
Abstract
Amino-acid starvation leads to an inhibition of cellular proliferation and the induction of programmed cell death (PCD) in the Drosophila ovary. Disruption of insulin signaling has been shown to inhibit the progression of oogenesis, but it is unclear whether this phenotype mimics starvation. Here, we investigate whether the insulin-mediated phosphoinositide kinase-3 pathway regulates PCD in mid oogenesis. We reasoned that under well-fed conditions, disruption of positive components of the insulin signaling pathway within the germline would mimic starvation and produce degenerating egg chambers. Surprisingly, mutants did not mimic starvation but instead produced many abnormal egg chambers in which the somatic follicle cells disappeared and the germline persisted. These abnormal egg chambers did not show an induction of caspases and lysosomes like that observed in wild-type (WT) degenerating egg chambers. Egg chambers from insulin signaling mutants were resistant to starvation-induced PCD, indicating that a complete block in insulin-signaling prevents the proper response to starvation. However, target of rapamycin (Tor) mutants did show a phenotype that mimicked WT starvation-induced PCD, indicating an insulin independent regulation of PCD via Tor signaling. These results suggest that inhibition of the insulin signaling pathway is not sufficient to regulate starvation-induced PCD in mid oogenesis. Furthermore, starvation-induced PCD is regulated by Tor signaling converging with the canonical insulin signaling pathway.
Similar content being viewed by others


Main
The insulin-mediated class I phosphoinositide kinase-3 (PI3K) pathway is a highly conserved nutrient-sensing pathway in diverse organisms. In mammals, insulin signaling affects oocyte maturation and fertilization.1, 2 Mutations in the insulin signaling pathway in C. elegans can lengthen life span but reduce fertility.3, 4 Improper insulin signaling in Drosophila leads to reduced body size and female sterility.5 Thus, insulin signaling is essential for reproduction in diverse organisms, but how this pathway regulates fertility is not fully understood.
Proper nutrition during Drosophila oogenesis is critical for egg chamber production.6, 7, 8 Depriving flies of yeast as a protein source leads to programmed cell death (PCD) at two stages: early oogenesis in the germarium and mid oogenesis during stages 7–9.7 Egg chambers undergoing PCD during mid oogenesis are characterized by nuclear condensation and fragmentation of germline-derived nurse cells (NCs), engulfment by somatic follicle cells (FCs), and ultimately FC death.5
During PCD in mid oogenesis, dying NCs show characteristics of both apoptotic and autophagic PCD.5 The effector caspase Death caspase-1 (Dcp-1) is essential for germline PCD in mid oogenesis. dcp-1 mutants that have been starved display mid-stage egg chambers that have a persistence of uncondensed NC nuclei but an absence of FCs.5 This phenotype indicates that the dcp-1 mutant germline is unable to die in response to starvation, although the FCs respond and undergo PCD. dcp-1 mutants show decreased autophagy,5 but autophagy appears to have a minor role during PCD in mid oogenesis. Autophagy-deficient egg chambers show normal chromatin condensation, but reduced levels of DNA fragmentation.5
The cell death pathway upstream of dcp-1 is unclear. Double mutants lacking the initiator caspases Strica and Dronc only partially disrupt PCD in mid oogenesis.5 Levels of the caspase inhibitor Drosophila inhibitor of apoptosis protein 1 (DIAP1) decline during mid oogenesis,5 which may be the mechanism that makes this stage of oogenesis more susceptible to PCD. However, mid oogenesis PCD is not regulated by the IAP-binding proteins Rpr, Hid, Grim and Skl,9 so the mechanism controlling DIAP1 levels is unknown. Recently, we have determined that mitochondrial events and the Bcl-2 family partially control PCD in mid oogenesis.10 However, how nutritional cues link to Dcp-1, DIAP1, or mitochondria has not been determined. One candidate for the regulation of starvation-induced PCD in the ovary is the insulin signaling pathway. The insulin signaling pathway is involved in the regulation of apoptosis and autophagic PCD at several points during Drosophila development.11, 12
In the Drosophila ovary, insulin signaling is important for germline stem cell division,7, 13 but evidence that it is involved in the regulation of PCD in mid oogenesis is limited. The insulin receptor (InR) and the InR substrate (Chico) are required for egg chamber progression and proliferation of FCs;5, 7 however, the phenotype of the terminal egg chambers has not been closely analyzed. The terminal egg chambers of target of rapamycin (Tor) mutant germline clones (GLCs) show an induction in apoptosis as visualized by acridine orange staining.14 Recently, rapamycin treatment has been shown to induce autophagy in the fly ovary.15 These studies suggest that the insulin signaling pathway may be involved in the regulation of starvation-induced PCD in mid oogenesis; however, detailed analysis of mid-stage egg chambers has not been reported.
In this study, we analyze multiple components of the insulin signaling pathway within the germline to determine whether their disruption mimics the effects of starvation in the ovary. Interestingly, we find that these mutants show an arrest early in oogenesis but most of these egg chambers do not mimic starvation-induced PCD. We provide evidence that caspases and lysosome formation are blocked in abnormal egg chambers produced by GLCs of InR, chico, and ribosomal protein S6 kinase (S6k). However, Tor GLCs do produce degenerating mid-stage egg chambers, mimicking starvation-induced PCD. These findings suggest that Tor signaling acts independently of the insulin pathway during mid oogenesis. To determine whether another pathway acts in parallel to insulin signaling during starvation, we starved the mutants but found that only Tor mutants resembled wild-type (WT)-starved flies. These findings indicate that the insulin and Tor signaling network are required in the ovary for proper progression of oogenesis and prevention of PCD.
