
Abstract
Granzyme M (GzmM), an orphan Gzm, is constitutively and abundantly expressed in innate effector natural killer cells. We previously demonstrated that GzmM induces caspase (casp)-dependent apoptosis and cytochrome c release from mitochondria. We also resolved the crystal structure for GzmM and generated its specific inhibitor. However, how GzmM causes casp activation has not been defined. Here we found that casp-8 is an initiator caspase in GzmM-induced casp cascade, which causes other casp activation and Bid cleavage. GzmM does not directly cleave procaspase-3 and Bid, whose processing is casp dependent. Casp-8 knockdown or deficient cells attenuate or abolish GzmM-induced proteolysis of procaspase-3 and Bid. Extrinsic death receptor pathway adaptor Fas-associated protein with death domain (FADD) contributes to GzmM-induced casp-8 activation. GzmM specifically cleaves FADD after Met 196 to generate truncated FADD (tFADD) that enhances its self-association for oligomerization. The oligomerized tFADD facilitates procaspase-8 recruitment to promote its auto-processing leading to casp activation cascade. FADD-deficient cells abrogate GzmM-induced activation of casp-8 and apoptosis as well as significantly inhibit lymphokine-activated killer cell-mediated cytotoxicity. FADD processing by GzmM can potentiate killing efficacy against tumor cells and intracellular pathogens.
Similar content being viewed by others


Main
Cytotoxic lymphocytes, such as natural killer (NK) and cytotoxic T-lymphocyte (CTL) cells, kill their target cells through two main pathways, death receptor pathway and granzyme (Gzm)/perforin pathway. Gzm/perforin-mediated cytotoxicity is a major pathway for cytotoxic lymphocytes to kill intracellular pathogens and transformed tumor cells.1 Perforin assists the entry of Gzms into the target cell cytosol, whose structural basis was just defined.2 Gzms are a number of serine proteases that induce distinct cell death pathways through proteolysis of different target cell substrates.3, 4, 5, 6, 7, 8, 9, 10 Granzyme M (GzmM) is a chymotrypsin-like serine protease that preferentially degrades its substrates after Met or Leu.11, 12 GzmM is constitutively abundantly expressed in innate effector NK cells, which have an essential role in eradication of tumors at early stage.13 It suggests that GzmM may exert a critical role in NK cell-mediated cytolysis of tumor cells.
We previously demonstrated that GzmM induces caspase (casp)-dependent cell death through cleaving the inhibitor of casp-activated DNase to release DNase for damaging chromatin DNA.8 GzmM also cleaves the DNA-damage sensor enzyme poly(ADP-ribose) polymerase (PARP) to abolish DNA repair. We also found GzmM can cause mitochondrial swelling and loss of mitochondrial transmembrane potential leading to cytochrome c (Cyt c) release.9 We have resolved the crystal structure of GzmM and determined that Asp86 and His41 in the catalytic triad contribute more to enzymatic activity than the attack residue Ser182.12 We also generated a specific inhibitor for GzmM. We just reported that GzmM can degrade the inhibitor of apoptosis gene family protein (IAP) Survivin after Leu138, which abolishes the stability of the Survivin-X-linked inhibitor-of-apoptosis protein (XIAP) complex to initiate XIAP degradation leading to free casp activity.14 However, how GzmM causes casp activation has not been elucidated.
Fas-associated protein with death domain (FADD) is a main adaptor protein to transduce apoptotic signals mediated by extrinsic death receptors such as Fas and tumor necrosis factor receptor 1 (TNF-R1).15 FADD harbors two structurally similar motifs, the death effector domain (DED) and the death domain (DD). Once bound with their ligands, death receptors can recruit FADD and initiator procaspase-8 and/or -10 to form a death-inducing signaling complex (DISC).16, 17 The DISC is thought to trigger casp cascade by inducing the proximity of initiator casp-8 or-10.18, 19 A report showed that procaspase-8 undergoes dimerization and autocatalysis in the DISC to cause casp-8 activation.20 However, how the FADD causes initiator casp activation has not yet been elucidated. Here we found that casp-8 is an initiator casp in GzmM-induced casp cascade.
Results
Casp activation cascade is required for GzmM-induced apoptosis
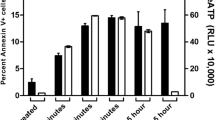
To determine whether or not GzmM causes casp cascade, the pan casp inhibitor Z-VAD (100 μM) was used to pretreat Jurkat cells before loading of GzmM plus Ad (adenovirus). Z-VAD could remarkably restrain GzmM-induced apoptosis at all time points (Figure 1A). Our previous report showed that the enzymatically inactive mutant D86N-GzmM (D-NGzmM) has no enzymatic activity.12 As expected, cells treated with D-NGzmM plus Ad did not cause cell death. GzmM caused activation of casp-8, -9 and -3 (Figure 1B). Z-VAD could completely block casp activation. Similar results were found through activity assay by proteolysis of casp-specific fluorogenic substrates (Figure 1C). Casp-8 processing started earlier than other detected initiator casp-9 and -2. Similar results were observed in GzmM-treated HeLa cells (data not shown). These results indicate that GzmM does not directly cleave these procaspases for casp activation during loading of GzmM.

Casp activation cascade is required for GzmM-induced apoptosis. (a) Casp pan inhibitor Z-VAD can restrain GzmM-induced apoptosis. Jurkat cells (2 × 105) were pretreated with 100 μM Z-VAD for 30 min before 1 μM GzmM plus Ad treatment with the indicated time points at 37 °C. Cells were stained with FITC-conjugated Annexin V and PI for flow cytometry. Cells treated with enzymatically inactive mutant D-NGzmM/Ad for 4 h served as a negative control. Total dead cells were calculated by Annexin V and PI single-positive as well as Annexin V/PI double-positive cells and shown as means±S.D. at the right panel. These data are representative of three independent experiments. (b) GzmM-induced casp cascade is blocked by Z-VAD. Treated Jurkat (2 × 105) cells as above were probed by specific antibodies against casp-8, -9 and -3. The same blot was stripped for immunoblotting. β-actin was detected as a loading control. (c) Casp activations are confirmed by casp-specific fluorogenic substrates. The cell lysates from the above treated Jurkat cells were incubated with Ac-IETD-AFC, Ac-LEHD-AFC, Ac-VDVAD-AFC or Ac-DEVD-AFC for assessment of casp-8, -9, -2 and -3/7 activity, respectively. Casp activity was shown as relative fluorescent light units (RFLU) change per minute
GzmM mediates casp-dependent Cyt c release but Bid independent
GzmM began to cause Cyt c release after 3 h treatment as shown in Figure 2A. Z-VAD could block GzmM-mediated release of Cyt c. We found casp-8 activation was earlier than Cyt c release (Figure 2B). Moreover, casp-3 was not activated at 1 h and casp-8 activation was earlier than casp-3 by immunofluorescence staining (Figure 2C). D-NGzmM plus Ad treated cells served as a negative control. Z-VAD pretreatment still blocked the activation of casp-8 and -3. These suggest that casp-8 activation is an upstream event to mitochondrial damage. We next examined whether Bid cleavage contributes to casp activation. Bid was processed in GzmM-treated cells (Figure 2D), whereas Bid cleavage could be blocked by Z-VAD. However, Bid cleavage by GzmB was not abolished by Z-VAD (Figure 2D). Casp-3 was activated after GzmM or GzmB treatment that was used as positive controls. The same blot was stripped for detection of β-actin, which served as a good loading control. To further confirm that GzmM does not directly cleave Bid, Jurkat cell lysates or recombinant Bid (rBid) was treated with different doses of GzmM or 1 μM GzmB. GzmM did not process native or recombinant Bid even with 2 μM GzmM (Figure 2E). Bid was cleaved by GzmB that was used as a positive control. GzmB directly processes Bid to target mitochondria resulting in Cyt c release.21 However, GzmM-induced Bid cleavage was casp dependent but not directly processed by GzmM.

