD and NID during clonal contraction.3, 4, 5 We mimicked AICD by restimulating the T cells with anti-CD3. SARM knockdown T cells showed significantly reduced cell death (Figure 5d). We mimicked NID by IL-2 deprivation28 and observed that SARM knockdown dose dependently rescued T-cell death by up to 50% (Figure 5e), indicating that SARM has a substantial proapoptotic role during T-cell termination.
SARM knockdown T cells show enhanced proliferation following influenza infection
We developed an appropriate ‘adoptive transfer mouse model’ system to substantiate the proapoptotic role of SARM in infection-activated T cells. The rationale for the choice of this system is described in the Materials and Methods section. In a typical experiment, naive OTI cells activated in vitro by SIINFEKL peptide were transduced with SARM-specific shRNA and GFP-containing retrovirus. Then, the cells were developed into memory T cells and adoptively transferred into congenic Thy1.1 mice, which were infected with a sublethal dose of WSN SIINFEKL (Figure 6a). On different days post infection (dpi), we monitored the proportion of GFP-positive SARM-knocked down OTI T cells in various organs, using the Thy1.2 marker. Initially, to test the efficacy of this system we adoptively transferred 5 × 105 naive OTI T cells into Thy1.1 mice and challenged the mice with WSN SIINFEKL. We observed OTI T cells in the mediastinal lymph node (MLN) of the infected mice but not in the uninfected recipient mice (Figure 6b). The detected OTI T cells were activated as indicated by the elevated expression of CD44 (Figure 6c). This data clearly demonstrate that the adoptively transferred OTI cells underwent activation-induced proliferation during influenza infection.

SARM knockdown T cells proliferated more than control T cells following influenza infection. (a) Schematic representation of the development of an adoptive transfer mouse model to study the effect of SARM knockdown during influenza infection. Boxes indicated by dotted arrows are the results of key assessments performed to ensure efficient response by the adoptive transfer mouse model system. (b) Thy1.2-positive OTI CD8 T cells in the mediastinal lymph node (MLN) of infected and uninfected recipient mice on 7 dpi. (c) CD44 expression in Thy1.2-positive OTI CD8 T cells. Grey line corresponds to naive OTI CD8 T cells from OTI+ RAG−/− mice and solid black line corresponds to OTI CD8 T cells from infected recipient mice. (d) Primary T cells were activated with 1 μg of SIINFEKL peptide and the cells were stained with activation markers (CD69, CD44 and CD25) on days 0 and 2 post activation. Post staining, the cells were washed and resuspended in FACS buffer and analyzed by flow cytometry. Black line corresponds to in vitro activated primary T cells and grey line corresponds to naive primary T cells. (e) Activated OTI CD8 T cells were cultured in the presence of IL-7 for more than 2 weeks to develop into memory T cells, and verified by staining with CCR7 and CD62L. Grey line corresponds to naive and activated T cells in CCR7 and CD62L staining, respectively. Solid black line corresponds to in vitro developed memory T cells. (f) OTI CD8 T cells in mediastinal lymph node (MLN), peripheral lymph node (PLN) and spleen of infected recipient mice at various dpi. The Thy1.2 versus CD8 profile was gated based on all live cells in the indicated tissues. (g) Lysates from retroviral-transduced in vitro memory T cells (non-targeting control and SARM-specific shRNA) were immunoblotted with anti-SARM. The same membrane was reprobed with anti-actin. (h) The proportion of GFP-positive non-targeting shRNA or SARM-specific shRNA expressing OTI CD8 T cells in MLN of infected recipient mice on various dpi. Data represent the means of results from four recipient mice. *P<0.0007; NS; non-significant
The upregulation of CD44, CD69 and CD25 following SIINFEKL peptide treatment indicates successful activation of OTI T cells in vitro (Figure 6d). Similarly, culturing of activated T cells in the presence of IL-7 upregulated memory T-cell markers, CCR7 and CD62L, indicating memory T-cell development in vitro (Figure 6e), which we had adoptively transferred into Thy1.1 mice. The mice were then challenged with WSN SIINFEKL virus. We investigated the dynamics of T-cell response to influenza infection by measuring the percentage of OTI T cells in MLN, peripheral lymph node (PLN) and spleen from 3–14 dpi. Lung draining MLN harbored a greater proportion of OTI T cells compared with the PLN and spleen. The highest number of OTI T cells was observed on 5 dpi. By 7 and 10 dpi, the OTI cells had started to contract and by 14 dpi, no distinct population of OTI cells was observed (Figure 6f). Thus, we have developed an efficient system to study the role of SARM during T-cell immune response.