Results
Insulin pathway mutants do not mimic starvation-induced cell death in the ovary
Given the known role of insulin signaling in controlling the response to nutrition, we reasoned that in the presence of nutrients, mutants of positive components of the insulin signaling pathway would mimic the PCD seen in starved WT flies. We chose to analyze two upstream genes, InR and chico, and two downstream genes, Tor and S6k (Figure 1a). All four alleles that were selected have been described as likely null alleles.16, 17, 18, 19 Because most of these mutants are homozygous lethal, we generated GLCs. Flies were well-fed, but mutant GLCs did not progress into mid oogenesis (Supplementary Figure 1), consistent with previous studies.7, 14, 20 To determine the stage of oogenesis in which growth arrested, the last healthy egg chamber at the end of an ovariole was scored (Supplementary Figure 1m). Stage 4 of oogenesis was the predominant stage at which oogenesis arrested for InR339 (38%), chico1 (50%), TorΔP (59%), and S6kl−1 (54%) GLCs.

Insulin signaling pathway mutants have two distinct phenotypes in the Drosophila ovary. All egg chambers were stained with DAPI to label DNA and Fas III to label the FC membranes. (a) The simplified Drosophila insulin signaling cascade. (b–d) WT well-fed stage 8 egg chambers display no NC nuclear fragmentation and FCs are still present. (e–g) WT starved stage 8 egg chamber undergoing degeneration (Deg), in which NC nuclei fragment and FCs are still present. (h–j) Starved dcp-1prev mutants display the Pwop phenotype in a stage 8 egg chamber with uncondensed NC nuclei but a disappearance of FCs. (k–m) Stage 4 egg chamber from InR339 GLCs shows Pwop phenotype like dcp-1. (n–p) Stage 5 Pwop from chico1 GLCs. (q–s) Stage 4 degenerating egg chamber from TorΔP GLCs. (t–v) Stage 4 Pwop from S6kl−1 GLCs. Scale bar: 20 μm. (w) Quantification of insulin signaling pathway mutant ovary phenotypes. Egg chambers classified as degenerating displayed fragmented NC nuclei (Black). Pwop egg chambers displayed uncondensed persisting NC nuclei and a premature disappearance of the FCs (Grey). Healthy egg chambers were not included because we wished to compare the frequency of degenerating vs. Pwop egg chambers. Degenerating egg chambers in WT ranged from stages 7 to 9 (mid oogenesis). Degenerating and Pwop egg chambers from insulin signaling mutants ranged from stages 2 to 6 (early oogenesis)
Surprisingly, we found that many of the mutant egg chambers did not mimic starvation-induced PCD (Figure 1e). All four mutants produced egg chambers that failed to degenerate, showing an aberrant morphology where the germline persisted and FCs disappeared (Figures 1k, n and t). This phenotype was reminiscent of that seen in dcp-1 mutants Figure 1h), where the germline is unable to die despite receiving a death signal. We refer to this phenotype as ‘peas without pods’ (Pwop), where the peas represent the uncondensed NCs and the pod represents the FC layer.5 Fas III labeling showed that most of the FCs were lost from the pwops, and some of the remaining FC membranes became stretched over the NCs (Figures 1i, l, o and u).
Some egg chambers from the insulin pathway mutants did appear to degenerate normally (Figure 1q), so the occurrence of Pwops and degenerating egg chambers was quantified (Figure 1w, Supplementary Figure 1n). Well-fed or starved WT flies produced degenerating egg chambers (100%) and did not produce any Pwops. As previously reported, starved WT flies produced more degenerating egg chambers than well-fed flies (data not shown).5 In contrast to WT, InR339 GLCs produced a large number of Pwops (97%) as did Akt1q (71%) and S6kl−1 (74%). In GLCs of the hypomorphic allele InRE19 (Supplementary Figure 1d), later stages of oogenesis were present but the major phenotype was Pwop (98%). Hemizygotes of the hypomorphic allele S6K07084 (Supplementary Figure 1j) produced mid-stage egg chambers that degenerated (54%) and almost an equal number of Pwops (46%). Similarly, chico1 GLCs produced an almost equal distribution of degenerating (48%) and Pwops (52%). To further analyze chico1, we looked at a potentially stronger allelic combination of chico1 in trans to a deficiency (Supplementary Figure 1f). This combination produced more Pwops (75%), suggesting that chico1 is not a null allele, or that the FC genotype influences germline survival in the chico1 mutant. TorΔP GLCs largely resembled WT starvation (85%) but still produced some Pwops (15%). However, Tor mutant egg chambers degenerated earlier in oogenesis than that normally occurs in WT (Figures 2e and q).