GzmM-mediated Cyt c release is casp-dependent but Bid-independent. (a) Z-VAD can block Cyt c release after loading of GzmM. Jurkat cells (4 × 105) were loaded with GzmM/Ad with the indicated times. Cytoplasmic and pellet fractions were separated and probed for Cyt c. β-actin served as a loading control. Supern, supernatant. (b) Casp-8 is activated earlier than Cyt c release. HeLa or Jurkat cells were treated with GzmM plus Ad for the indicated time points. Cells were fixed, permeabilized and stained with antibodies against Cyt c and activated casp-8. D-NGzmM/Ad treated cells were used as a negative control. Z-VAD blocked the activation of casp-8. (c) Casp-3 is activated when Cyt c is released. Cells were treated as above and stained with antibodies against active casp-3 and Cyt c. Z-VAD blocked the activation of casp-3. (d) Bid cleavage can be blocked by Z-VAD. Jurkat cells (2 × 105) were pretreated with Z-VAD for 30 min and then treated with 0.5 and 1 μM GzmM, 1 μM D-NGzmM or 1 μM GzmB plus Ad for 4 h. Bid or casp-3 processing was determined by western blotting. Casp-3 was used as activity controls for GzmM and GzmB. β-actin was used as a loading control. The same blot was stripped for immunoblotting. (e) GzmM does not degrade Bid directly. Jurkat cell lysates (left panel) or recombinant Bid (rBid) (right panel) was incubated with the indicated concentrations of GzmM or 1 μM GzmB at 37 °C for 2 h. Cell lysates were probed with anti-Bid antibody by immunoblotting and rBid processing was stained with Coomassie brilliant blue. D-NGzmM (1 μM) was treated as a negative control
Casp-8 is an initiator casp for GzmM-induced casp cascade
To verify which casp initiates casp cascade, different casp inhibitors were preincubated with HeLa cells before loading of GzmM. Through careful titration of used casp inhibitors, we determined that 25 μM was the optimal concentration for inhibition experiments. We observed that the inhibitor for casp-8 (Z-IETD) completely blocked the activation of casp-8, -9 and -3 (Figure 3A, left panel). The casp-9 and -3 inhibitors (Z-LEHD and Z-DEVD) only partially inhibited the activation of casp-8, -9 and -3. The casp-2 inhibitor Z-VDVAD did not significantly inhibit activation of casp-8, -9 and -3. β-actin was unchanged as a good loading control. These results indicate that casp-8 is an initiator casp for GzmM-induced casp cascade. Moreover, capapse-8 inhibitor Z-IETD significantly attenuated GzmM-induced cytolysis compared with that of GzmM/Ad (30.3 versus 67.3%, P<0.01) (Figure 3A, right panel). Casp-2, -3 and -9 inhibitors could also partially decrease GzmM-induced cell death. Similar results were obtained in Jurkat cells. These data suggest that casp activation cascade has a major role in GzmM-induced cytotoxicity and casp-8 is an initiator casp.

Casp-8 activation is an initial step for casp cascade. (a) Specific casp-8 inhibitor Z-IETD significantly blocks other casp activation induced by GzmM. HeLa cells (2 × 105) were incubated with the indicated casp inhibitors for 30 min before GzmM/Ad loading for 4 h. Treated cells were harvested for casp processing by immunoblotting (left panel). β-actin served as a loading control. The same blot was stripped for immunoblotting. Total dead cells were calculated as above (right panel). **P<0.01. The data represent three independent experiments. (b) Casp-8 knockdown restrains casp activation. Casp-8 was knocked down in HeLa cells by transfection of pSUPER-shCasp-8 and stably silenced cell lines were established by puromycin selection (upper panel). Casp-8-silenced HeLa cells were treated with GzmM/Ad or D-NGzmM/Ad for 4 h followed by immunoblotting (lower panel). β-actin was used as a loading control. (c) Casp-9 silencing does not affect casp activation. Casp-9-silenced cell lines were established through transfection of pSUPER-shCasp-9 (upper panel). Casp-9-silenced HeLa cells were treated with GzmM/Ad or D-NGzmM/Ad for 4 h followed by immunoblotting (lower panel). (d) Casp-3 silencing does not impair the activation of casp-8 and casp-9. Casp-3 was silenced in HeLa cells by transfected pSUPER-shCasp-3 (upper panel). The cells were treated with GzmM plus Ad for 4 h and probed by immunoblotting. (e) Casp-8 deficiency blocks GzmM-mediated casp activation and cell death. Wild-type (WT) and casp-8 deficient Jurkat cells (Jurkat Casp-8−/−) were treated with 1 μM GzmM/Ad, 1 μM GzmB /Ad or 100 ng/ml TNF-α plus 10 μg/ml cycloheximide (CHX) at 37 °C for 4 h. Treated cells were harvested for immunoblotting (left panel) and flow cytometry (right panel). Dead cells were calculated as described as above. **P<0.01
To confirm that casp-8 initiates casp activation, casps-8 and -9 were knocked down in HeLa cells and their stably silenced cell lines were established by puromycin selection (Figures 3B and C, upper panel). Two shRNA sequences against casp-8 effectively silenced its expression. Casp-8 depletion could restrain activation of casp-9 and -3 (Figure 3B, lower panel). The empty pSUPER vector-transfected cells were used as a control. D-NGzmM/Ad treated cells served as a negative control. β-actin was unchanged as a loading control. However, casp-9 knockdown cells could not reduce activation of casps compared with the cells transfected with empty pSUPER vector (Figure 3C, lower panel). D-NGzmM/Ad treated cells were used as a control. Furthermore, we silenced casp-3 and established its stably silenced lines (Figure 3D, upper panel). Casp-3 knockdown did not apparently affect GzmM-induced activation of casp-8 and -9 (Figure 3D, lower panel). In order to further verify the initial casp-8 for GzmM-induced casp cascade clearly, casp-8-deficient Jurkat cells (clone I9.2) were treated with GzmM/Ad for detection of casp activation. Casp-8 deficiency completely ablated casp-3 and Bid processing (Figure 3E, left panel). In contrast, GzmB still proteolyzed procaspase-3 and Bid. As TNF-α/cycloheximide (CHX) causes casp-8-dependent apoptosis,15 casp-8 deficiency almost abolished casp-3 processing and completely blocked Bid proteolysis (Figure 3E, left panel). β-actin was used as a loading control. Furthermore, casp-8 deficiency significantly decreased GzmM-induced cytolysis compared with that of wild-type (WT) cells (25±5 versus 64±6%, P<0.01) (Figure 3E, right panel). However, casp-8 deficiency had no apparent effect on GzmB-induced apoptosis (60±7 versus 66±7%, P>0.05). In contrast, TNF-α/CHX treatment caused much less cell death in casp-8 deficient cells compared with in WT cells (34±4 versus 72±9%, P<0.01). Taken together, casp-8 is an initiator casp in GzmM-induced casp cascade.
