
Abstract
In type II diabetes (T2DM), there is a deficit in β-cells, increased β-cell apoptosis and formation of intracellular membrane-permeant oligomers of islet amyloid polypeptide (IAPP). Human-IAPP (h-IAPP) is an amyloidogenic protein co-expressed with insulin by β-cells. IAPP expression is increased with obesity, the major risk factor for T2DM. In this study we report that increased expression of human-IAPP led to impaired autophagy, due at least in part to the disruption of lysosome-dependant degradation. This action of IAPP to alter lysosomal clearance in vivo depends on its propensity to form toxic oligomers and is independent of the confounding effect of hyperglycemia. We report that the scaffold protein p62 that delivers polyubiquitinated proteins to autophagy may have a protective role against human-IAPP-induced apoptosis, apparently by sequestrating protein targets for degradation. Finally, we found that inhibition of lysosomal degradation increases vulnerability of β-cells to h-IAPP-induced toxicity and, conversely, stimulation of autophagy protects β-cells from h-IAPP-induced apoptosis. Collectively, these data imply an important role for the p62/autophagy/lysosomal degradation system in protection against toxic oligomer-induced apoptosis.
Similar content being viewed by others


Main
In type II diabetes (T2DM), hyperglycemia is because of inadequate insulin secretion in response to relative insulin resistance. The islet in T2DM is characterized by a deficit in β-cells,1, 2 increased β-cell apoptosis attributable to endoplasmic reticulum (ER) stress3, 4 and intracellular β-cell toxic aggregates of the amyloidogenic protein islet amyloid polypeptide (IAPP),5 the expression of which increases with insulin resistance.6 IAPP is co-expressed and co-secreted with insulin by β-cells7 with its best-characterized physiological role being to inhibit insulin secretion through a direct paracrine effect on β-cells.8 IAPP has the propensity to form amyloid fibrils in species at risk of T2DM (humans, non-human primates and cats).9 In contrast to human-IAPP (h-IAPP), the rodent form of IAPP (r-IAPP) is non-amyloidogenic. Transgenic expression of h-IAPP in β-cells of rodents at rates present in insulin resistance leads to the development of diabetes because of ER stress-induced β-cell apoptosis with formation of membrane-damaging intracellular IAPP oligomers comparable to those present in humans with T2DM.3, 5, 10, 11, 12
Conserved mechanisms protect long-lived cells with a high protein synthetic burden (such as β-cells) from accumulation of intracellular protein aggregates and the adverse consequences termed proteotoxicity.9 A quality control system in the ER recognizes misfolded proteins and targets them for degradation by the ubiquitin/proteasome system.13 A second pathway of protein degradation, autophagy, also implies a role for ubiquitin in removal of misfolded proteins.14 Macroautophagy (hereafter referred to as autophagy) permits selective autodigestion of aged or abnormal organelles and clearance of ubiquitinated proteins.14 An isolation membrane forms in the cytoplasm to surround such targets through the activity of specific autophagy effectors.15 Among such effectors, microtubule-associated protein 1 light chain 3 (LC3) is cleaved into a soluble form known as LC3-I, which undergoes covalent conjugation to phosphatidylethanolamine to form an autophagosome-bound LC3-II.16 This lipidated form of LC3-I allows the nascent double membrane to wrap around intracellular cargo to form autophagosomes that then fuse with the lysosomes where the sequestered material is degraded by hydrolytic enzymes.
Recognition of polyubiquitinated proteins destined for degradation by autophagy is mediated by the p62/sequestosome 1 (SQSTM1) through its ubiquitin-binding UBA domain.17 Also, p62 possesses a LC3 interaction region (LIR) that facilitates direct interaction with LC3 in the autophagosome and causes p62 to be itself degraded by autophagy. Thus, p62 serves to target ubiquitinated proteins or organelles to autophagy.17 Predictably, p62 accumulates and forms cytoplasmic inclusions in autophagy-deficient mice (e.g., in liver and neurons) as the mechanism to remove accumulating p62 and its sequestrated target is lost. Based on these observations, accumulation of p62 has become established as a marker for impaired autophagy and/or defects in lysosomal degradation.18, 19
Given the presence of membrane-permeant toxic oligomers in β-cells in humans with T2DM as well as in h-IAPP transgenic rodents, by definition the mechanisms to remove intracellular protein oligomers under these conditions are either defective or overwhelmed. In the present studies we approached the question of whether the increased h-IAPP expression present under conditions of insulin resistance compromises autophagy, and if so, whether this increases the vulnerability of β-cells to apoptosis.
To address this, we first tested the hypothesis that high expression of amyloidogenic h-IAPP (either by gene dosage or by obesity) leads to impaired autophagy. We studied rodent models that collectively permitted us to affirm the hypothesis, independent of the confounding effects of hyperglycemia or nonspecific overload of the ER. We next hypothesized that suppression of the p62-mediated removal of ubiquitinated proteins leads to increased vulnerability of β-cells to h-IAPP-induced apoptosis. Conversely, we hypothesized that enhanced p62 availability decreases β-cell vulnerability to h-IAPP toxicity. Finally, we hypothesized that inhibition of the degradation of proteins sequestered by autophagy increases vulnerability of β-cells to h-IAPP-induced apoptosis and that enhanced autophagy protects β-cells from h-IAPP-induced apoptosis.