WT progression of starvation-induced PCD at mid oogenesis. All egg chambers were stained with DAPI (blue) to label DNA, anti-cleaved caspase-3 (green) and LT (red) to label lysosomes. (a–a″) A healthy egg chamber from a well-fed fly. (b–e″) Egg chambers from starved flies in the process of degeneration. (b–e) In degenerating egg chambers, NC nuclei become condensed and fragment into smaller pieces that are engulfed by the surrounding FCs. (a′–e′) As the NC nuclei fragment, caspase activity increases. (a″–e″) LT positive spots increase, indicating an increase in lysosomes and/or autolysosomes. All egg chambers are at stage 8. All images were taken at the same magnification and are projections of 15–25 images. Scale bar is 15 μm
To further investigate how insulin signaling is involved in starvation-induced PCD, we compared the progression of apoptotic and autophagic events in WT flies and insulin signaling mutants. One of the hallmarks of PCD in the ovary is nuclear condensation and fragmentation, so we used the extent of nuclear breakdown to stage degenerating WT egg chambers (Figures 2a–e).5 Apoptotic events were marked with anti-cleaved caspase-3 to label active Drosophila caspases21 (Figures 2a′–e′). Active caspases were first detected when NC nuclei began to fragment, and caspase activity increased as NC nuclei fragmented into smaller pieces. Lysosome formation was visualized with LysoTracker (LT) (Figures 2a″–e″). LT positive spots appeared at the start of NC nuclear fragmentation and increased as nurse nuclei fragmented into smaller pieces, similar to anti-cleaved caspase-3 staining.
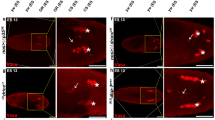
However, Pwops produced by InR339, chico1 and S6kl−1 GLCs displayed reduced punctate caspase-3 staining (Figure 3a, b and e) compared with WT degenerating egg chambers (Figure 2d′). The caspase-3 staining in the Pwop egg chambers may be associated with the dying FCs (Supplementary Figure 2). In InR339 and chico1 GLC Pwops, there were few LT positive spots, and there were larger domains, suggesting acidification of the NC cytoplasm (Figures 3a and b). In S6kl−1 GLCs, LT staining was not detectable (Figure 3e′), suggesting that S6k has a positive role in autophagy induction. This is similar to observations in the fat body of starved S6Kl−1 homozygotes in which LT staining is not present.11 Degenerating egg chambers from chico1 and TorΔP GLCs (Figures 3c and d) mimicked the caspase-3 staining seen in degenerating WT egg chambers. The LT staining of chico1 and TorΔP GLC degenerating egg chambers showed a mix of spots similar to WT starved and large domains similar to InR339. We were unable to visualize autophagy more directly using the autophagosome marker GFP-LC322 in early stage (1–6) egg chambers of WT or chico1 mutants. We found that chico mutant egg chambers showed elevated levels of the autophagic adaptor protein, p62 (Supplementary Figure 3). Elevated levels of p62 have previously been reported in autophagy-deficient egg chambers.23 Taken together, these data indicate that mutant egg chambers that degenerate like WT display a similar induction in apoptosis and autophagy, whereas Pwop egg chambers are defective in caspase activation and lysosome formation. Furthermore, Tor mutants more closely resemble WT starved flies than the other components of the pathway.

Insulin pathway mutant degenerating egg chambers have caspase and lysosome activity, whereas Pwops show weak caspase activation and acidification. All egg chambers were stained with DAPI (blue), anti-cleaved caspase-3 (green) and LT (red). (a–a″) InR339 GLC Pwop stage 6 egg chamber. (b–b″) A stage 5 Pwop from chico1 GLCs. (c–c″) A degenerating stage 4 egg chamber from chico1 GLCs. (d–d″) A degenerating stage 5 egg chamber from TorΔP GLCs. (e–e″) Pwop stage 4 egg chamber from S6kl−1 GLCs. Pwop and degenerating egg chamber stages are approximate due to difficulties in staging abnormal egg chambers. All images were taken at the same magnification and are projections of 15–20 images. Scale bar is 15 μm
DNA fragmentation is blocked in Pwop egg chambers from insulin signaling pathway mutants
To further investigate a disruption in apoptotic events, we used TUNEL to see whether we could detect DNA fragmentation in Pwops. WT starved degenerating egg chambers produced numerous TUNEL-positive spots indicating that DNA fragmentation had occurred (Figures 4a–c). Likewise, mutant degenerating egg chambers displayed TUNEL-positive spots similar to starved WT egg chambers (Figures 4g–l). However, Pwops had no TUNEL-positive spots (Figures 4d–f). This trend was most apparent in mutants with mixed phenotypes like S6kl−1 GLCs: degenerating egg chambers were TUNEL-positive and Pwops were TUNEL-negative (Figures 4m–o). The lack of DNA fragmentation in Pwops provides further evidence that apoptotic events are blocked.