FADD is indispensable for GzmM-induced casp activation cascade
FADD is an adaptor protein that recruits procaspase-8 to create the DISC complex in the extrinsic death receptor pathway.22 However, it is unknown how FADD triggers casp-8 activation in GzmM-induced apoptosis. The dominant negative form of FADD (DN-FADD), without DED, can inhibit apoptosis mediated by death receptors.23 We found that DN-FADD overexpression was able to suppress activation of casp-8, -9 and -3 (Figure 4A). TNF-α/CHX was treated as a positive control. These data are representative of at least three separate experiments. Surprisingly, FADD deficiency completely abolished GzmM-induced casp activation (Figure 4B). However, FADD deficiency did not affect GzmB-induced casp processing. Moreover, FADD deficiency remarkably repressed GzmM-induced cytolysis (34±3 versus 60±6%, P<0.01) (Figure 4C). FADD deficiency had no apparent effect on GzmB- or TNF-α/CHX-induced cell death (69±5 versus 73±8%, P>0.05; 80±2 versus 81±9%, P>0.05). The intrinsic pathway is another classical death pathway that needs apoptotic protease-activating factor 1(Apaf-1), procaspase-9, Cyt c and dATP to form an apoptosome to initiate casp cascade.24 Apaf-1 is an essential protein for apoptosome formation. We wanted to see whether Apaf-1 participates in GzmM-induced cell death. Apaf-1 was knocked down in HeLa cells and stably silenced cell lines were established (Figure 4D, upper panel). Apaf-1 knockdown did not impair GzmM-induced activation of casp-8, -9 and -3 compared with those of the empty vector-transfected HeLa cells (Figure 4D, lower panel). Staurosporine served as a positive control. β-actin was unchanged as a loading control. These data imply that the intrinsic death pathway does not contribute to activation of casp-8. To further confirm the physiological relevance of these observations, rIL-2-activated lymphokine-activated killer (LAK) cells were incubated with 51Cr-labeled WT or FADD−/− Jurkat cells followed by a 4 h 51Cr release assay. LAK cells showed good killing activity against WT Jurkat cells (Figure 4E). However, FADD deficiency significantly reduced LAK cell-induced cytotoxicity. The inhibitor of cytotoxic granule pathway concanamycin A (CMA) could inhibit but not block LAK cell-induced cytolysis, which is in agreement with our previous reports.25, 26 These data indicate that the extrinsic death pathway is involved in LAK cell-mediated cytolysis in addition to a major granule pathway.

FADD is indispensable for GzmM-mediated casp activation. (a) FADD dominant negative form (DN-FADD) inhibits casp activation. HeLa cells were transfected with Flag-DN-FADD (upper panel) and treated with GzmM/Ad or TNF-α/CHX for 4 h. Casp activation was assayed by immunoblotting. The same blot was stripped for detection of β-actin. Similar results were obtained by using Jurkat cells (data not shown). (b) FADD deficiency restrains GzmM-mediated casp activation. FADD deficient-Jurkat cells (Jurkat FADD−/−) were treated with GzmM/Ad, D-NGzmM/Ad or GzmB/Ad and assayed as above. (c) FADD deficiency inhibits GzmM-mediated apoptosis but not GzmB. FADD-deficient Jurkat cells were treated with 1 μM GzmM/Ad, 1 μM D-NGzmM/Ad, 1 μM GzmB /Ad or TNF-α/CHX for 4 h, and the dead cells were calculated as above. **P<0.01. (d) Apaf-1 knockdown does not suppress GzmM-mediated casp activation. Apaf-1 was knocked down in HeLa cells by transfection of pSUPER-shApaf-1 and stably silenced cell lines were established by puromycin selection (upper panel). Apaf-1-silenced HeLa cells were treated with GzmM plus Ad followed by immunoblotting. Staurosporine (10 μM) was treated as a positive control. (e) FADD deficiency attenuates LAK cell-mediated cytolysis. 51Cr-labeled WT or FADD-deficient Jurkat cells were incubated with rIL-2-activated LAK cells with different E/T ratios for 4 h at 37 °C. Concanamycin (0.5 μM) was used to block cytotoxic granule pathway. Cytolysis of target cells was calculated by 51Cr release as means±S.D. These data were representative of at least three independent experiments. **P<0.01
GzmM proteolyzes FADD at Met196 to generate truncated FADD
To determine whether FADD is directly processed by GzmM, recombinant FADD (rFADD) was incubated with different doses of GzmM at 37 °C for 2 h or with 1 μM D-NGzmM for 2 h. rFADD began to degrade at 0.1 μM and was almost proteolyzed at 0.5 μM of GzmM (Figure 5A). rFADD was cleaved as early as 15 min and completely processed after 60 min. The inactive D-NGzmM did not degrade rFADD. These results indicate that FADD processing needs enzymatic activity of GzmM. To observe whether GzmM can cleave native FADD, Jurkat cell lysates (2 × 105 cell equivalent) were incubated with 1 μM GzmM at 37 °C for 15–60 min. Native FADD started to proteolyze by 15 min and was completely cleaved after 45 min (Figure 5B). Casp-8 and Bid were not directly cleaved by GzmM as negative controls. In contrast, 1 μM GzmB did not proteolyze FADD even by 1 h treatment. Casp-8 and Bid were directly cleaved by GzmB as positive controls. The same membrane was stripped and probed for β-actin as a loading control.