To investigate the proapoptotic role of SARM during/after a viral infection, we transduced the SIINFEKL-activated OTI T cells with retrovirus expressing SARM-specific shRNA or non-targeting control shRNA with GFP reporter. We observed a significant knockdown of SARM in memory T cells (Figure 6g). The SARM knockdown (∼20% GFP+ cells) and control (∼10% GFP+ cells) memory T cells were separately adoptively transferred into Thy1.1 mice. The mice were then challenged with influenza. The proportion of GFP+ transduced OTI T cells was analyzed in MLN on various dpi corresponding to the activation (3 dpi), peak response (5 dpi) and contraction phase (7 and 10 dpi) of T-cell immune response. Non-targeting control-transduced T cells gradually expanded from 10–24% by 5 dpi, were maintained at 26% by 7 dpi, and significantly contracted to 14% by 10 dpi (Figure 6h). Interestingly, SARM-specific shRNA-transduced T-cell population expanded from 20–40% by 5 dpi and continued to expand to 56% by 7 dpi, and then contract to 40% by 10 dpi (Figure 6h). Thus, by 7 dpi SARM-knocked down T-cell numbers increased significantly compared with non-targeting control T cells. This in vivo data further support the proapoptotic role of SARM during T-cell contraction following an influenza infection.
SARM is downregulated in NK/T-cell lymphoma patient tissue
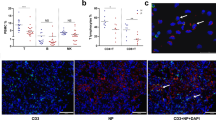
Following pathogen eradication, apoptosis of the activated T cells shuts down T-cell immune response. Failure of this results in a concomitant risk of developing lymphoma or autoimmune disease. Hence, we examined how the levels of proapoptotic SARM is altered in NK/T-cell lymphoma compared with healthy tissue. We found a nine-fold decrease in the expression of SARM mRNA in NK/T-cell lymphoma tissues (n=6) compared with healthy NK cells (Figure 7a). Coincidently, healthy NK cells stimulated with IL-2 also expressed lesser SARM than the unstimulated cells (Figure 7a). NK/T lymphoma cells and stimulated NK cells proliferate significantly faster than unstimulated healthy cells. Hence, it is evident that decrease in SARM expression supports NK/T-cell proliferation. Further analysis of NK/T-cell lymphoma-derived cell lines also showed markedly lower levels of SARM mRNA compared with healthy NK cells (Figure 7b). Consistently, SARM overexpression sensitizes NK-YS (a NK/T-lymphoma tissue-derived cell line29), to apoptosis (Supplementary Figure S3e). Thus, the lowered expression of proapoptotic SARM is probably linked to the proliferative and anti-apoptotic phenotype of NK/T lymphoma pathogenesis.

SARM is downregulated in NK/T-cell lymphoma patient tissues. RNA was extracted either from formalin-fixed paraffin-embedded patient extranodal tissues (n=6) or healthy NK cells (n=2) or IL-2 stimulated healthy NK cells (n=2) or NK/T-cell lymphoma cell lines (n=6). cDNA, reverse-transcribed from RNA was used as template for real-time PCR and data were normalized to human glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Relative quantification (RQ) of SARM expression (a) between healthy NK cells, stimulated NK cells and NK/T-cell lymphoma tissue, and (b) between healthy NK cells and NK/T-cell lymphoma-derived cell lines. Each sample is represented by a triangle. Black line indicates the mean of each group. *P<0.005; **P<0.0005
Discussion
In this study, we demonstrated the role of SARM in restoring activated T cells homeostasis. In mammals, SARM is proposed to be a common regulator of neuronal and immune response to danger.30 Here we demonstrate that mitochondria-localized SARM mediates intrinsic apoptosis in cytotoxic T cells (Figures 2 and 4). The proapoptotic function of SARM is mapped to the C-terminal SAM and TIR domains (Figures 1b and 4i). Kaiser et al.31 reported that cells expressing a SARM clone lacking the N-terminal region did not exhibit apoptosis. The first 27 amino acids of SARM are the mitochondria-targeting signal sequence. Furthermore, SARM mutant lacking the mitochondria-targeting ability exhibits reduced apoptotic phenotype compared with the wild type.27 Thus, it is evident that the mitochondrial localization of SARM is linked to its proapoptotic role. SARM overexpression caused mitochondrial clustering (Figure 4), which has also been observed in several cell death phenomenon.32 Being involved in regulating microtubule stability,33 SARM might control the mitochondrial traffic in microtubules. However, further investigations are necessary to show how SARM induces mitochondrial clustering. Nevertheless, as mitochondrial clustering precedes cytochrome c release,34 it is likely that SARM-induced mitochondrial aggregation triggers intrinsic apoptosis.