DNA fragmentation is blocked in insulin signaling mutant Pwop egg chambers. All egg chambers were assayed for fragmented DNA using the TUNEL ApopTag Plus Fluorescein In Situ Detection Kit (green), and stained with DAPI (blue). (a–c) WT degenerating egg chamber, in which NC nuclear fragmentation has occurred, shows many TUNEL-positive spots. (d–f) InR339 GLC stage 5 Pwop egg chamber. (g–i) chico1 GLC healthy early stage egg chambers and a degenerating stage 2 egg chamber. (j–l) TorΔP GLC healthy (stage 4) and degenerating (stages 4 and 5) egg chambers. (m–o) S6kl−1 GLC degenerating (stage 1) and Pwop stage 4 egg chambers. Arrows indicate degenerating egg chambers and arrowheads indicate Pwops. All images were taken at the same magnification and are projections of 15–20 images. Scale bar is 15 μm
DIAP1 levels remain moderate in Pwop egg chambers of insulin signaling mutants
The insulin pathway mutant Pwop egg chambers resembled egg chambers of dcp-1 mutants, suggesting that PCD had initiated, but was unable to progress. DIAP1 is a potent caspase inhibitor, and its expression level has been shown to decrease in mid-stage egg chambers,5 perhaps to lower the threshold for caspase activation and make this stage more sensitive to starvation-induced PCD. To determine whether DIAP1 could be protecting the germline of Pwops in the insulin pathway mutants, we examined DIAP1 protein levels with immunocytochemistry. In healthy WT egg chambers, DIAP1 staining was seen in the NCs of early stage egg chambers but levels decreased in the NCs of mid-stage egg chambers (Figures 5a and b), as previously shown.5 Degenerating WT egg chambers had greatly reduced levels of DIAP1 in the NCs (Figures 5c and d). Pwops retained moderate levels of DIAP1 in and surrounding the persisting NC nuclei (Figures 5e, f and k). However, in TorΔP GLC-degenerating egg chambers, DIAP1 levels were depleted similar to WT (Figures 5i and j). The moderate levels of DIAP1 that remained in the Pwops of insulin signaling mutants suggests that DIAP1 could indeed be protecting the germline.

DIAP1 levels are maintained in insulin signaling mutant Pwop egg chambers. All egg chambers were stained with DAPI (Blue) and Anti-DIAP1 (Red). (a and b) WT healthy stage 5 egg chamber (*) and healthy stage 8 egg chamber. Boxed region from b shows DIAP1 in the NC and FC of an early stage egg chamber. (c and d) WT starved degenerating stage 9 egg chamber. Boxed region from d shows that DIAP1 is depleted in the NC. (e and f) InR339 GLC Pwop stage 5 egg chamber. Boxed region in f shows that the NC retains DIAP1 surrounding the nucleus. (g and h) chico1 GLC Pwop stage 4 egg chamber. Similar to InR mutants, the boxed region from k displays DIAP1 surrounding a NC nucleus. (i and j) TorΔP GLC-degenerating stage 4 egg chamber. Similar to the degenerating WT egg chamber, the boxed region from j displays reduced DIAP1 staining in the NC. (k and l) S6kl−1 GLC Pwop stage 4 egg chamber. A NC from the boxed region in l displays DIAP1 surrounding a NC nucleus. Boxed regions are cropped zooms. Scale bar is 20 μm
PI3K activity is lost in InR and S6k Pwops
PI3K is a target of InR that phosphorylates inositol lipids at the plasma membrane, which in turn recruit proteins with phosphoinositide-interacting domains called Pleckstrin Homology (PH) domains.24 We used a tubulin-green fluorescent protein PH reporter (tGPH)25 to visualize PI3K activity in egg chambers of WT and insulin pathway mutants. In well-fed control flies, egg chambers displayed the tGPH reporter at the plasma membrane of both the NCs and the FCs, demonstrating PI3K activity (Figures 6a–d). Degenerating egg chambers from starved control flies lost tGPH from the plasma membrane of the FCs and the NCs (Figures 6e–h). Thus, our results indicate that PI3K activity is lost in degenerating egg chambers.

PI3K activity is lost in InR and S6k Pwops. All egg chambers expressing the tGPH reporter (green) were stained with DAPI (blue) and Fas III (red) to label FCs. FasIII is enriched in the polar cells in mid-stage egg chambers. (a) tGPH mid-stage egg chamber from well-fed fly displayed (b) PI3K activity at the plasma membranes of FCs and NCs. (c) Fas III and (d) Merge indicates co-localization at the FC plasma membrane of tGPH reporter and Fas III. (e) tGPH-degenerating mid-stage egg chamber from starved fly displayed (f) no PI3K activity at the plasma membranes of the FCs or NCs. (g) Fas III staining shows that FCs increased in size during engulfment of NC remnants. (h) Merge. (i–k) tGPH;InR339 GLC Pwop stage 4 egg chambers. (l) Merge indicates co-localization at the FC plasma membrane of tGPH reporter and Fas III. (m–o) tGPH;S6kl−1 GLC Pwop stage 3 egg chamber. (p) The merge displays co-localization at the FC plasma membrane of tGPH reporter and Fas III. Scale bar is 20 μm
To determine whether PI3K activity was reduced in the mutant egg chambers, flies were generated to carry the tGPH reporter in InR339 or S6kl−1 GLCs. GLCs of S6k, which acts downstream of PI3K, would be expected to display normal recruitment of tGPH. S6kl−1 GLCs displayed membrane localization of the tGPH reporter in 67% (n=15) of Pwop egg chambers (Figures 6m and n). Most of the tGPH labeling in Pwop egg chambers of S6kl−1 GLCs localized with the FC marker Fas III, indicating that the remaining PI3K activity was not in the NCs (Figures 6n–p). Similarly, InR339 GLCs showed the tGPH reporter at the plasma membrane in 40% (n=25) of Pwops (Figures 6i and j), but the reporter was not localized in the NCs of anterior healthy egg chambers (data not shown). These data indicate that the insulin signaling pathway is not intact in InR or S6k GLCs, because we do not see PI3K activity in healthy NCs. Furthermore, PI3K activity is observed in many of the Pwops; however, this activity localized to the membranes of the remaining FCs that have stretched to surround the NCs.