FADD is cleaved by Gzm M at Met196. (a) Recombinant FADD (rFADD) is processed by GzmM. rFADD was incubated with different concentrations of GzmM at 37 °C for 2 h or with 0.5 μM GzmM for the indicated times. Samples were analyzed by immunoblotting with anti-FADD antibody. (b) Native FADD is cleaved by GzmM. Jurkat cell lysates (2 × 105 cell equivalents) were treated with 1 μM GzmM for the indicated time points, and probed with anti-FADD antibody. The same immunoblots were stripped and reprobed with anti-casp-8 and anti-Bid antibodies. GzmB was loaded as a positive control, and β-actin was used as a loading control. (c) FADD cleavage starts earlier than casp-8 activation. HeLa cells were loaded with GzmM plus Ad for the indicated times followed by immunoblotting. Z-VAD (100 μM) was used to block casp activation. (d) FADD is cleaved by GzmM at Met196. Recombinant full-length FADD (FL-FADD) and point mutant M196A-FADD were incubated with 0.5 μM GzmM for 1 h followed by immunoblotting (upper panel). HEK293A cells were transfected with 3 × Flag-CMV-M196A-FADD. After 24 h, cells were harvested and lysed. Cell lysates were treated with 1 μM GzmM for 1 h and probed with anti-FADD antibody (lower panel). (e) M196A-FADD mutant impairs GzmM-mediated casp-8 activation and apoptosis. FADD-deficient cells were infected with lentivirus encoding wild-type FL-FADD or M196A FADD mutant for 18 h, and loaded with 1 μM GzmM plus Ad for 4 h at 37 °C. Cells were harvested for immunoblotting (upper panel) and flow cytometry (lower panel). Dead cells were counted as described above. **P<0.01. (f) FADD is processed in LAK cell-mediated cytotoxicity and blocked by the GzmM specific inhibitor. 51Cr-labeled HeLa cells were incubated with LAK cells (E/T ratio 10 : 1) for 4 h at 37 °C. For GzmM blocking, LAK cells were pretreated with 100 μM GzmM inhibitor (Ac-KVPL-CMK) before incubation with HeLa cells. FADD and β-actin were detected by immunoblotting (upper panel). Specific lysis was calculated by 51Cr release as shown means±S.D. (lower panel). *P<0.05; **P<0.01. (g) Rescued FL-FADD expression, but not M196A-FADD, can restore killing activity by LAK cells. FL-FADD or M196A-FADD expression was rescued in FADD-deficient Jurkat cells as 5E (upper panel). FL-FADD- or M196A-FADD-expressed Jurkat cells were incubated with LAK cells at the indicated E/T ratios. Specific cytotoxicicity was tested as above. *P<0.05; **P<0.01
To further verify the cleavage of FADD in a more physiological condition, HeLa cells were loaded with 1 μM GzmM/Ad for 30–120 min. FADD was proteolyzed after 30 min and almost processed by 90 min (Figure 5C). The pan casp inhibitor Z-VAD could not block FADD cleavage, which indicates that FADD was directly cut by GzmM but not by casps. The same membrane was stripped and probed for casp-8, casp-3 and β-actin. Moreover, FADD processing was much earlier than procaspase-8 processing. Casp-3 processing appeared after casp-8 activation. These data suggest that FADD processing is an earlier event than procaspase-8 processing. The cleaved products (truncated FADD (tFADD)) were cutoff 2 kDa from the full-length FADD (FL-FADD). The DED of FADD locates at the N-terminus, whose structure is rigid and unsuitable for proteolysis.17 Therefore, the cleavage site was supposed to locate at the C-terminus of FADD. Because GzmM prefers Leu or Met at the P1 site, and Pro at P2 site,11 Met196 was predicted as the candidate cleavage site. As expected, Met196Ala-FADD (M196A-FADD) mutant was not proteolyzed by GzmM (Figure 5D, upper panel). To further confirm this cleavage site, 3 × Flag-M196A-FADD was overexpressed in HEK293A cells and their lysates were treated by GzmM. We found that GzmM failed to cleave 3 × Flag-M196A-FADD (Figure 5D, lower panel), whereas endogenous FADD was still cleaved by GzmM in cells expressing 3 × Flag-M196A-FADD or empty vector control. β-actin was unchanged as a loading control. As the truncated form of FADD was not able to be sequenced by the N-terminal sequencing technique, we generated recombinant GST-FADD(181–208)–maltose-binding protein (MBP) fusion protein and incubated it with GzmM. GzmM cleaved the GST-FADD(181–208)–MBP fusion protein to generate two major bands (Supplementary Figure 1). We analyzed the two bands by immunoblotting and determined the C-terminal cleavage product as the middle band. We sequenced the C-terminal band through the N-terminal sequencing and identified the N-terminal sequence as SWNSD, which is consistent with the inferred cleavage site by GzmM (193-MSPM↓SWNSD-201).
To define the relationship between FADD processing and casp activation in GzmM-induced cytolysis, FL-FADD or M196A-FADD mutant was overexpressed in FADD-deficient Jurkat cells. M196A-FADD mutant was not proteolyzed by GzmM and was incapable of fully restoring casp-8 activation (Figure 5E, upper panel). GzmM could process FL-FADD to form tFADD leading to casp-8 activation. FADD deficiency failed to cause activation of casp-8. M196A-FADD-overexpressed cells also impaired GzmM-induced cytotoxicity compared with that of FL-FADD-transfected cells (Figure 5E, lower panel). These data indicate that FADD processing contributes to casp-8 activation. Importantly, FADD was cleaved in LAK cell-mediated cytolysis (Figure 5F, upper panel). We previously designed a tetrapeptide GzmM inhibitor (Ac-KVPL-CMK) and verified this inhibitor was cell permeable.12 The GzmM inhibitor (100 μM) could block GzmM-induced cell death and FADD cleavage (Supplementary Figure 2). Notably, the GzmM inhibitor was also able to block LAK cell-mediated FADD processing (Figure 5F, upper panel). Moreover, the GzmM inhibitor could significantly suppress LAK cell-mediated cytotoxicity. Identical results were obtained in Jurkat cells (data not shown). To further confirm the critical role of FADD processing in LAK cell-mediated cytotoxicity, FADD−/− Jurkat cells with rescued FL-FADD and M196A-FADD expression (Figure 5G, upper panel) were incubated with rIL-2 activated LAK cells. As expected, the rescued FL-FADD expression restored killing activity by LAK cells (Figure 5G, lower panel). In contrast, the GzmM-non-cleavable form M196A-FADD still resisted LAK cell-mediated cytolysis. Taken together, these results imply that FADD is a specific physiological substrate for GzmM and its processing enhances LAK cell-mediated cytotoxicity.