The Bcl-2 family members are important for T-cell homeostasis.35, 36, 37 SARM expression significantly reduced the level of the anti-apoptotic Bcl-xL (Figure 3), which is known to normally translocate BAX and BAK from the mitochondrial outer membrane to prevent mitochondrial outer membrane permeabilization.38, 39 Bcl-xL has been reported to inhibit intrinsic apoptosis.40 Consistently, we found that overexpression of Bcl-xL and BAX, BAK double knockout (KO) significantly reduced SARM-induced apoptosis (Figure 3), suggesting that SARM mediates intrinsic apoptosis via Bcl-2 family members (Supplementary Figure S8). Furthermore, we found that SARM expression downregulates ERK phosphorylation (Figure 2g). pERK is known to inhibit caspase-9 activity.41 It is likely that inactivation of ERK by SARM contributes to increased caspase activity.
SARM expression also induced apoptosis in different cell types like neuronal, NK and B cells (Supplementary Figure S3). Consistently SARM has been implicated in neuronal survival.9 C. elegans Tir-1 is reported to mediate anoxic death.17 Thus, the proapoptotic function of SARM is evolutionarily conserved from C. elegans to human, and its role is not limited to T cells.
Immediately post activation, the expression of SARM drastically fell and rose with increase in Fas and FasL (Supplementary Figure S7e), two key mediators of T-cell homeostasis.42 We showed that increased SARM expression significantly reduced the level of Bcl-xL (Figure 3a). The stoichiometry between the pro- and anti-apoptotic Bcl-2 proteins dictates the cellular commitment to apoptosis.43 SARM mRNA drastically dropped during activation-induced proliferation of NK and T cells in vitro (Figures 5a and 7a), and also during T-cell proliferation in vivo (Figure 5b). Consistently, the expression of SARM was reduced in actively proliferating NK/T-cell lymphoma tissues (Figure 7a). Hence, differential expression of SARM during T-cell activation, proliferation and contraction modulates its proapoptotic function.
The biological importance of SARM during T-cell contraction was confirmed by influenza infection in vivo. We tracked SARM-knocked down OT T cells on different dpi and found that the cell population expanded more than the wild-type cells during the contraction phase (Figure 6), clearly supporting the pathophysiological role of SARM. By 10 dpi, SARM-knocked down T-cell population also started to contract, indicating that other pathways are simultaneously operational. Although SARM knockdown significantly reduced NID and AICD (Figure 5), it did not completely abolish these cell death pathways. Failure in T-cell contraction is known to cause autoimmunity and lymphoproliferative diseases like lymphoma, with significant increase in T-cell count.6 Coincidentally, compared with normal cells, NK/T-cell lymphoma patient tissues have significantly lower levels of SARM (Figure 7), which support cell proliferation, indicating how SARM contributes to NK/T-cell lymphoma pathogenesis.
In conclusion, we have demonstrated how SARM mediates intrinsic apoptosis to terminate activated T cells (Figure 8). We have provided compelling evidence that mitochondria-localized SARM perturbs the mitochondrial integrity by altering the levels of Bcl-xL and blocking the cell survival mediator, ERK. We recapitulated the physiological significance of SARM-induced apoptosis during T-cell contraction following an influenza infection in our model mouse system. Furthermore, we discovered that a decrease in SARM expression correlates with NK/T-cell lymphoma.