Pwop egg chambers cannot undergo germline PCD in response to starvation
The block in egg chamber degeneration in insulin pathway mutants could be because another pathway acts in parallel to insulin signaling to control egg chamber survival. Therefore, we reasoned that starvation of the insulin signaling mutants would affect such a parallel pathway and the mutants would show an increase in degenerating egg chambers when they were starved. Surprisingly, GLCs from starved insulin pathway mutants still showed the Pwop phenotype (Figure 7). InR339 and Aktq GLCs, and chico1 hemizygotes showed slight increases in the frequency of degenerating egg chambers, but the majority were still Pwops and there was no statistically significant change. chico1 GLCs did show a significant change between well-fed and starved, suggesting either that this is not a null allele or the surrounding WT tissue in the GLC-carrying flies could be contributing to the degeneration of the NCs. Starved TorΔP and S6kl−1 GLCs also showed no significant difference between well-fed and starved conditions. These data indicate that the abnormal Pwop phenotype seen in insulin signaling mutants cannot be explained simply by a parallel pathway acting during starvation.

Quantification of starved insulin signaling pathway mutant ovary phenotypes. Insulin mutants were deprived of amino acids similar to WT starved flies. Bar graphs shows the percentage of degenerating egg chambers compared with Pwops in well-fed versus starved flies. WT, InR339 GLC, chico1/Df, Aktq GLC, TorΔP GLC, and S6kl−1 GLC comparison of well fed versus starved had a P-value >0.05 indicating there was no significant difference, whereas chico1 GLCs had a P-value <0.05. (WT P-value=1, InR339 GLCs P-value=0.064, chico1 GLCs P-value=0.039, chico1/Df P-value=0.6234, Aktq GLCs P-value=0.308, TorΔP GLCs P-value=0.1471, and S6kl−1 GLCs P-value=1). The total numbers of egg chambers (degenerating+Pwop) for starved flies were: 54 (WT), 192 (InR339 GLCs), 61 (Aktq GLCs), 47 (chico1 GLCs), 49 (chico1 /Df), 55 (TorΔP GLCs), 46 (S6kl−1 GLCs). Data for the well-fed flies were the same as in Figure 1 or Supplementary Figure 1. Error bars represent S.E. calculated using a standard two-tailed t-test
Discussion
Following starvation, mid-stage egg chambers degenerate via PCD that has hallmarks of both apoptosis and autophagic PCD. In the Drosophila salivary gland and midgut, autophagic PCD is regulated by the insulin signaling pathway.11, 12 Results from our study indicate that the insulin signaling pathway may regulate PCD in the ovary similar to other Drosophila tissues, but additionally there is cross-talk from other pathways. This cross-talk could come from Tor signaling that is independent of InR. Interestingly, Tor has recently been shown to also have insulin-independent roles in germline stem cell proliferation in the Drosophila ovary.13
We expected that disruption of the insulin signaling pathway would lead to degenerating egg chambers, resembling those seen in starved WT flies. Surprisingly, we found many abnormal Pwop egg chambers in which the NCs persisted and the FCs disappeared, indicating that PCD was initiated but incomplete. These Pwop egg chambers exhibited a block in DNA fragmentation, weak to no caspase activation, moderate levels of DIAP1 protein, and large domains or a lack of LT staining. Some of the mutant egg chambers did appear to be degenerating normally and these displayed PCD hallmarks such as DNA fragmentation, active caspases, reduced DIAP1 protein, and LT staining. It is also possible that the Pwop phenotype arises because insulin signaling in the NCs is required for FC survival, and viable FCs are needed for germline death.26
WT egg chambers that degenerate due to starvation occur specifically during mid oogenesis.7 InR, chico, Akt, Tor, and S6k GLCs have a growth delay in which they produce only early stage egg chambers, indicating that insulin signaling is required to progress into mid oogenesis.7, 13 Because insulin signaling mutant egg chambers arrest before entering mid oogenesis, the early stages may be incompatible for the induction of PCD. DIAP1 levels remain high in early stage WT egg chambers but are reduced in mid-stage egg chambers, likely making them more vulnerable to starvation-induced PCD.5 Perhaps insulin signaling mutant egg chambers at early stages are still protected by DIAP1 and do not reach the susceptible mid-stage. Pwops from InR, chico, and S6k GLCs did retain moderate levels of DIAP1, suggesting that DIAP1 could be protecting these abnormal egg chambers from PCD. However, early stage degenerating egg chambers with a depletion of DIAP1 were observed in Tor GLCs, suggesting that Tor mutants can overcome this block, and a pathway in addition to insulin signaling acts to regulate Tor. Consistent with this model, we found that chico Tor double GLCs mimicked Tor single GLCs (Supplementary Figure 1).
To determine whether an alternative pathway acts in parallel to insulin signaling, we starved the mutant GLCs. If a parallel survival pathway was acting, we expected that egg chambers from starved insulin pathway mutants would degenerate in a manner similar to egg chambers of starved WT flies. Interestingly, InR, chico, Akt, and S6k mutants still produced the Pwop phenotype. This could be because early stage egg chambers are resistant to PCD. An alternative possibility is that complete loss of the insulin pathway leads to an aberrant state. Perhaps, dying egg chambers require a certain threshold of insulin signaling to enter into starvation-induced PCD. This hypothesis is supported by our observations of InR and S6K hypomorphic alleles (InR05545 and S6K07084), which produce mid-stage egg chambers that degenerate in a manner similar to egg chambers of starved WT flies (Supplementary Figure 1). These findings support the notion that a small amount of InR activity permits progression into mid oogenesis but also promotes PCD.