Results
Increased expression of h-IAPP in β-cells in vivo induces defective autophagy that precedes development of hyperglycemia
The number of autophagosomes was assessed by the conversion of LC3-I to LC3-II. In h-IAPP transgenic rat (HIP rat) islets, a twofold increase in LC3-II was observed in comparison to wild-type (WT) rats (Figure 1a), indicating an increased number of autophagosomes, which could be because of either enhanced autophagosome formation or a decrease in lysosomal degradation. To resolve this issue, p62 protein levels were examined. The protein levels of p62 were increased in HIP rat islets (1.8±0.4-fold versus WT rats, P<0.05; Figure 1a). In contrast, p62 mRNA levels were comparable in islets of HIP and WT rats (Figure 1b), indicating that the increase in p62 protein levels in HIP rats is most probably because of a decrease in p62 lysosomal degradation.

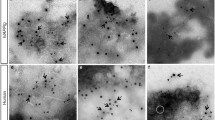
High expression of h-IAPP increases LC3-II and p62 protein levels and induces p62-positive cytoplasmic inclusions in HIP rats. (a) Protein levels of LC3, p62 and IAPP were assessed by western blot using islet protein lysates obtained from 4- to 6-month-old wild type (WT, n=6) and HIP rats (HIP, n=6). GAPDH was used as loading control. The graph represents the quantification of p62 protein levels. (b) Levels of p62 mRNA were evaluated by RT-qPCR in islets isolated from WT (n=3) and HIP rats (n=3). Data are expressed as mean±S.E.M.; *P<0.05. (c) p62 protein levels were assessed by immunofluorescence (p62, red; insulin, green; nuclei, blue) in pancreatic tissue from 4- to 6-month-old WT and HIP rats. High magnification of p62-positive structures found in HIP rats (i, ii and iii)
Immunostaining for p62 revealed the presence of p62-positive cytoplasmic inclusions in β-cells of HIP but not WT rats (Figure 1c). Different sizes of cytoplasmic inclusions were found, ranging from punctuate aggregates (Figure 1c, i and ii) to large globular structures (Figure 1c, iii).
These data suggest that high expression of h-IAPP leads to increased number of autophagosomes, accumulation of p62 and formation of p62-positive cytoplasmic inclusions, which may be due, at least in part, to impairment of lysosomal degradation in β-cells in vivo.
The action of h-IAPP to induce defective autophagy in vivo depends on its oligomeric properties
To investigate whether disruption of autophagy by h-IAPP depends on its propensity to form toxic oligomers, we examined islets of mice with comparable transgenic β-cell expression of the oligomeric human form of IAPP (h-TG) versus the soluble rodent form of IAPP (r-TG). Toxic oligomers of h-IAPP form intracellularly in β-cells of h-TG but not r-TG mice, and h-TG but not r-TG mice develop diabetes.12 LC3-II levels in islets isolated from h-TG mice were increased (5.9±1.9-fold, P<0.05) when compared with r-TG mice (Figure 2a), indicating an increased number of autophagosomes. The p62 levels in islets from h-TG mice were also increased (8.4±2.8-fold, P<0.05) when compared with r-TG mice (Figures 2a and b). The mRNA levels of p62 were increased by 1.2-fold in islets of h-TG mice versus r-TG mice (P<0.05, Figure 2c), indicating that the 8.4-fold increase in p62 protein levels in h-TG mice was most likely because of a decrease in p62 lysosomal degradation.

High expression of h-IAPP, but not r-IAPP, increases LC3-II and p62 protein levels and induces formation of p62-positive cytoplasmic inclusions in h-IAPP homozygous transgenic mice. (a) Protein levels of LC3 and p62 were assessed by western blot using islet protein lysates obtained from WT (WT, n=4), r-IAPP transgenic (r-TG, n=3) and h-IAPP transgenic mice (h-TG, n=4). GAPDH was used as loading control. The graph represents the quantification of LC3-II protein levels. (b) The graph represents the quantification of p62 protein levels. (c) Levels of p62 mRNA were evaluated by RT-qPCR in islets isolated from WT (n=3), r-TG (n=3) and h-TG mice (n=3). Data are expressed as mean±S.E.M.; *P<0.05. (d) p62 protein levels were assessed by immunofluorescence (p62, red; insulin, green; nuclei, blue) in pancreatic tissue from WT, r-TG and h-TG mice. High magnification of p62-positive structures found in h-TG mice (i, ii and iii)
In h-TG but not r-TG mice, we detected by western blot a high molecular weight band corresponding to that previously reported to be composed of tightly aggregated p62.19 Consistent with the western blots, intracellular deposits of p62 were identified by immunohistochemistry in β-cells of h-TG mice, but not in r-TG or WT mice (Figure 2d). These inclusions ranged from small punctuate aggregates to filamentous structures and large globular inclusions (Figure 2d, i, ii and iii), comparable to those noted in HIP rats (Figure 1c). We conclude that the action of IAPP to disrupt lysosomal clearance of p62 depends on the property of h-IAPP to form toxic oligomers, and not just the synthetic burden of IAPP.
Obesity exacerbates h-IAPP-induced defective autophagy
We examined changes in autophagy in the context of islet adaptation to obesity without development of diabetes (obese non-transgenic (ONT) versus lean non-transgenic (LNT)), as well as in the context of failed adaptation to obesity with development of diabetes (obese transgenic (OT) versus obese non-transgenic (ONT)) (Supplementary Table 1).
First, we examined islets from obese versus lean mice to investigate the effect of obesity on autophagy in the absence of oligomeric h-IAPP. In islets from ONT mice, there was a 1.7-fold increase in LC3-II levels in comparison to LNT mice (Figure 3a). Although p62 protein levels remained unchanged (Figures 3a and b), p62 mRNA expression was increased in ONT versus LNT mice (Figure 3c). This suggests that in the presence of obesity-induced insulin resistance, β-cells stimulate autophagic flux to manage the increased synthetic burden of insulin and IAPP.6 In these mice, the increased p62 mRNA expression might be crucial to deal with the consequences of the increased protein burden.