FADD cleavage enhances its self-association to recruit procaspase-8 for auto-processing
Overexpressed FL-FADD has been reported to form death effector filaments, which efficiently recruits and activates casp-8. Self-association of FADD is crucial for both death receptor pathway and formation of death effector filaments to activate casp-8.27 Therefore, we hypothesized that tFADD might be more susceptible to self-association than that of FL-FADD for recruiting procaspase-8. To verify this hypothesis, we utilized the yeast two-hybrid system to analyze self-association of FADD. cDNAs for the FL-FADD or tFADD was inserted into the plasmids carrying the Gal4 activation domain (AD) (pGADT7) and DNA-binding domain (BD) (pGBKT7). pGADT7-FL-FADD/pGBKT7-FL-FADD or pGADT7-tFADD/pGBKT7-tFADD were co-transformed into an AH109 yeast strain. The BD-murine p53 and AD-SV40 large T antigen fusions were performed as a positive control. Interestingly, tFADD exhibited much stronger self-association than FL-FADD (Figure 6A). FL-FADD just showed weak self-interaction. Next, we coexpressed Flag-FL-FADD/hemagglutinin (HA)-FL-FADD or Flag-tFADD/HA-tFADD in HEK293A cells for co-immunoprecipitation assay. We observed that tFADD showed much more enhanced self-interaction than FL-FADD (Figure 6B). The expression levels of FL-FADD and tFADD had no significant differences. Control IgG failed to precipitate either Flag- or HA-tagged proteins.

Truncated FADD (tFADD) exhibits enhanced self-association to recruit procaspase-8 resulting in casp-8 activation. (a) tFADD exerts stronger self-association than FL-FADD. AH109 yeast strain was co-transformed with the indicated constructs and the transformants were plated on synthetic defined medium with or without histidine (−His) and adenine (−Ade). −His−Ade plates were supplemented with 1.5 mM 3-AT to suppress background growth. +His+Ade plates were served as identical amount controls of yeasts. AH109 cotransformed with pGADT7-SV40 large T-antigen and pGBKT7-murine p53 was used as a positive control (left panel). Interaction activity was quantified by β-galactosidase assay and shown as means±S.D. (right panel). **P<0.01. 1: AD-FL-FADD+BD-FL-FADD; 2: AD-tFADD+BD-tFADD; 3: AD-SV40 large T antigen+BD-murine p53; 4: AD vector+BD vector; 5: AD-FL-FADD+BD-murine p53; 6: AD-SV40 large T antigen+BD-FL-FADD; 7: AD-tFADD+BD-murine p53; 8: AD-SV40 large T antigen+BD-tFADD. (b) Self-association of tFADD was confirmed by Co-IP assay. Flag-FL-FADD and HA-FL-FADD or Flag-tFADD and HA-tFADD were cotransfected into HEK293A cells. After 24 h, cells were harvested and lysed for immunoprecipitation with anti-Flag antibody and visualized by immunoblotting with anti-HA and anti-Flag antibodies. Mouse IgG was used as a negative control. (c) tFADD facilitates its oligomerization. GST-FL-FADD or GST-tFADD recombinant proteins were analyzed by gel filtration (Superdex 200 column) (left panel). GST-FL-FADD or GST-tFADD was observed on reduced (R, lower panel) and native (N, upper panel) gels. D, dimmer; M, monomer; P, polymer. (d) tFADD efficiently recruits procaspase-8. HEK293A cells were transfected with Flag-tagged FL-FADD or tFADD for 24 h followed by casp-8 immunoprecipitation. (e) Overexpression of tFADD causes casp-8 activation. HEK293A cells were infected with lentivirus encoding FL-FADD, GFP, M196A-FADD or tFADD for 24 h followed by immunoblotting. The same blot was stripped for western blotting. (f) tFADD overexpression causes apoptosis. Cells overexpressing with the indicated proteins for 24 h were analyzed by Annexin V/PI staining. The data are representative of at least three independent experiments
To further confirm self-association of tFADD, we expressed recombinant GST-FL-FADD or GST-tFADD and then fractionated by gel filtration followed by native gel resolution. FL-FADD could form some dimmers (Figure 6C). Notably, GST-tFADD was able to produce dimmers and polymers, which was more prone to form oligomers than GST-FL-FADD. We next wanted to determine whether tFADD self-association efficiently recruits procaspase-8 for its auto-processing. Interestingly, anti-casp-8 antibody could precipitate a large amount of tFADD from Flag-tFADD-overexpressed HEK293A cells (Figure 6D). However, anti-casp-8 antibody just precipitated a little amount of FL-FADD from Flag-FL-FADD-overexpressed HEK293A cells. Input of cell lysates was identical for each sample that was confirmed by immunoblotting. Surprisingly, tFADD itself was able to cause casp-8 activation (Figure 6D). We then wanted to confirm direct activation of casp-8 by tFADD in intact cells. FL-FADD, M196A-FADD or tFADD was overexpressed in HEK293A cells followed by probing for casp-8. We observed that tFADD was able to cause casp-8 activation (Figure 6E). By contrast, FL-FADD or M196A-FADD failed to activate casp-8. The expression levels of the three proteins were almost identical. Additionally, tFADD itself could trigger apparent apoptosis (Figure 6F), whereas FL-FADD or M196A-FADD overexpression did not cause significant cell death. Taken together, tFADD triggers casp-8 activation to initiate GzmM-induced casp activation cascade.
Discussion
GzmM is constitutively and abundantly expressed in the innate effector NK cells, which act as the first line of defense against virus-infected or transformed tumor cells.13 Human NK cell line KHYG-1 with high expression of GzmM showed a great capacity to kill tumor cells, although GzmA and GzmB were not detectable in this killer cell line.28 It suggests that GzmM may have a critical role in NK cell-mediated cytotoxicity in innate immunity. We previously demonstrated that GzmM induces casp-dependent apoptosis of target tumor cells, whose death features are similar to GzmB.8 GzmB-mediated casp activation has been well defined. As GzmB cuts after aspartate, several procaspases have been reported as substrates for GzmB, including procaspase-3, -7, -8 and -10.29 Direct cleavage of procaspase-3 and -7 by GzmB causes casp activation cascade leading to degradation of hundreds of protein substrates.30 Additionally, GzmB can directly cleave Bid to a truncated Bid (tBid) to damage mitochondria resulting in Cyt c release.21 However, herein we found that GzmM causes casp-dependent Bid cleavage and Cyt c release, but it does not directly cleave procaspases or Bid.