A model to illustrate the proapoptotic potential of SARM following T-cell immune response. Antigen presenting cells (APC) present the epitopes of the foreign pathogens to the T cells. Antigen-specific T cells undergo clonal expansion and differentiate into effector T cells. These T cells mediate pathogen clearance. Later, apoptosis specifically contracts the expanded T cells in a well-orchestrated manner. Our influenza infection mouse model demonstrates the proapoptotic function of SARM during T-cell contraction. SARM expression is significantly decreased during activation-induced T-cell clonal expansion to support proliferation. During T-cell contraction, SARM is elevated and it acts proapoptotically via intrinsic pathway. SARM seems to oppose the survival signal mediated by ERK phosphorylation, and modulates the levels of the anti-apoptotic Bcl-xL, leading to the loss of mitochondrial integrity via BAX BAK and ROS. The release of the mitochondrial content leads to activation of the initiator caspase-9 and effector caspase-3. Activated caspases cause apoptosis, thereby bringing down the number of T cells. Failure of T-cell contraction may result in lymphoproliferative diseases and autoimmunity. Coincidently, the expression of proapoptotic SARM is reduced in NK/T lymphoma tissues compared with the healthy cells
Materials and Methods
Mice and viruses
Transgenic TCR OTI+ RAG−/−, were from Taconic Farms, Inc (Hudson, NY, USA). These mice express transgenic TCR on CD8 T cells, recognizing SIINFEKL peptide presented by the major histocompatiblity complex class I molecule H2-Kb. B6.PL-Thy1a/CyJ mice (The Jackson Laboratory, Bar Harbor, ME, USA) expressing the congenic Thy1.1 marker, were used as recipients at 8–12 weeks of age. The animals were bred and handled in SPF environment under Institutional Animal Care and Use Committee guidelines (NUS protocol, BR023/08 and IRB 09-018E). The recombinant WSN SIINFEKL influenza virus encoding the SIINFEKL epitope in the neuraminidase stalk was constructed by reverse genetics.44 The viruses were grown in Madin-Darby canine kidney cells.
Transfection
HEK 293T cells were transfected using Lipofectamine LTX (Life Technologies, Carlsbad, CA, USA). Primary T cells were nucleofected using Amaxa nucleofector device. Details on cell culture, plasmids and reagents are in the Supplementary Methods online.
Apoptosis assays
Hoechst and propidium iodide assay
Transfected cells were resuspended in ice-cold PBS containing 5 μg/ml Hoechst 33342 and 1 μg/ml of propidium iodide. The cells were then incubated on ice for 30 min. Stained cells were analyzed by flow cytometry, using UV488 nm dual excitation and measuring the fluorescence emission at ∼460 and >575 nm.
TUNEL assay:
Transfected HEK 293T cells were fixed with 4% formaldehyde and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice. After washing with PBS, 50 μl of TUNEL reaction mixture (Roche, Penzberg, Germany) was added. Cells were incubated in a humidified dark chamber at 37 °C for 1 h, and washed thrice and analyzed by flow cytometry (λex 540 nm/λem 580 nm). Cells treated with DNase I for 10 min at room temperature served as positive control.
Analysis of MPTP formation (MPTP ΔΨm):
Following transfection/nucleofection at indicated time intervals, 10 nM of TMRE was added to cell culture medium. After 20 min incubation at 37 °C, cells were washed and resuspended in FACS buffer containing 0.5% BSA and 0.1% sodium azide in PBS. Positive control cells were treated with 50 μM CCCP for 1 h. FACS analysis was performed at λex 540 nm/λem 595 nm.
Caspase activity assay:
After transfection/nucleofection, cells were washed in FACS buffer and centrifuged. Cell pellets were resuspended in 30 μl of caspase-3 substrate (phiphilux), or caspase-9 (caspalux) or caspase-8 (caspalux) substrate, and incubated for 40 min at 37 °C. After washing in FACS buffer, the cells were analyzed at λex 552/λem 580 nm.
ROS assay:
To transfected HEK 293T cells, 10 μM freshly prepared CM-H2DCFDA was added and incubated for 30 min at 37 °C. Cells were washed, resuspended in FACS buffer and flow cytometry (λex 515 nm/λem 488 nm) was performed. Cells treated with 100 mM H2O2 for 2 h act as a positive control.