Tor can be regulated by nutrients through multiple pathways. The presence of amino acids promotes Tor activity via Rag small guanosine triphosphatases (Rag GTPases) and MAP4K3.27, 28, 29, 30 In Drosophila, MAP4K3 and Rag GTPases are required for growth in response to amino acids and over-expression of Rag GTPases suppresses starvation-induced autophagic PCD in the fat body.29, 30 Furthermore, MAP4K3 has been shown to physically interact with Rag GTPases and amino acids regulate their association.30 Thus, Tor is an integration point for multiple nutrient sensing pathways, which may explain why Tor mutant egg chambers resemble degenerating egg chambers seen in WT starved flies.
Tor is found in two distinct complexes, TORC1 and TORC2,31, 32 which leads to further complexity in the Tor signaling network. Rag GTPases, MAP4K3 and the insulin signaling pathway converge on TORC1, which then phosphorylates downstream targets such as S6K.33 The regulation and activity of TORC2 is less well understood. However, the Tor mutants we analyzed would be disrupted for both TORC1 and TORC2, which could explain why Tor mutants showed a different phenotype than the other mutants. In one possible scenario, complete removal of Tor leads to PCD in the germline, whereas disruption of insulin signaling or S6K is not sufficient. Because S6K is regulated only by TORC1,33 this model explains why the phenotype of S6K more closely resembles chico and InR mutants than Tor mutants.
Using a reporter for PI3K activity, we determined that PI3K activity was associated with healthy egg chambers and lost from degenerating egg chambers in WT flies. Healthy egg chambers from S6k or InR GLCS did not display the tGPH reporter in NCs, indicating that both InR and S6K are required to maintain PI3K activity. Surprisingly, PI3K activity appeared to be restored in many of the Pwop egg chambers from both S6k and InR GLCs; however, this staining was predominantly associated with FC membranes that had stretched over the NCs. One possible explanation for residual PI3K activity is that PI3K is activated by an alternative mechanism in Pwop egg chambers. The regulatory subunit of PI3K binds to a SRC homology-2 (SH2) domain on Chico, but recently the adaptor protein Lnk has been shown to have an SH2 domain that may interact with PI3K.34 Mutants of Lnk are reduced in size and produce abnormal egg chambers remarkably similar to those seen in InR, chico, Akt, and S6k GLCs.34
The pattern of LT suggested that the insulin signaling mutant egg chambers cannot induce autophagy properly. WT starved mid-stage egg chambers that are dying display LT positive spots, whereas InR and chico Pwop egg chambers display LT staining as large domains that partially overlap with NC nuclei. Interestingly, similar large domains of LT staining occur during developmental PCD of NCs, and may represent large lysosomes, or an acidification of NC remnants.35 Disruption of insulin signaling may not be sufficient to induce apoptosis and autophagic PCD, but the presence of acidified persisting NCs may indicate necrosis.36 It may be that these abnormal egg chambers progress to a necrotic state because they cannot induce apoptosis.
Our results indicate that the insulin signaling pathway is an important factor for cell survival in the Drosophila ovary, similar to its role in mammals. Mammalian ovaries cultured in serum-free media show an induction in apoptotic and autophagic PCD.37 Survival of mouse primordial follicles requires PI3K, phosphoinositide-dependent protein kinase-1 (PDK1), and S6k1, which are members of the insulin signaling cascade.38 Treatment with rapamycin, a drug known to block Tor activity, inhibits oocyte growth in cultured Drosophila and mammalian ovaries, and leads to apoptosis and autophagy.37, 39, 40, 26 Taken together, these findings suggest an evolutionarily conserved role for insulin and Tor signaling in promoting survival in the ovary. Furthermore, characterization of starvation-induced PCD in the Drosophila ovary may give insight into the mechanisms of degeneration of defective oocytes in mammalian systems during reproductive aging and fertility disorders.
Materials and Methods
Drosophila stocks
Drosophila stocks were maintained at 25 °C. All stocks were obtained from the Bloomington Drosophila Stock Center unless noted. tubulin-GPH was a gift from Bruce Edgar.25 This reporter for PI3K activity has the PH domain of the Drosophila homolog of the general receptor for phosphoinositides-1 (GRP1) fused to GFP and placed under the control of the β-tubulin promoter (tGPH).25 GRP1 binds to phosphatidylinositol-3,4,5-P3 (PIP3) generated by PI3K activity on phospholipids at the plasma membrane; thus, this GFP fusion construct is localized to the plasma membrane when PI3K is active. Df(2L)J2 is a deficiency that removes chico. chico116 was recombined onto the FRT40A chromosome. InR339FRT82B was a gift from Daniela Drummond-Barbosa.17 S6kl−1 41 was a gift from Tom Neufeld and recombined onto the FRT82B chromosome. GLCs were generated with these FRT recombinants as well as TorΔPFRT4018 using the FLP/FRT/ovoD system as described.9 To induce clones, larvae were heat shocked for 2 days at 37 °C for 1 h. Under these conditions, <20% of egg chambers have large FC clones (data not shown).