Casp-8 exits within cells as an inactive zymogen. This zymogen is activated by auto-processing or cleavage by other proteases. As an initiator casp, procaspase-8 requires several kinds of molecular platforms to facilitate its autocatalysis, such as DISC in the death receptor pathway.16 Death receptors, such as Fas, TNF-R1, TRAIL-R1 and TRAIL-R2, are characterized by multiple conserved cysteine-rich domains within their extracellular domain and an intracellular DD. The intracellular DD motif can recruit the DD-containing adaptor protein FADD through homophilic DD/DD interactions.16, 17, 31 FADD possesses an N-terminal DED and a C-terminal DD domains, which recruits procaspase-8 to generate the DISC.
A recent report demonstrated that the conformational and cleavage status of procaspase-8 determines its proteolytic activity and substrate preference in the DISC.32 In vitro DISC reconstitution model shows a two-step activation mechanism, which results in a critical switch in both the catalytic activity and substrate repertoire of casp-8. Without the second-step cleavage event, Fas-DISC-mediated activation of procaspase-8 causes cell survival. In this study, GzmM cleaves FADD after Met196 to form tFADD that remains an intact DD motif. Intriguingly, tFADD exhibits much stronger self-association than FL-FADD. It suggests that removal of the C-terminus of FADD may promote conformational changes of tFADD. Conformation of tFADD might fully activate the DISC to drive the second-step cleavage event of casp-8. Actually, processed FADD by GzmM or tFADD overexpression can generate p41 product of casp-8 resulting in casp-3 activation and Bid cleavage, which is consistent with the second-step cleavage event to drive the cell to death.32
NK/CTL cells attack their target cells through two main pathways, death receptor pathway and granule-mediated pathway.30 Granule killer cells can express ligands of the death receptors to initiate death receptor-mediated cell death, which is termed as an extrinsic death pathway. The granule-mediated death pathway is a major mechanism for granule killer cells to eradicate intracellular pathogens and tumor cells.1 Here we found that GzmM can activate casp-8 through cleavage of death receptor adaptor FADD. Thereafter, casp-8 causes activation of other downstream casps and Bid cleavage. Activated casp-3 can enhance Bid processing to generate tBid, which targets mitochondria to liberate Cyt c. Once Cyt c releases into cytosol, it binds to Apaf-1, procaspase-9 and dATP to form an apoptosome,23 which subsequently activates casp-3 to execute casp activation cascade. GzmM initiates casp activation cascade by connection of both extrinsic and intrinsic death pathways through initial activation of casp-8. Notably, different Gzms in killer lymphocytes may exert their killing roles with different dynamics and efficiency. FADD−/− and M196A-FADD Jurkat cells showed killing resistance by LAK cells compared with the WT Jurkat cells within 4 h incubation (Figures 4E and 5G). However, when we prolonged the incubation time up to 8 h, there was no significant killing difference between the WT and FADD−/− or M196A-FADD Jurkat cells (data not shown). These data suggest that other Gzms like GzmB can bypass the FADD-casp 8 pathway to mediate target cell death with longer incubation. We hypothesized that GzmM may exert killing functions earlier than GzmB in physiological conditions or GzmM might be more effective than GzmB for tumor clearance at early time. The physiological roles of different Gzms in tumor clearance need to be further investigated.
Materials and Methods
Cell lines, antibodies and reagents
HeLa and human embryonic kidney epithelial 293A (HEK293A) cells were cultured in DMEM (Gibco, Carlsbad, CA, USA), supplemented with 10% (vol/vol) fetal bovine serum (FBS), 50 μM β–Mercaptoethanol, 100 U/ml penicillin and 100 μg/ml streptomycin. Jurkat wild-type, Jurkat casp-8-deficient (clone I9.2) and Jurkat FADD-deficient (clone I2.1) cells were grown in RPMI1640 medium supplemented with 10% FBS and 50 μM β–Mercaptoethanol. Commercial antibodies were mouse monoclonal antibodies against casp-8, casp-9 (Cell Signaling Technology, Danvers, MA, USA), β-actin, Flag (Sigma-Aldrich, St. Louis, MO, USA), Cyt c, FADD (BD Pharmingen, San Jose, CA, USA), rabbit monoclonal antibody against HA (Cell Signaling Technology), rabbit polyclonal antibody against caspase-3 and active casp-3 (Cell Signaling Technology), Alexa488-conjugated donkey anti-rabbit IgG, Alexa594-conjugated donkey anti-mouse IgG (Molecular Probes, Carlsbad, CA, USA) and HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Casp inhibitors Z-VDVAD-FMK, Z-DEVD-FMK, Z-IETD-FMK, Z-LEHD-FMK and Z-VAD-FMK (all O-methylated) were purchased from Calbiochem (Darmstadt, Germany). Fluorogenic substrates Ac-VDVAD-AFC, Ac-DEVD-AFC, Ac-IETD-AFC and Ac-LEHD-AFC were from Calbiochem. TNF-α, CHX, staurosporine and o-nitrophenyl β-D-galactopyranoside (ONPG) were from Sigma-Aldrich. Superdex 200 column was from GE Healthcare (Uppsala, Sweden).
Cellular casp activity assay
Casp activity was determined by following manufacturer's instruction. Briefly, 4 × 105 Jurkat cells were treated as the indicated times, washed once with PBS and lysed with 25 μl lysis buffer (50 mM HEPES, 100 mM NaCl, 0.1% CHAPS, 5 mM DL-dithiothreitol (DTT), 0.1 mM EDTA, pH7.4). Cell lysates were incubated with 30 μM casp substrates (Ac-VDVAD-AFC, Ac-DEVD-AFC, Ac-IETD-AFC and Ac-LEHD-AFC) at 37 °C in assay buffer (50 mM HEPES, 100 mM NaCl, 0.1% CHAPS, 10 mM DTT, 1 mM EDTA, 10% glycerol, pH7.4). The 7-amino-4-trifluoromethyl coumarin (AFC) was measured by excitation at 390 nm and emission at 485 nm by Fluoroskan Ascent (Type 374, ThermoLabsystems, Waltham, MA, USA).