Bcl-xL cotransfection assay:
The HEK 293T cells were cotransfected with SARM and Bcl-xL in a ratio of 1 : 5. At 24 h post-transfection, the cells were resuspended in FACS buffer. 7-Aminoactinomycin D (7-AAD; 5 μl) was added and cell death was enumerated by flow cytometry.
Transmission electron microscopy
SARM-transfected HEK 293T cells were fixed with 4% paraformaldehyde and 1% glutaraldehyde. The cells were dehydrated in ethanolic series, gradually infiltrated with LR white resin and polymerized at 50 °C for 2 days. Following ultramicrotomy, the thin gold sections were transferred on to the nickel grids and stained with rabbit anti-GFP and 10 nm gold-conjugated anti-rabbit secondary antibody (Ted Pella, Inc., Redding, CA, USA). The grids were treated with osmium tetroxide, uranyl acetate and lead citrate. The samples were observed under JEM 1010 TEM (Jeol Ltd, Tokyo, Japan).
siRNA-mediated knockdown of SARM
Activated CD8 T cells (2 × 106) were nucleofected with 2 μg of pEGFPN3 plasmid and increasing amounts of SARM-specific/non-targeting siRNA (Life Technologies). At 24 h post-nucleofection, cells were either cultured in the presence or absence of 100 units/ml IL-2 to induce NID. Twenty-four hours later, dead cells were enumerated by flow cytometry based on GFP-positive cells that were 7-AAD-positive.
In vivo activation of T cells
The SIINFEKL peptide was thoroughly mixed with complete Freund’s adjuvant. Each OTI+ RAG−/− mouse was injected s.c. in the hind footpad with 30 μl of complete Freund’s adjuvant emulsion containing 5 μg of SIINFEKL. The corresponding draining popliteal and inguinal lymph nodes were checked for T-cell activation marker, CD69. The lymph node cells were pooled and stained with trypan blue dye, and live cells were enumerated using a Neubauer hemocytometer chamber. The RNA was extracted from the lymph node cells and SARM expression profile was analyzed (Supplementary Methods online). The experiment was terminated at a humane endpoint based on IACUC guidelines.
Adoptive transfer mouse model system
Rationale:
We have developed a new and relevant mouse model to study the effect of SARM knockdown in T cells during influenza infection. During respiratory influenza infection, the major lymphocyte population that contributes to the cell-mediated inflammatory immune response is CD8 T cells.45 Hence, we employed influenza infection model system to follow the immune response of CD8 T cells. The motivation for innovating this novel model system is that the existing SARM KO mice harbor a heterogenous T-cell population expressing a wide repertoire of TCRs, hence the number of virus-specific CD8 T cells will be too low to study the recruitment of naive T cells in response to an influenza infection. Endogenous CD8 T cells specific for immunodominant epitopes of influenza virus are usually below detection limit in tissues before clonal expansion and during clonal contraction. Hence, it will be difficult to follow the T-cell immune response in SARM KO mice. The expression of diverse TCR by the responding polyclonal CD8 T cells would complicate direct comparison of their persistence at different time points during T-cell immune response in the SARM KO mice. In view of these considerable setbacks with the use of SARM KO mice, we strategically planned and developed a novel in vivo adoptive transfer mouse model system to study the effect of SARM knockdown during an experimental influenza infection. The ‘adoptive transfer’ mouse model takes advantage of two major components: (i) CD8 T cells expressing a transgenic TCR, called OTI and (ii) a recombinant influenza virus that expresses the SIINFEKL epitope recognized by the OTI TCR – WSN SIINFEKL.44 The adoptive transfer mouse allowed us to monitor a cohort of CD8 T cells specific for the same epitope to accurately follow T-cell immune response. Using our adoptive transfer model system, we have studied the secondary response rather than primary response. Secondary immune response is more rapid and intense allowing us to track the T-cell immune response effectively.