Ovary staining
Flies were conditioned on yeast paste for 2–3 days. For starvation experiments, flies were conditioned 2–3 days and then moved to apple juice agar for 1–2 days and dissected. Ovaries were dissected in Drosophila Ringers (130 mM NaCl, 4.7 mM KCl, 1.9 mM CaCl2, 10 mM Hepes, pH 6.9). Well-conditioned WT ovaries were included with mutant ovaries as carrier to retain mutant ovaries that were greatly reduced in size. Ovaries were fixed for 10 min in one part Buffer B (100 mM KH2 PO4 /K2HPO4 (pH 6.8), 450 mM KCl, 150 mM NaCl, 20 mM MgCl2 6H2O), 1 part 36% formaldehyde, 4 parts dH2O, and an equal volume heptane, then washed in PBS plus 0.1% TritonX (PBT). LysoTracker Red (Invitrogen, Grand Island, NY, USA) was used at 1:50 for 3 min and washed with PBS before fixation. Anti-cleaved caspase-3 (Cell Signaling, Danvers, MA, USA) was used at 1 : 100. Anti-DIAP1 was a gift from Bruce Hay and used at 1 : 200.42 Anti- Ref(2)P was a gift from Ioannis Nezis and used at 1 : 1000.23 FC-specific antibody anti-Fas III43 (Developmental Studies Hybridoma Bank, Iowa City, IA, USA) was used at 1 : 50. Secondary antibodies Goat-anti-rabbit Alexa Fluor 488 (Invitrogen), Goat-anti-rat DyLight 488 and Goat anti-mouse Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) were all used at 1 : 200 for 2 h. TUNEL was performed using the ApopTag Plus Fluorescein In Situ Detection Kit (Millipore, Billerica, MA, USA).44 For the TUNEL assay, ovaries were treated with 20 μg/ml of Proteinase K for 20 min, then post fixed for 5 min. Ovaries were washed after staining three times for 10 min each in PBT to reduce background, and mounted onto microscope slides in Vectashield mountant with DAPI (Vector Labs, Burlingame, CA, USA). Images were taken using an Olympus BX51 microscope with DSU spinning disc attachment, except anti-DIAP1, tGPH, anti-Ref(2)P, and anti-Fas III images, which were taken on an Olympus FluoView 10i Confocal microscope. All images were prepared in Adobe Photoshop.
Abbreviations
- Dcp-1:
-
death caspase-1
- DIAP1:
-
Drosophila inhibitor of apoptosis protein 1
- Dronc:
-
Drosophila nedd-2 like caspase
- FC:
-
follicle cell
- GLC:
-
germline clone
- Hid:
-
head involution defective
- InR:
-
insulin receptor
- LT:
-
LysoTracker
- NC:
-
nurse cell
- PCD:
-
programmed cell death
- PI3K:
-
phosphoinositide kinase-3
- Pwop:
-
peas without pods
- Rpr:
-
Reaper
- Skl:
-
Sickle
- S6k :
-
Ribosomal protein S6 kinase
- Strica:
-
Serine threonine rich caspase
- tGPH:
-
tubulin-Green Fluorescent Protein Plextrin Homology
- Tor :
-
target of rapamycin
- WT:
-
wild-type
References
Yoshimura Y, Ando M, Nagamatsu S, Iwashita M, Adachi T, Sueoka K et al. Effects of insulin-like growth factor-I on follicle growth, oocyte maturation, and ovarian steroidogenesis and plasminogen activator activity in the rabbit. Biol Reprod 1996; 55: 152–160.
Acevedo N, Ding J, Smith GD . Insulin signaling in mouse oocytes. Biol Reprod 2007; 77: 872–879.
Tissenbaum HA, Ruvkun G . An insulin-like signaling pathway affects both longevity and reproduction in Caenorhabditis elegans. Genetics 1998; 148: 703–717.
Lin K, Hsin H, Libina N, Kenyon C . Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 2001; 28: 139–145.
Pritchett TL, Tanner EA, McCall K . Cracking open cell death in the Drosophila ovary. Apoptosis 2009; 14: 969–979.
Sang JH, King RC . Nutritional requirements of axenically cultured Drosophila melanogaster adults. J Exp Biol 2010; 38: 793–809.
Drummond-Barbosa D, Spradling AC . Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev Biol 2001; 231: 265–278.
Buszczak M, Cooley L . Eggs to die for: cell death during Drosophila oogenesis. Cell Death Differ 2000; 7: 1071–1074.
Peterson JS, Bass BP, Jue D, Rodriguez A, Abrams JM, McCall K . Noncanonical cell death pathways act during Drosophila oogenesis. Genesis 2007; 45: 396–404.
Tanner EA, Blute TA, Brachmann CB, McCall K . Bcl-2 proteins and autophagy regulate mitochondrial dynamics during programmed cell death in the Drosophila ovary. Development 2011; 138: 327–338.
Berry DL, Baehrecke EH . Autophagy functions in programmed cell death. Autophagy 2008; 4: 359–360.
Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol 2009; 19: 1741–1746.
LaFever L, Feoktistov A, Hsu HJ, Drummond-Barbosa D . Specific roles of Target of rapamycin in the control of stem cells and their progeny in the Drosophila ovary. Development 2010; 137: 2117–2126.
Zhang Y, Billington Jr CJ, Pan D, Neufeld TP . Drosophila target of rapamycin kinase functions as a multimer. Genetics 2006; 172: 355–362.