Knockdown of casp-8, -9 and Apaf-1
RNA sequences against casp-8, casp-9 and Apaf-1 were designed based on pSUPER system instructions (Oligoengine, Seattle, WA, USA) and cloned into pSUPER-puro vector. The target sequences were as follows: casp-8 (shRNA#1: 5′-AAAGGGAACTTCAGACACCAG-3′, shRNA#2: 5′-GGAACAACTGGACAGTGAA-3′),33 casp-9 (5′-ACAGATGCCTGGTTGCTTTAA-3′),34 casp-3 (5′-AGGTGGCAACAGAATTTGAGT-3′)34 and Apaf-1 (5′-AATTGGTGCACTTTTACGTGA-3′).35 The constructs were transfected into HeLa cells by using Lipofectamine2000 (Invitrogen, Carlsbad, CA, USA) as described.25 Stable clone pools were selected in the presence of 1 μg/ml puromycin.
Cyt c release assay
Jurkat cells were harvested and washed twice with PBS after treatment with GzmM/Ad for the indicated times as described.7 Cells were lysed with 0.025% digitonin buffer for 5 min on ice. The pellet and supernatant fractions were obtained for detection of Cyt c.
Immunoprecipitation
For immunoprecipitation of casp-8 and FADD, 5 × 106 HEK293A cells were transfected with the indicated constructs for 24 h, and lysed with lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 1% Triton X-100 and protease inhibitor cocktail, pH 7.4). After centrifugation, the supernatants were incubated with 1 μg/ml of anti-casp-8 antibody or anti-Flag antibody overnight at 4 °C. Cell lysates were immunoprecipitated with 30 μl of protein A/G plus agarose (Santa Cruz Biotechnology) at 4 °C for 4 h. The precipitates were washed three times with wash buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, pH 7.4) followed by immunoblotting.
Yeast two-hybrid assay
AH109 yeast strain was co-transformed with the indicated constructs (pGBKT7-FL-FADD, pGADT7-FL-FADD, pGBKT7-tFADD, pGADT7-tFADD, pGADT7-SV40 large T-antigen and pGBKT7-murine p53) following the guidelines in the manufacturer's instructions (Clontech, Palo Alto, CA, USA). Double transformants were selected on synthetic defined (SD) medium lacking leucine and tryptophan. Interaction of fusion proteins was monitored by spotting the transformants on plates lacking histidine, adenine, leucine and tryptophan in the presence of the competitive inhibitor of the His3 protein 3-amino-1,2,4-triazole (3-AT) or analyzed by using a liquid β-galactosidase assay.
Protein expression and mutagenesis
Recombinant human active GzmM and enzymatically inactive mutant D86N-GzmM were expressed and purified as previously described.12 The cDNA encoding full-length FADD (FL-FADD) (residues 1–208) or truncated FADD (tFADD) (residues 1–196) was cloned into pET-28a or pGEX-6P-1. FADD fragment (181–208) was fused with MBP, and cloned into PGEX-6P-1. His-tag-fused FL-FADD and tFADD were purified by using Ni-NTA columns, and GST-tag-fused FL-FADD, tFADD and FADD(181–208)-MBP were purified with GST columns. For mammalian cell expression, FL-FADD, M196A-FADD (point mutation at Met196 to Ala), DN-FADD (residues 80–208) and tFADD were inserted into 3 × Flag-CMV-10 or pCMV-HA vectors, which fused FADD with N-terminal Flag tag or N-terminal HA tag, respectively. The constructs were transfected into HeLa or HEK293A cells for 24 h, and the expression levels of each construct was determined by immunoblotting.
Casp cleavage and apoptosis assay
HeLa or Jurkat cells were loaded with Gzms and replication-deficient Ad type V as described.9, 14 After treatment with the indicated times, cells were harvested and washed once with PBS. The samples were added with 5 × SDS loading buffer to stop the reaction and casp cleavage was monitored by immunoblotting. For apoptosis assay, cells were washed twice with PBS, and stained with FITC-conjugated Annexin V and propidium iodide followed by flow cytometry (FACSCalibur, BD Biosciences, San Jose, CA, USA). All the statistical data were analyzed by Student's t-test.
Cleavage assay
Jurkat or HeLa cells were treated with lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM EDTA, 1% Triton X-100, pH7.4) for 30 min. Cell lysates (equivalent to 2 × 105 cells) or 1 μM rFADD was incubated with different concentrations of GzmM or D86N-GzmM for the indicated time points in 20 μl cleavage buffer (150 mM NaCl, 50 mM Tris-HCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.4). Reactions were stopped by 5 × SDS loading buffer and resolved by SDS-PAGE followed by immunoblotting.
Immunofluorescence
HeLa cells were grown on coverslips for 24 h, and then treated with GzmM/Ad at 37 °C. Jurkat cells were treated with GzmM/Ad and plated on coverslips that were pretreated with 0.01% poly-L-lysine (Sigma-Aldrich). Next, cells were fixed, permeabilized and blocked for 30 min with 10% donkey serum in PBS as a previous report.36 Coverslips were incubated with anti-active casp-8, anti-active casp-3 or anti-Cyt c antibody overnight at 4 °C, washed with PBS and stained with Alexa488-conjugated donkey anti-rabbit IgG and Alexa594-conjugated donkey anti-mouse IgG. Images were visualized with laser scanning confocal microscopy (Olympus FV500, Tokyo, Japan).
LAK cell-mediated cytolysis assay
PBMCs from healthy donors (Beijing Red Cross Blood Center, Beijing, China) were separated as described.36 LAK cells were achieved by incubation with human rIL-2 (1000 U/ml) in 15% FBS/RPMI1640. For inhibition assay, 51Cr(chromium)-labeled HeLa or Jurkat cells were pretreated with 500 nM CMA or 100 μM GzmM-specific inhibitor (Ac-KVPL-CMK) before incubation with LAK cells. Lysis percentage was assayed by release of 51Cr as described.3
Abbreviations
- Ad:
-
adenovirus
- casp:
-
caspase
- CMA:
-
concanamycin A
- CTL:
-
cytotoxic T-lymphocyte
- Cyt c:
-
cytochrome c
- DD:
-
death domain
- DED:
-
death effector domain
- DISC:
-
death-inducing signaling complex
- DN-FADD:
-
dominant negative FADD
- D-NGzmM:
-
D86N-mutated granzyme M
- FADD:
-
Fas-associated protein with death domain
- Gzm:
-
granzyme
- HA:
-
hemagglutinin
- LAK:
-
lymphokine-activated killer
- NK:
-
natural killer
- XIAP:
-
X-linked inhibitor-of-apoptosis protein.
References
Fan Z, Zhang Q . Molecular mechanisms of lymphocyte-mediated cytotoxicity. Cell Mol Immunol 2005; 2: 259–264.
Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 2010; 468: 447–451.
Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J . Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 2003; 112: 659–672.
Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A et al. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol 2003; 4: 145–153.
Trapani JA, Sutton VR . Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol 2003; 15: 533–543.