Interaction of obesity and h-IAPP expression, adaptive and maladaptive changes in autophagy. (a) Protein levels of LC3 and p62 were assessed by western blot using islet protein lysates obtained from lean non-transgenic (LNT, n=5), lean transgenic (LT, n=4), obese non-transgenic (ONT, n=5) and obese transgenic (OT, n=4–6) 10- to 12-week-old mice. Insulin and GAPDH were used as control. The graph represents the quantification of LC3-II protein levels. (b) The graph represents the quantification of p62 protein levels. (c) Levels of p62 mRNA were evaluated by RT-qPCR in islets isolated from LNT (n=5), LT (n=4), ONT (n=5) and OT mice (n=5). Data are expressed as mean±S.E.M.; *P<0.05; **P<0.01; ***P<0.001. (d) p62 protein levels were assessed by immunofluorescence (p62, red; insulin, green; nuclei, blue) in pancreatic tissue from LNT, LT, ONT and OT 10- to 12-week-old mice. (e) The graph represents the quantification of β-cells positive for p62 in each group (expressed in percentage). (f) The graph represents the quantification of β-cell area positive for p62 aggregates in each group (expressed in percentage). Data are expressed as mean±S.E.M.; *P<0.05; **P<0.01. The mathematical transformation ‘logarithm (value+1)’ was applied before statistical analysis of LC3-II, p62 protein levels and β-cells positive for p62. (g) Fluorescence confocal images of p62 at magnification × 63 (p62, red; insulin, green) in pancreatic tissue from LNT, LT, ONT and OT 10- to 12-week-old mice
We then investigated the effect of hemizygous expression of h-IAPP on lean mice. In islets from lean transgenic mice (LT), we detected an increase in LC3-II levels (2.9±0.5-fold, P<0.01; Figure 3a) associated with an increase in p62 protein levels (∼3-fold, P<0.05; Figures 3a and b) when compared with LNT mice. The levels of p62 mRNA were comparable in islets from LNT and LT mice (Figure 3c). These data show that hemizygous expression of h-IAPP, although insufficient to induce diabetes, is sufficient to impair lysosome-dependent degradation.
Finally, to investigate the interactive effects of obesity and h-IAPP on autophagy, we examined islets from OT versus LT mice. In islets from OT mice, we detected a 1.8-fold increase in LC3-II (Figure 3a) and a twofold increase in p62 protein levels (Figures 3a and b) when compared with LT. Thus, obesity exacerbates the impairment of autophagic clearance already present in lean h-IAPP transgenic animals.
To complement western blot results, we assessed p62 protein expression using immunofluorescence. Although p62 protein levels measured by western blot were similar in islets from ONT versus LNT mice, the number of β-cells containing detectable labeling for p62 was higher in ONT mice (1.5±0.2 versus 0.5±0.2%, P<0.05; Figures 3d and e). This difference might be explained by the changes in distribution of p62 within cells. In contrast to islets from LNT mice containing weak punctuated p62 labeling, in islets from ONT we detected bright clusters of p62 punctuates (Figure 3g), which occupied 0.03% of the β-cell area (Figure 3f).
In lean mice, hemizygous expression of h-IAPP caused an increase in the number of β-cells positive for p62 (1.6±0.5 versus 0.5±0.2% in LNT, P<0.05; Figures 3d and e), as well as the formation of large p62-positive inclusions taking up 0.35±0.05% of the β-cell area (Figure 3f). In islets from OT mice, we not only detected an increase in the number of β-cells stained for p62 (4±0.8%, P<0.05 versus LT; Figure 3e), but also an increase in the number and size of p62-positive inclusions, which occupied 0.7±0.15% of the β-cell area (P<0.01 versus LT; Figure 3f). The presence of p62-positive inclusions in islets from LT mice, which was exacerbated by obesity, further supports a defect in lysosomal degradation.
We conclude that the healthy islet adaptation to obesity includes enhanced autophagy to compensate for increased protein demand, which is linked to efficient lysosomal clearance. However, in mice prone to the islet phenotype of T2DM because of the expression of oligomeric h-IAPP, obesity leads to defective autophagy and exacerbates impaired lysosomal degradation already apparent at lower expression levels of h-IAPP in lean mice.
h-IAPP expression increases autophagosomes and p62 levels in a β-cell line, with p62 levels subsequently declining in relation to h-IAPP-induced apoptosis
To further investigate the role of p62 accumulation in β-cells of h-IAPP transgenic rodents, we used the INS 832/13 (rat insulinoma cell line) cells transduced with adenoviruses expressing h-IAPP or the non-amyloidogenic and non-toxic r-IAPP for comparable load of protein.20 Cells transduced with h-IAPP exhibited a ∼5-fold increase in LC3-II levels after 36 and 48 h (P<0.01), suggesting either an induction of autophagy or a decrease in autophagosome clearance (Figure 4), comparable to that reported above in vivo. In cells transduced with r-IAPP, LC3-II levels were also increased at 48 h (3.2±0.3-fold, P<0.001), but to a much lesser extent than in cells transduced with h-IAPP (Figure 4). Although transduction of cells with h-IAPP adenovirus induced a transient increase in p62 protein levels, with a maximal effect observed at 24 h (1.6±0.1-fold, P<0.05; Figure 4), transduction of cells with r-IAPP adenovirus did not affect p62 protein levels. Of interest, the increase in p62 levels detected in h-IAPP-transduced cells was followed by a rapid decline in p62 protein levels at 36 and 48 h. This decline in p62 protein levels was coincident with h-IAPP-induced apoptosis shown by the cleavage of caspase-3 (Figure 4 and Supplementary Figure 2), implying a role for p62 in β-cell survival.