In vitro T-cell activation, retroviral packaging and spinoculation:
Naive OTI CD8 T cells obtained from the spleen and lymph nodes of OTI+ RAG−/−mice were stimulated using 1 μg/ml SIINFEKL peptide. pCL-ECO packaging plasmid and pMKO.1 GFP plasmids containing SARM-specific shRNA and non-targeting control shRNA were cotransfected into HEK 293T cells. Retrovirus-containing supernatant was collected 2 days post-transfection. The retrovirus supernatant was directly added to the activated OTI CD8 T cells in 24-well plates. The cells were spinoculated at 1500 × g for 120 min at 32 °C. Two days post-retroviral transduction, the T cells were resuspended in media containing 5 ng/ml rIL-7. The cells were cultured for more than 2 weeks to develop in vitro memory T cells, and analyzed for memory T-cell markers, CCR7 and CD62L. The in vitro developed memory T cells were adoptively transferred to the recipient mice as described below.
In vivo adoptive transfer, influenza infection and tissue preparation:
Donor cells (5 × 105 retrovirally transduced in vitro, developed memory T cells) were injected i.v. into B6-Thy1.1 recipient mice. The mice were then infected with a sublethal dose of WSN SIINFEKL virus (75 PFU) by intratracheal instillation under anesthesia. To analyze the OTI T cell response, the lung draining mediastinal lymph nodes were crushed with the frosted glass slides and filtered through 80 μm nylon filter. The cells were then stained with anti-CD8 PE and anti-Thy1.2 PE-Cy7, and analyzed by flow cytometry.
RNA extraction from extranodal nasal tissues of NK/T-lymphoma patients, normal NK cells and NK/TL cell lines
Patients diagnosed with NK/T lymphoma were identified from the 1990 to 2009 archives of the Department of Pathology, National University Hospital, Singapore.7 The cases were classified according to the 2008 WHO lymphoma classification. This study was approved by the ‘Domain Specific Review Board,’ National Healthcare Group, Singapore. Total RNA from NK/TL formalin-fixed paraffin-embedded tissues was isolated using a High Pure RNA Paraffin Kit following the manufacturer’s instructions (Roche Applied Science, Penzberg, Germany). Normal NK cells were obtained from buffy coat of whole blood samples obtained from healthy donors from the Blood Donation Center, National University Hospital. Normal NK cells were isolated using the NK cell isolation kit (Miltenyi Biotec, Bergish Gladbach, Germany). The NK cells were stimulated with IL-2. The NK/T lymphoma cell lines used in this study included NK-92, KHYG-1, Hank-1, SNK-6, SNT-8 and NK-YS. The culture conditions, phenotypic and genotypic characteristics of these NK cell lines are as described in Ng et al.7 Total RNA was extracted from the freshly isolated NK cells and NK cell lines using the miRNeasy Mini Kit (Qiagen, Hilden, Germany). Real-time PCR was performed as described in Supplementary Methods online.
Statistical significance was analyzed by two-tailed Student’s t-test. The corresponding P-values are indicated in the figure legends.
Abbreviations
- 7-AAD:
-
7-aminoactinomycin D
- AICD:
-
activation-induced cell death
- ARM:
-
Armadillo motif
- BAK:
-
Bcl-2 homologous antagonist killer
- BAX:
-
Bcl-2 associated X protein
- Bcl-2:
-
B cell lymphoma-2
- Bcl-xL:
-
B cell lymphoma-extra large
- dpi:
-
days post infection
- ERK:
-
extracellular signal-regulated kinase
- FL:
-
full length
- GFP:
-
green fluorescent protein
- IL-2:
-
interleukin-2
- MEF:
-
mouse embryo fibroblast
- MLN:
-
mediastinal lymph node
- MPTP:
-
mitochondria permeability transition pore
- NAC:
-
N-acetyl cysteine
- NID:
-
neglect-induced cell death
- NK:
-
natural killer
- PLN:
-
peripheral lymph node
- ROS:
-
reactive oxygen species
- SAM:
-
sterile alpha motif
- SARM:
-
sterile α- and heat armadillo-motif-containing protein
- TCR:
-
T-cell receptor
- TIR:
-
Toll/IL-1 receptor domain
- TLR:
-
Toll-like receptor
References
Kumar H, Kawai T, Akira S . Toll-like receptors and innate immunity. Biochem Biophys Res Commun 2009; 388: 621–625.
Elmore S . Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35: 495–516.
Brenner D, Krammer PH, Arnold R . Concepts of activated T cell death. Crit Rev Oncol Hematol 2008; 66: 52–64.