Barth JM, Szabad J, Hafen E, Kohler K . Autophagy in Drosophila ovaries is induced by starvation and is required for oogenesis. Cell Death Differ 2011; 18: 915–924.
Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF et al. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 1999; 97: 865–875.
Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E . An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol 2001; 11: 213–221.
Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP . Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 2000; 14: 2712–2724.
Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E . Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 2000; 14: 2689–2694.
Richard DS, Rybczynski R, Wilson TG, Wang Y, Wayne ML, Zhou Y et al. Insulin signaling is necessary for vitellogenesis in Drosophila melanogaster independent of the roles of juvenile hormone and ecdysteroids: female sterility of the chico1 insulin signaling mutation is autonomous to the ovary. J Insect Physiol 2005; 51: 455–464.
Fan Y, Bergmann A . The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ 2010; 17: 534–539.
Rusten TE, Lindmo K, Juhasz G, Sass M, Seglen PO, Brech A et al. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell 2004; 7: 179–192.
Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol 2010; 190: 523–531.
Rameh LE, Cantley LC . The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem 1999; 274: 8347–8350.
Britton JS, Lockwood WK, Li L, Cohen SM, Edgar BA . Drosophila′s insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell 2002; 2: 239–249.
Thomson TC, Johnson J . Inducible somatic oocyte destruction in response to rapamycin requires wild-type regulation of follicle cell epithelial polarity. Cell Death Differ 2010; 17: 1717–1727.
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320: 1496–1501.
Findlay GM, Yan L, Procter J, Mieulet V, Lamb RF . A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J 2007; 403: 13–20.
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL . Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10: 935–945.
Bryk B, Hahn K, Cohen SM, Teleman AA . MAP4K3 regulates body size and metabolism in Drosophila. Dev Biol 2010; 344: 150–157.
Soulard A, Cohen A, Hall MN . TOR signaling in invertebrates. Curr Opin Cell Biol 2009; 21: 825–836.
Cybulski N, Hall MN . TOR complex 2: a signaling pathway of its own. Trends Biochem Sci 2009; 34: 620–627.
Lee G, Chung J . Discrete functions of rictor and raptor in cell growth regulation in Drosophila. Biochem Biophys Res Commun 2007; 357: 1154–1159.
Werz C, Kohler K, Hafen E, Stocker H . The Drosophila SH2B family adaptor Lnk acts in parallel to chico in the insulin signaling pathway. PLoS Genet 2009; 5: e1000596.
Bass BP, Tanner EA, Mateos San Martin D, Blute T, Kinser RD, Dolph PJ et al. Cell-autonomous requirement for DNaseII in nonapoptotic cell death. Cell Death Differ 2009; 16: 1362–1371.
Golstein P, Kroemer G . Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 2007; 32: 37–43.
Rodrigues P, Limback D, McGinnis LK, Plancha CE, Albertini DF . Multiple mechanisms of germ cell loss in the perinatal mouse ovary. Reproduction 2009; 137: 709–720.
Reddy P, Adhikari D, Zheng W, Liang S, Hamalainen T, Tohonen V et al. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum Mol Genet 2009; 18: 2813–2824.
Lobascio AM, Klinger FG, Scaldaferri ML, Farini D, De Felici M . Analysis of programmed cell death in mouse fetal oocytes. Reproduction 2007; 134: 241–252.
Yaba A, Bianchi V, Borini A, Johnson J . A putative mitotic checkpoint dependent on mTOR function controls cell proliferation and survival in ovarian granulosa cells. Reprod Sci 2008; 15: 128–138.
Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G . Drosophila S6 kinase: a regulator of cell size. Science 1999; 285: 2126–2129.
Yoo SJ, Huh JR, Muro I, Yu H, Wang L, Wang SL et al. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat Cell Biol 2002; 4: 416–424.
Kugler JM, Lem C, Lasko P . Reduced cul-5 activity causes aberrant follicular morphogenesis and germ cell loss in Drosophila oogenesis. PLoS One 2010; 5: e9048.
McCall K, Peterson JS, Pritchett TL . Detection of cell death in Drosophila. Methods Mol Biol 2009; 559: 343–356.
Acknowledgements
We thank Thomas Neufeld, Daniela Drummond-Barbosa, Armen Manoukian, Bruce Edgar, and the Bloomington Stock Center for flies. We thank Bruce Hay, Ioannis Nezis, and the Developmental Studies Hybridoma Bank for antibodies. We thank members of the lab, Caryn Navarro, Rob Hausman, John Celenza, and Horacio Frydman for helpful suggestions. We apologize to authors whose work we could not cite due to space limitations. This work was supported by NIH R01 GM060574 to KM and the NSF Northeast Alliance for Graduate Education and the Professoriate HRD-0450339 (TP).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E Baehrecke
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Pritchett, T., McCall, K. Role of the insulin/Tor signaling network in starvation-induced programmed cell death in Drosophila oogenesis. Cell Death Differ 19, 1069–1079 (2012). https://doi.org/10.1038/cdd.2011.200
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.200
Keywords
This article is cited by
-
The ovary and its genes—developmental processes underlying the establishment and function of a highly divergent reproductive system in the female castes of the honey bee, Apis mellifera
Apidologie (2018)
-
The TORC1 inhibitors Nprl2 and Nprl3 mediate an adaptive response to amino-acid starvation in Drosophila
Cell Death & Differentiation (2014)

{kind=link}
{kind=link}
{kind=link}