High expression of h-IAPP in INS 832/13 cells increases LC3-II and affects p62 protein levels. INS 832/13 cells were transduced at 400 MOI with r-IAPP or h-IAPP adenoviruses and cell lysates were collected at different time points as indicated (time 0, non-transduced cells). Protein levels of LC3, p62, cleaved caspase-3 (Cl. Casp-3) and IAPP were assessed by western blot. GAPDH was used as loading control. The graphs represent the quantification of LC3-II and p62 protein levels (n=4). Data are expressed as mean±S.E.M.; *P<0.05; **P<0.01; ***P<0.001, significant differences versus non-transduced cells (Ctrl)
Loss of p62 renders β-cells more vulnerable to h-IAPP-induced toxicity
To evaluate whether p62 has a role in β-cell survival, the expression of p62 was specifically silenced by shRNA lentivirus in INS 832/13 cells. Increasing concentrations of shRNA lentivirus for 72 h resulted in a dose-dependent knockdown of p62 protein content and gradual emergence of cleaved caspase-3 (Figure 5a).

Downregulation of p62 by shRNA lentivirus induces apoptosis in INS 832/13 cells and exacerbates h-IAPP-induced β-cell apoptosis. (a) INS 832/13 cells were transduced for 72 h with control shRNA or p62 shRNA lentivirus at increasing concentrations. Levels of p62 and cleaved caspase-3 (Cl. Casp-3) were analyzed by western blot. GAPDH was used as loading control. The graphs represent the quantification of p62 protein levels and the cleaved form of caspase-3 (n=3). Data are expressed as mean±S.E.M.; *P<0.05; **P<0.01; ***P<0.001, significant differences versus Ctrl shRNA-transduced cells (at the corresponding concentration). (b) INS 832/13 cells were transduced for 48 h with control shRNA or p62 shRNA lentivirus (0.1 μg/ml) and transduced at 400 MOI with r-IAPP (R) or h-IAPP (H) adenoviruses for the last 27 h (C, non-transduced cells). Levels of cleaved caspase-3 and p62 were analyzed by western blot. GAPDH was used as loading control. The graph represents the quantification of the cleaved form of caspase-3 (n=4). Data are expressed as mean±S.E.M.; ***P<0.001
To determine whether a decrease in p62 expression exacerbates h-IAPP-induced apoptosis, we transduced INS 832/13 cells with p62 shRNA or control shRNA (48 h) and transduced these cells with h-IAPP or r-IAPP adenoviruses for 27 h. As expected, knockdown of p62 increased the cleavage of caspase-3 in comparison to control shRNA-transduced cells (Figure 5b). Furthermore, the knockdown of p62 in the presence of high expression of h-IAPP induced a significant increase in caspase-3 cleavage versus r-IAPP-transduced cells (Figure 5b, P<0.001). This shows that p62 knockdown exacerbates h-IAPP toxicity, supporting the hypothesis that p62 accumulation serves a protective role in β-cells stressed by h-IAPP expression.
Increased p62 protein expression reduces h-IAPP-induced β-cell apoptosis
To further evaluate the protective role of p62 against h-IAPP-induced β-cell apoptosis, we overexpressed p62 in INS 832/13 cells transduced with h-IAPP adenovirus. The p62-GFP fusion protein was efficiently expressed in cells and the transfected cells displayed cytoplasmic inclusions that appeared as two distinct types based on size (Figure 6a, right panels), comparable to those in β-cells of h-IAPP transgenic rodents. The size, number and intensity of labeling of these inclusions increased with increasing p62 expression levels (data not shown). p62-GFP colocalized with the autophagic marker LC3-RFP (red fluorescent protein fused to LC3) in these cytoplasmic inclusions (Supplementary Figure 1), suggesting that p62-GFP behaves as endogenous p62 in INS 832/13 cells. Expression of p62-GFP in cells caused a 26% decrease in h-IAPP-induced apoptosis in comparison to GFP plasmid-transfected cells used as control (P<0.05; Figure 6b). Thus, increased expression of p62 reduces h-IAPP toxicity and this protective effect might be mediated by the formation of large p62-cytoplasmic inclusions in β-cells.

Overexpression of p62 in INS 832/13 cells reduces h-IAPP-induced β-cell apoptosis. (a) INS 832/13 cells were transfected with GFP plasmid or p62-GFP plasmid (0.1μg) for 24 h and observed under fluorescence microscope (p62, green; nuclei, blue). (b) INS 832/13 cells were transduced at 400 MOI with r-IAPP (R) or h-IAPP (H) adenoviruses for 36 h (C, non-transduced cells). Cells were transfected with GFP plasmid or p62-GFP plasmid (0.1 μg) for the last 22 h. Levels of p62 and cleaved caspase-3 (Cl. Casp-3) were analyzed by western blot. GAPDH was used as loading control. The graph represents the quantification of the cleaved form of caspase-3 (n=4). Data are expressed as mean±S.E.M.; *P<0.05
Inhibition of lysosomal degradation exacerbates h-IAPP-induced β-cell apoptosis
The accumulation of p62-positive inclusions and the increase in autophagosome number (increased LC3-II) induced by h-IAPP expression implies that a high expression of h-IAPP impairs lysosome function. To test whether inhibition of lysosomal degradation exacerbates h-IAPP-induced β-cell death, we treated INS 832/13 cells (transduced or not with h-IAPP) with E-64-d/pepstatin A, the inhibitors of proteases in the lysosome. As control, LC3-II and p62 protein levels were increased in cells treated with these inhibitors, confirming the accumulation of autophagosomes and p62 (data not shown). Inhibition of lysosomal degradation increased h-IAPP-induced apoptosis measured by the cleavage of caspase-3 (1.6-fold increased versus non-treated h-IAPP-transduced cells, P<0.05; Figure 7a).