Krammer PH, Arnold R, Lavrik IN . Life and death in peripheral T cells. Nat Rev Immunol 2007; 7: 532–542.
Arnold R, Brenner D, Becker M, Frey CR, Krammer PH . How T lymphocytes switch between life and death. Eur J Immunol 2006; 36: 1654–1658.
Bidere N, Su HC, Lenardo MJ . Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol 2006; 24: 321–352.
Ng SB, Selvarajan V, Huang G, Zhou J, Feldman AL, Law M et al. Activated oncogenic pathways and therapeutic targets in extranodal nasal-type NK/T cell lymphoma revealed by gene expression profiling. J Pathol 2011; 223: 496–510.
Huang Y, de Reyniès A, de Leval L, Ghazi B, Martin-Garcia N, Travert M et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal type. Blood 2010; 115: 1226–1237.
Kim Y, Zhou P, Qian L, Chuang JZ, Lee J, Li C et al. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med 2007; 204: 2063–2074.
Couillault C, Pujol N, Reboul J, Sabatier L, Guichou JF, Kohara Y et al. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol 2004; 5: 488–494.
Liberati NT, Fitzgerald KA, Kim DH, Feinbaum R, Golenbock DT, Ausubel FM et al. Requirement for a conserved Toll/interleukin-1 resistance domain protein in the Caenorhabditis elegans immune response. Proc Natl Acad Sci USA 2004; 101: 6593–6598.
Carty M, Goodbody R, Schröder M, Stack J, Moynagh PN, Bowie AG . The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol 2006; 7: 1074–1081.
Peng J, Yuan Q, Lin B, Panneerselvam P, Wang X, Luan XL et al. SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur J Immunol 2010; 40: 1738–1747.
Belinda LW, Wei WX, Hanh BT, Lei LX, Bow H, Ling DJ . SARM: a novel Toll-like receptor adaptor, is functionally conserved from arthropod to human. Mol Immunol 2008; 45: 1732–1742.
Zhou X, Jiang T, Du X, Zhou P, Jiang Z, Michal JJ et al. Molecular characterization of porcine SARM1 and its role in regulating TLRs signaling during highly pathogenic porcine reproductive and respiratory syndrome virus infection in vivo. Dev Comp Immunol 2012 (in press).
Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS . The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile Virus pathogenesis. J Virol 2009; 83: 9329–9338.
Hayakawa T, Kato K, Hayakawa R, Hisamoto N, Matsumoto K, Takeda K et al. Regulation of anoxic death in Caenorhabditis elegans by mammalian apoptosis signal-regulating kinase (ASK) family proteins. Genetics 2011; 187: 785–792.
Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012; 337: 481–484.
Yang S, Thor AD, Edgerton S, Yang X . Caspase-3 mediated feedback activation of apical caspases in doxorubicin and TNF-alpha induced apoptosis. Apoptosis 2006; 11: 1987–1997.
Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F et al. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci 2001; 21: 9519–9528.
Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL . Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 2009; 1787: 1395–1401.
Knudson CM, Brown NM . Mitochondria potential, bax “activation,” and programmed cell death. Methods Mol Biol 2008; 414: 95–108.
Zhang W, Liu HT . MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002; 12: 9–18.
Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P . Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc Natl Acad Sci USA 2010; 107: 726–731.
Kohno M, Tanimura S, Ozaki K . Targeting the extracellular signal-regulated kinase pathway in cancer therapy. Biol Pharm Bull 2011; 34: 1781–1784.
Martinou JC, Youle RJ . Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 2011; 21: 92–101.
Panneerselvam P, Singh LP, Ho B, Chen J, Ding JL . Targeting of pro-apoptotic TLR adaptor SARM to mitochondria: definition of the critical region and residues in the signal sequence. Biochem J 2012; 442: 263–271.
Gronski MA, Weinem M . Death pathways in T cell homeostasis and their role in autoimmune diabetes. Rev Diabet Stud 2006; 3: 88–95.
Tsuchiyama J, Yoshino T, Mori M, Kondoh E, Oka T, Akagi T et al. Characterization of a novel human natural killer-cell line (NK-YS) established from natural killer cell lymphoma/leukemia associated with Epstein-Barr virus infection. Blood 1998; 92: 1374–1383.