Zhao T, Zhang H, Guo Y, Zhang Q, Hua G, Lu H et al. Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ 2007; 14: 489–499.
Zhao T, Zhang H, Guo Y, Fan Z . Granzyme K directly processes bid to release cytochrome c and endonuclease G leading to mitochondria-dependent cell death. J Biol Chem 2007; 282: 12104–12111.
Lu H, Hou Q, Zhao T, Zhang H, Zhang Q, Wu L et al. Granzyme M directly cleaves inhibitor of caspase-activated DNase (CAD) to unleash CAD leading to DNA fragmentation. J Immunol 2006; 177: 1171–1178.
Hua G, Zhang Q, Fan Z . Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J Biol Chem 2007; 282: 20553–20560.
Hou Q, Zhao T, Zhang H, Lu H, Zhang Q, Sun L et al. Granzyme H induces apoptosis of target tumor cells characterized by DNA fragmentation and Bid-dependent mitochondrial damage. Mol Immunol 2008; 45: 1044–1055.
Mahrus S, Kisiel W, Craik CS . Granzyme M is a regulatory protease that inactivates proteinase inhibitor 9, an endogenous inhibitor of granzyme B. J Biol Chem 2004; 279: 54275–54282.
Wu L, Wang L, Hua G, Liu K, Yang X, Zhai Y et al. Structural basis for proteolytic specificity of the human apoptosis-inducing granzyme M. J Immunol 2009; 183: 421–429.
Fan Z, Yu P, Wang Y, Fu ML, Liu W, Sun Y et al. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood 2006; 107: 1342–1351.
Hu D, Liu S, Shi L, Li C, Wu L, Fan Z . Cleavage of survivin by Granzyme M triggers degradation of the survivin-XIAP complex to free caspase activity leading to cytolysis of target tumor cells. J Biol Chem 2010; 285: 18326–18335.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Strasser A, Jost PJ, Nagata S . The many roles of FAS receptor signaling in the immune system. Immunity 2009; 30: 180–192.
Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol 2010; 17: 1324–1329.
Donepudi M, Mac Sweeney A, Briand C, Grutter MG . Insights into the regulatory mechanism for caspase-8 activation. Mol Cell 2003; 11: 543–549.
Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM et al. A unified model for apical caspase activation. Mol Cell 2003; 11: 529–541.
Chang DW, Xing Z, Capacio VL, Peter ME, Yang X . Interdimer processing mechanism of procaspase-8 activation. EMBO J 2003; 22: 4132–4142.
Sutton VR, Wowk ME, Cancilla M, Trapani JA . Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity 2003; 18: 319–329.
Boldin MP, Goncharov TM, Goltsev YV, Wallach D . Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996; 85: 803–815.
Chinnaiyan AM, Tepper CG, Seldin MF, O'Rourke K, Kischkel FC, Hellbardt S et al. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J Biol Chem 1996; 271: 4961–4965.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–489.
Guo Y, Chen J, Shi L, Fan Z . Valosin-containing protein cleavage by granzyme K accelerates an endoplasmic reticulum stress leading to caspase-independent cytotoxicity of target tumor cells. J Immunol 2010; 185: 5348–5359.
Hua G, Wang S, Zhong C, Xue P, Fan Z . Ignition of p53 bomb sensitizes tumor cells to granzyme K-mediated cytolysis. J Immunol 2009; 182: 2152–2159.
Muppidi JR, Lobito AA, Ramaswamy M, Yang JK, Wang L, Wu H et al. Homotypic FADD interactions through a conserved RXDLL motif are required for death receptor-induced apoptosis. Cell Death Differ 2006; 13: 1641–1650.
Suck G, Branch DR, Smyth MJ, Miller RG, Vergidis J, Fahim S et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol 2005; 33: 1160–1171.
Afonina IS, Cullen SP, Martin SJ . Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev 2010; 235: 105–116.
Cullen SP, Martin SJ . Mechanisms of granule-dependent killing. Cell Death Differ 2008; 15: 251–262.
Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E et al. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009; 457: 1019–1022.
Hughes MA, Harper N, Butterworth M, Cain K, Cohen GM, MacFarlane M . Reconstitution of the death-inducing signaling complex reveals a substrate switch that determines CD95-mediated death or survival. Mol Cell 2009; 35: 265–279.
Chandra D, Choy G, Deng X, Bhatia B, Daniel P, Tang DG . Association of active caspase 8 with the mitochondrial membrane during apoptosis: potential roles in cleaving BAP31 and caspase 3 and mediating mitochondrion-endoplasmic reticulum cross talk in etoposide-induced cell death. Mol Cell Biol 2004; 24: 6592–65607.
Rahmani M, Davis EM, Crabtree TR, Habibi JR, Nguyen TK, Dent P et al. The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol 2007; 27: 5499–5513.
Lassus P, Opitz-Araya X, Lazebnik Y . Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 2002; 297: 1352–1354.
Shi L, Wu L, Wang S, Fan Z . Granzyme F induces a novel death pathway characterized by Bid-independent cytochrome c release without caspase activation. Cell Death Differ 2009; 16: 1694–1706.
Acknowledgements
We thank G Hua, L Wu, X Yang, L Wang and J Chen for their helpful advice, and Y Teng and CC Liu for their technical support. We thank Dr. G Zhang for critical editing. We thank Dr. CF Ware for providing pcDNA3-FADD plasmid. This work was supported by the National Natural Science Foundation of China (30830030, 30972676), 973 Programs (2010CB911902) and the Innovative Program of CAS (XDA01010407, KSCX2-YW-R-42).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Author contributions
SW designed and performed research, analyzed data and wrote the paper; PX and LS performed research; ZF initiated the study, designed, organized and wrote the paper.
Additional information
Edited by SJ Marin
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Wang, S., Xia, P., Shi, L. et al. FADD cleavage by NK cell granzyme M enhances its self-association to facilitate procaspase-8 recruitment for auto-processing leading to caspase cascade. Cell Death Differ 19, 605–615 (2012). https://doi.org/10.1038/cdd.2011.130
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.130
Keywords
This article is cited by
-
Tumor-targeted delivery of a C-terminally truncated FADD (N-FADD) significantly suppresses the B16F10 melanoma via enhancing apoptosis
Scientific Reports (2016)
-
Perforin and granzymes: function, dysfunction and human pathology
Nature Reviews Immunology (2015)
-
Granzyme M targets topoisomerase II alpha to trigger cell cycle arrest and caspase-dependent apoptosis
Cell Death & Differentiation (2014)
-
Granzyme M: behind enemy lines
Cell Death & Differentiation (2014)
-
NK cell intrinsic regulation of MIP-1α by granzyme M
Cell Death & Disease (2014)

{kind=link}
{kind=link}