Inhibition of lysosomal degradation exacerbates h-IAPP-induced β-cell apoptosis in INS 832/13 cells and rat islets. (a) INS 832/13 cells were transduced at 400 MOI with r-IAPP (R) or h-IAPP (H) adenoviruses for 36 h (C, non-transduced cells) and treated with lysosomal inhibitors (Lyso I) (E-64-d, 10 μg/ml and pepstatin A, 10 μg/ml) for the last 24 h. Levels of cleaved caspase-3 (Cl. Casp-3) were analyzed by western blot. GAPDH was used as loading control. The graph represents the quantification of the cleaved form of caspase-3 (n=4). Data are expressed as mean±S.E.M.; *P<0.05. (b) Isolated islets obtained from wild-type (WT: n=3) and HIP rats (HIP: n=5) were treated with lysosomal inhibitors (E-64-d, 10 μg/ml and pepstatin A, 10 μg/ml) for 24 h. Levels of cleaved caspase-3 and ubiquitin were analyzed by western blot. GAPDH was used as loading control. The graph represents the quantification of the cleaved form of caspase-3. Data are expressed as mean±S.E.M.; **P<0.01
To further confirm the observation that lysosomal function is important to protect β-cells from h-IAPP toxicity in primary cells, we repeated these studies in islets isolated from HIP rats and WT rats. In WT rat islets treated with lysosomal inhibitors, an accumulation of LC3-II and p62 was clearly detected, demonstrating that p62 is degraded by lysosomes in pancreatic islets (data not shown), similar to other cell types.21 Inhibition of lysosomal degradation in HIP rat islets further increased the accumulation of polyubiquitinated proteins, and this was not observed in treated WT rat islets (Figure 7b). Inhibition of lysosomal activity in HIP rat islets led to increased cleavage of caspase-3 (1.7-fold increased versus non-treated HIP rat islets, P<0.01; Figure 7b and Supplementary Figure 3). These data show that h-IAPP-induced apoptosis is exacerbated by inhibition of lysosome-dependent clearance in INS 832/13 cells as well as in primary β-cells that express h-IAPP.
Enhanced autophagy decreases h-IAPP-induced β-cell apoptosis
Having established that inhibition of autophagy/lysosomal degradation exacerbates h-IAPP-induced β-cell apoptosis, we next tested whether enhancement of autophagy can rescue cells from h-IAPP-induced apoptosis. We stimulated autophagy with rapamycin, an inhibitor of the mammalian target of rapamycin (m-TOR) signaling pathway. Cells treated with rapamycin showed decreased phosphorylation of m-TOR that indicates a stimulation of autophagosome formation (Figure 8, top panel). Treatment of h-IAPP-transduced cells with rapamycin significantly reduced the cleavage of caspase-3 (66% decrease versus non-treated h-IAPP-transduced cells, P<0.001; Figure 8). It also decreased the modest cleavage of caspase-3 induced by r-IAPP transduction (Figure 8). These data demonstrate that rapamycin, by enhancing the autophagy pathway, protects β-cells against h-IAPP-induced apoptosis.

Enhanced autophagy by treatment with rapamycin decreases h-IAPP-induced apoptosis in INS 832/13 cells. INS 832/13 cells were transduced at 400 MOI with r-IAPP (R) or h-IAPP (H) adenoviruses for 48 h (C, non-transduced cells) and treated with rapamycin (Rapa, 10 nM) for the last 40 h. Levels of P-mTOR and cleaved caspase-3 (Cl. Casp-3) were analyzed by western blot. GAPDH was used as loading control. The graph represents the quantification of the cleaved form of caspase-3 (n=4). Data are expressed as mean±S.E.M.; **P<0.01; ***P<0.001
Discussion
These studies provide evidence that increased expression of h-IAPP, as occurs with insulin resistance, impairs autophagy/lysosome-dependent degradation in β-cells, and that this action of h-IAPP to perturb lysosomal clearance of misfolded proteins in vivo depends on its propensity to form toxic oligomers. As we established that inhibition of lysosomal degradation increases vulnerability of β-cells to h-IAPP-induced toxicity and, conversely, stimulation of autophagy protects β-cells from h-IAPP-induced apoptosis, the property of h-IAPP to disrupt this protection against proteotoxicity is revealed as a potentially adverse self-sustaining cycle. The p62 scaffold protein is revealed to have an important role in sequestering misfolded proteins in β-cells and as such has a protective role against proteotoxicity induced by misfolded amyloidogenic proteins.
The potential role of p62 in defense against h-IAPP-induced toxicity was examined in detail after we noted accumulation of p62 protein in β-cells expressing h-IAPP. Similar p62-positive inclusions have been reported in brains from individuals with neurodegenerative diseases, including Alzheimer and Parkinson's diseases.22, 23 In β-cells as in neurons,24 these intracytoplasmic inclusions appeared as either small punctate spots (∼0.5 μm) or in much larger globular structures (1 to 2 μm). Typically, the larger p62-positive inclusions (called sequestosomes) are not surrounded by a membrane, and the smaller structures are surrounded by membranes and have thus been termed autophagosomes.25
The role of the p62-positive inclusions and their relationship to cell survival appears to be condition and/or cell-type specific. Besides its role in selective delivery of ubiquitinated proteins to the autophagosome, p62 acts as a scaffold in several signaling pathways crucial for cell survival and/or apoptosis.26 Mice deficient in p62 develop an Alzheimer-like phenotype with neurodegeneration.27 Huntingtin-induced neuronal death in cells was increased after depletion of p62.25 In contrast, removal of p62 in autophagy-deficient hepatocytes attenuates liver injury.18 Additionally, accumulation of p62 can also compromise proteasomal degradation, ultimately leading to cell death.28 In our studies, we found that decreased p62 availability led to increased vulnerability of β-cells to h-IAPP-induced apoptosis. Enhanced p62 levels and p62 inclusion formation were associated with reduced h-IAPP-induced β-cell apoptosis.