Dalod M . Studies of SARM1 uncover similarities between immune and neuronal responses to danger. Sci STKE 2007; 2007: pe73.
Kaiser WJ, Offermann MK . Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol 2005; 174: 4942–4952.
Aslan JE, Thomas G . Death by committee: organellar trafficking and communication in apoptosis. Traffic 2009; 10: 1390–1404.
Chen CY, Lin CW, Chang CY, Jiang ST, Hsueh YP . Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J Cell Biol 2011; 193: 769–784.
Haga N, Fujita N, Tsuruo T . Mitochondrial aggregation precedes cytochrome c release from mitochondria during apoptosis. Oncogene 2003; 22: 5579–5585.
Rathmell JC, Lindsten T, Zong WX, Cinalli RM, Thompson CB . Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat Immunol 2002; 3: 932–939.
Dunkle A, Dzhagalov I, He YW . Mcl-1 promotes survival of thymocytes by inhibition of Bak in a pathway separate from Bcl-2. Cell Death Differ 2010; 17: 994–1002.
Wojciechowski S, Tripathi P, Bourdeau T, Acero L, Grimes HL, Katz JD et al. Bim/Bcl-2 balance is critical for maintaining naive and memory T cell homeostasis. J Exp Med 2007; 204: 1665–1675.
Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ et al. Sphingolipid metabolism ooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell 2012; 148: 988–1000.
Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011; 145: 104–116.
Cory S, Huang DC, Adams JM . The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 2003; 22: 8590–8607.
Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR . Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol 2003; 5: 647–654.
Nagata S, Suda T . Fas and Fas ligand: lpr and gld mutations. Immunol Today 1995; 16: 39–43.
Chipuk JE, Green DR . How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 2008; 18: 157–164.
Topham DJ, Castrucci MR, Wingo FS, Belz GT, Doherty PC . The role of antigen in the localization of naive, acutely activated, and memory CD8(+) T cells to the lung during influenza pneumonia. J Immunol 2001; 167: 6983–6990.
Ingulli E, Funatake C, Jacovetty EL, Zanetti M . Cutting edge: antigen presentation to CD8 T cells after influenza A virus infection. J Immunol 2009; 182: 29–33.
Acknowledgements
We thank Professor Aihao Ding (Cornell University, NY, USA) for plasmids pEGFPN1–SARM AST, ST, SAM and TIR; Professor Yi-Ping Hsueh (Academia Sinica, Taiwan) for SARM antibody; Professor Victor Yu (NUS, Singapore) for wild type and BAX−/−BAK−/− MEF cell lines; Dr. Guangan Hu for discussion regarding in vivo experiments; Ms Sun Ye and Mr Glenn Wong for help with some experiments; Drs Ruijuan Du, Jing Zhang and Jason ZS Ho for proofreading. PP is a graduate scholar of the Singapore-MIT Alliance. We thank SMART ID-IRG for the usage of facilities. This work was financed by SMA-CSB and A*STAR BMRC (grant number: 08/1/21/19/574).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by JA Cidlowski
Authors’ contribution
PP conceived the ideas, designed and performed the experiments, and prepared the manuscript. LPS and VS performed some experiments. WJC and SBN contributed patient tissues and discussions. NST, BH and JC provided suggestions and manuscript preparation. JLD conceived the ideas, designed the experiments, and supervised the studies and preparation of the manuscript.
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Panneerselvam, P., Singh, L., Selvarajan, V. et al. T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell Death Differ 20, 478–489 (2013). https://doi.org/10.1038/cdd.2012.144
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.144
Keywords
This article is cited by
-
NAD+ metabolism and its roles in cellular processes during ageing
Nature Reviews Molecular Cell Biology (2021)
-
Prognostic Biomarker-Based Identification of Drugs for Managing the Treatment of Endometrial Cancer
Molecular Diagnosis & Therapy (2021)
-
Programmed axon degeneration: from mouse to mechanism to medicine
Nature Reviews Neuroscience (2020)
-
Mitochondria-localised ZNFX1 functions as a dsRNA sensor to initiate antiviral responses through MAVS
Nature Cell Biology (2019)
-
The mitochondrial Nod-like receptor NLRX1 modifies apoptosis through SARM1
Molecular and Cellular Biochemistry (2019)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}