Given these findings and the similarities between pancreatic β-cells in T2DM and neurons in neurodegenerative diseases, we propose that p62-positive inclusions may sequester misfolded proteins or damaged organelles to protect β-cells from toxicity and may thereby delay (or abolish) β-cell dysfunction and apoptosis induced by misfolded h-IAPP. Although we propose that p62 has a protective role against misfolded h-IAPP, it can be predicted that marked accumulation of p62 may be associated with β-cell dysfunction and apoptosis, as noted by use of lysosomal inhibitors. Thus, although p62 sequestration of misfolded proteins may serve as a buffer against proteotoxicity, ultimately the p62 protein-bound targets for degradation must be removed and degraded through the autophagy/lysosomal pathway to effect this protection over the longer term.
It is therefore not surprising that β-cell-specific knockout of the key autophagy initiator gene Atg7 (autophagy-related protein 7) in mice led to glucose intolerance, reduced insulin secretion and diminished β-cell mass because of increased β-cell apoptosis.29, 30 The accumulation of large aggregates positive for p62 and ubiquitin was also reported in β-cells of these autophagy-deficient mice.29, 30 Both increased number of autophagosomes and intracellular h-IAPP oligomers have been noted in β-cells in humans with T2DM.5, 31 In h-IAPP transgenic rodents, we noted increased autophagosomes that coincided with an accumulation of p62 protein levels, implying that the action of h-IAPP to increase the number of autophagosomes was secondary to impaired lysosomal clearance. However, initiation of autophagic vacuole formation and lysosomal clearance are inextricably linked and we cannot exclude a simultaneous role of h-IAPP in enhancement of autophagosome formation and alteration of lysosomal degradation.
The most common risk factor for T2DM is obesity that, by its actions to induce insulin resistance, leads to a marked increase in IAPP and insulin expression.6 We show in this study that obesity in the absence of oligomeric h-IAPP prompts an increase in autophagosome formation, consistent with a previous report.29 We extend those findings to note that increased autophagosome formation is accompanied by prompt delivery of p62 targets to autophagosomes from which there is efficient lysosomal clearance. In contrast, in mice expressing h-IAPP this β-cell adaptation to obesity fails, reproducing the findings in humans with T2DM.5 We conclude that h-IAPP-induced proteotoxicity may be exacerbated by an action of h-IAPP oligomers targeted for destruction to disrupt their own clearance by the autophagy/lysosomal pathway.
Toxic h-IAPP oligomers, like other soluble amyloid oligomers in protein misfolding diseases, have been shown to impose cellular toxicity by formation of nonselective ion channels, resulting in disruption of membranes.32, 33 h-IAPP toxic oligomers may disrupt autophagosomal and/or lysosomal membranes. This loss of membrane fidelity may impair transport of autophagosomes to lysosomes and/or disrupt autophagosome/lysosome fusion. Also, toxic membrane disrupting oligomers may impair function of the lysosomal proton pump and induce missorting or leakage of lysosomal enzymes, leading to failure to effectively degrade lysosomal contents because of impaired enzyme activation.34, 35, 36, 37
In conclusion, the p62/autophagy/lysosomal pathway, which is shown to be important in protection of β-cells against proteotoxicity, is impaired by expression of h-IAPP at rates sufficient to induce apoptosis. This action of h-IAPP to render β-cells more vulnerable to toxic oligomer-induced toxicity is a potential target for β-cell protection in T2DM.
Materials and Methods
Animal models
Rats and mice were bred and housed at the University of California, Los Angeles (UCLA) animal housing facility. The institutional animal care and use committee of the UCLA approved all surgical and experimental procedures. Animals were maintained on a 12-h day/night cycle with Harlan Teklad Rodent Diet 8604 (Madison, WI, USA) and water ad libitum. Males were used for the experiments.
Rats
The generation of h-IAPP transgenic rats (HIP rats) has been described in detail previously.10 In this study, we used 4- to 6-month-old WT (n=6) and pre-diabetic HIP rats (n=6) (fasting glucose=87±4 mg per 100 ml).
Mice
The generation and characterization of transgenic mice homozygous for human-IAPP (h-TG: FVB-Tg(IAPP)6Jdm/Tg(IAPP)6Jdm) and rodent-IAPP (r-TG: FVB/N-Tg(Iapp)6Wcs/Tg(Iapp)6Wcs) have been described previously.12 Control WT FVB mice were originally purchased from Charles Rivers Laboratory (Wilmington, MA, USA) and bred at UCLA. LNT, lean mice transgenic/hemizygous for h-IAPP (LT), ONT and obese mice transgenic/hemizygous for h-IAPP (OT) were obtained by crossbreeding FVB mice and homozygous h-IAPP transgenic mouse model (h-TG: FVB-Tg(IAPP)6Jdm/Tg(IAPP)6Jdm) with the Avy/agouti mouse model on the C57BL/6 background.38, 39 Fasting blood glucose concentrations and body weight on the day of tissue collection are presented in Supplementary Table 1.
We used mice and rats in a pre-diabetic state to avoid the confounding effect of glucose toxicity.
Islet isolation
After an overnight fast, animals were killed using isoflurane. The bile duct was clamped at the entrance to duodenum, cannulated, and pancreas was perfused with 2 ml of collagenase solution (HBSS (Invitrogen, Carlsbad, CA, USA) supplemented with 25 mM HEPES (Invitrogen), 0.23 mg/ml liberase (Roche, Penzberg, Germany) and 0.1 mg/ml DNase (Roche)). The pancreas was removed, transferred into a glass vial containing 2 ml of ice-cold collagenase solution, digested for 19 min at 37°C and then dispersed by shaking for 30 s. Islets were manually picked, washed with ice-cold PBS and lysed in lysis buffer (50 mM HEPES, 1% Nonidet P-40, 2 mM Na3VO4, 100 mM NaF, 10 mM PyrPO4, 4 mM EDTA, 1 mM PMSF, 1 μg/ml leupeptin and 1 μg/ml aprotinin). After 20 min of incubation in lysis buffer on ice, islets were sonicated for 10 s and centrifuged at 10 000 r.p.m. at 4°C for 10 min. Supernatant was stored at −20°C until use for subsequent protein determination by BCA assay (Bio-Rad, Hercules, CA, USA) and western blotting.
Adenovirus generation
The complementary cDNA encoding the full-length human and rat preproIAPP were used as templates to generate the human and rat IAPP adenoviruses, as described previously.20 Recombinant adenovirus expressing human and rat preproIAPP (h-IAPP and r-IAPP, respectively) were produced and purified according to the manufacturer's instructions (Clontech, Palo Alto, CA, USA).
Plasmids
We obtained LC3-RFP plasmid from the plasmid repository at Addgene (Cambridge, MA, USA; Addgene plasmid 21075) and this plasmid has been described previously.36 The pEGFPC1-p62 (p62-GFP) plasmid was a generous gift provided by Dr. J Moscat (Cincinnati, OH, USA).40 For plasmid amplification, 50 μl of DH5α competent cells (Invitrogen) were transformed with either LC3-RFP or p62-GFP plasmid. Tubes were incubated on ice for 30 min, followed by a heat shock step at 42°C for 30 s and incubated on ice for an additional 2 min. To each tube, 250 μl of SOC (Super Optimal broth with Catabolite repression) media (Invitrogen) was added and tubes were incubated at 37°C for 1 h. Following incubation, 50 μl of DH5α in SOC media was plated on a pre-warmed LB plate with the selective antibiotic kanamycin (Sigma, St Louis, MO, USA). For purification, 50 ml of LB medium with kanamycin was inoculated with a single colony from LB-kanamycin plate. The bacterial culture was grown overnight at 37°C at 300 r.p.m. Bacterial cells were harvested and purified according to the manufacturer's instructions for the Qiagen EndoFree Plasmid Maxi Kit (Valencia, CA, USA). Plasmid sequences were verified by DNA sequencing at the UCLA Sequencing Core Facility.
Lentivirus
p62 shRNA vector containing the 21-nt sequence 5′-GAGGTTGACATTGATGTGGAA-3′ was purchased from Open Biosystems (Huntsville, AL, USA). shRNA control vector containing the 21-nt sequence 5′-CAACAAGATGAAGAGCACCAA-3′ was purchased from Sigma. This shRNA control vector produces a negative control shRNA that does not target any known human, mouse or rat gene, but will activate the RNA-induced silencing complex (RISC). shRNA vectors were used to generate shRNA lentivirus at the UCLA Vector Core Facility. Lentivirus-based vectors encoding control and p62 shRNA were generated by transient co-transfection of 293T cells with a three-plasmid combination, as described previously, with slight modifications.41 The construct pMD.G was used for the production of the VSV-G viral envelope in combination with the packaging constructs pMDLg/pRRE and pRSV–REV, whereas the pLKO.1-puro constructs correspond to the different transfer vectors. Briefly, 100 mm dishes of nonconfluent 293T cells were co-transfected with 6.5 μg of pMDLg/pRRE, 3.5 μg of pMDG (encoding the VSV-G envelope), 2.5 μg of pRSV–REV and 10 μg of pLKO.1-p62shRNA or pLKO.1-control shRNA, by the calcium phosphate-DNA co-precipitation method. Next day, the cells were incubated for 8 h to obtain high-titer virus production, washed and incubated in fresh medium without sodium butyrate. Conditioned medium was harvested 16 h later. For transduction of 293T cells, cells were seeded at 2 × 105 cells per well in six-well plates (Corning, NY, USA) and incubated at 37°C overnight in their regular medium. Transductions were carried out in 1 ml of DMEM, including serial dilutions of lentiviral vector supernatant. Viral titer was determined by assessing viral p24 antigen concentration by ELISA (the Alliance HIV-I p24 ELISA Kit, PerkinElmer, Boston, MA, USA) and hereafter expressed as μg of p24 equivalent units per ml.
Cell culture
Rat insulinoma cell line INS 832/13 was kindly provided by Dr. C Newgard (Durham, NC, USA).42 INS 832/13 cells were grown in RPMI-1640 medium supplemented with 10 mM HEPES, 1 mM sodium pyruvate, 100 IU/ml penicillin and 100 μg/ml streptomycin (Invitrogen), 10% heat-inactivated FBS (Gemini, West Sacramento, CA, USA) and 50 μ M β-mercaptoethanol (Sigma) at 37°C in a humidified 5% CO2 atmosphere.
For adenovirus transduction experiments, INS 832/13 cells were plated on six-well plates at a density of 106 cells/well, and cultured for 24 h. Cells were transduced with r-IAPP or h-IAPP adenoviruses at 400 MOI in complete RPMI medium for the indicated time.
For lentiviral transduction experiments, INS 832/13 cells were plated on 12-well plates at a density of 500 000 cells/well, and cultured for 24 h. Cells were transduced with control shRNA or p62 shRNA lentivirus at different concentrations ranging from 0.01 to 0.1 μg/ml for 48 or 72 h in complete RPMI medium supplemented with 1 μg/μl protamine sulfate (Sigma).
For plasmid transfection experiments, INS 832/13 cells were plated on 12-well plates at a density of 300 000 cells/well. The next day, lipofectAMINE2000-plasmid complexes were prepared according to the manufacturer's instructions. Cells were transfected in Opti-MEM (Invitrogen) for 6 h and then cultured in complete RPMI medium for the indicated time.
Rapamycin, E-64-d and pepstatin A were purchased from Sigma. At the end of the experiment, cells were washed with cold PBS and lysed for 30 min at 4°C in a solubilizing buffer (50 mM HEPES, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 1 mM Na3VO4, 1 mM PMSF, 30 mM PyroPO4, 10 mM NaF and 1 mg/ml bacitracin) and centrifuged at 14 000 r.p.m. for 30 min to remove insoluble materials.
Western blotting
Proteins (25–50 μg/lane) were separated on a 4–12% Bis-Tris NuPAGE gel and blotted onto a PVDF membrane (FluoroTrans; Pall Life Sciences, Ann Arbor, MI, USA). Membranes were probed overnight at 4°C with primary antibodies against the cleaved form of caspase-3, LC3B, phospho-mTOR, ubiquitin, GAPDH and insulin (Cell Signaling Technology, Beverly, MA, USA), p62 (GP62-C; Progen Biotechnik, Heidelberg, Germany) and IAPP (25–37 aa; Peninsula Laboratories, San Carlos, CA, USA). Horseradish peroxidase-conjugated secondary antibodies were from Zymed Laboratories (San Francisco, CA, USA). Proteins were visualized by enhanced chemiluminescence (Millipore, St Louis, MO, USA) and protein expression levels were quantified using the Labworks software (UVP, Upland, CA, USA).
RNA isolation, RT-PCR, real-time quantitative PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) performing on-column DNase digestion with RNase-Free DNase Set (Qiagen) according to the manufacturer's instructions. RNA (0.25 μg) was used for preparation of single-stranded cDNA using Superscript III Reverse transcriptase (Invitrogen) by the oligo-dT priming method. Real-time quantitative PCR was performed with 700 ng cDNA using the LightCycler FastStart DNA Master SYBR Green I kit (Roche) and the LightCycler PCR equipment (Roche). The oligonucleotide primers were: 5′-GCCCTGTACCCACATCTCC-3′ and 5′-CCATGGACAGCATCTGAGAG-3′ for rat p62/SQSTM1 and 5′-GCTGCCCTATACCCACATCT-3′ and 5′-CGCCTTCATCCGAGAAAC-3′ for mouse p62/SQSTM1. All measurements were normalized to the housekeeping gene Cyclophilin.
Immunofluorescent staining
Pancreas was fixed in 4% paraformaldehyde for 24 h at 4°C and embedded in paraffin. Sections (4 μm) were stained as described.3 The slides were incubated overnight at 4°C with anti-p62 antibody diluted in 3% BSA/TBS/0.2% Tween-20, followed by 1 h treatment at room temperature (RT) with secondary antibody conjugated to Cy3 (1 : 200, Jackson ImmunoResearch Laboratories, West Grove, PA, USA); slides were then stained for insulin using rabbit anti-insulin antibody (1 : 200, RT, 1 h) and donkey-derived secondary antibody conjugated to FITC (1 : 200, RT, 1 h; Jackson ImmunoResearch Laboratories). Slides were mounted with Vectashield with DAPI (Vector Laboratories, Burlingame, CA, USA) and viewed using a Leica DM6000 microscope (Leica Microsystems, Bannockburn, IL, USA). Images were acquired using Openlab software (Improvision, Lexington, MA, USA).
Image analysis
For detailed evaluation of islet β-cell area and number of β-cell containing p62 aggregates, 15 islets per animal (with a minimum of 20 β-cells per islet in plane of section) were selected randomly. The β-cell was considered to be positive if it had any p62 labeling with intensity above background staining of exocrine pancreas. For the computation of the area of p62 aggregates/insulin+ area, 15 representative islets were identified and analyzed. For each islet, the islet cross-sectional area positive for p62 was quantified and divided by the islet cross-sectional area positive for insulin using Image Pro Plus (Media Cybernetics, Silver Spring, MD, USA).
Statistical analysis
Results are expressed as the means±S.E.M. for n independent experiments, as indicated in figure legends. Statistical analyses were carried out using Student's t-test or ANOVA followed by Fisher's post hoc test for multiple comparisons using Statistica (Statsoft, Tulsa, OK, USA). A P-value of <0.05 was taken as evidence of statistical significance (*P<0.05, **P<0.01, ***P<0.001).
Abbreviations
- Atg7:
-
autophagy-related protein 7
- ER:
-
endoplasmic reticulum
- h-IAPP:
-
human islet amyloid polypeptide
- HIP rat:
-
h-IAPP transgenic rat
- h-TG mouse:
-
h-IAPP transgenic mouse
- IAPP:
-
islet amyloid polypeptide
- INS 832/13:
-
rat insulinoma cell line
- LC3:
-
microtubule-associated protein 1 light chain 3
- LIR:
-
LC3 interaction region
- LNT:
-
lean non-transgenic
- LT:
-
lean transgenic
- m-TOR:
-
mammalian target of rapamycin
- ONT:
-
obese non-transgenic
- OT:
-
obese transgenic
- r-IAPP:
-
rodent islet amyloid polypeptide
- r-TG mouse:
-
r-IAPP transgenic mouse
- SQSTM1:
-
p62/sequestosome 1
- T2DM:
-
type II diabetes mellitus
- UCLA:
-
University of California, Los Angeles
- WT:
-
wild type
- RISC:
-
RNA-induced silencing complex
- RT:
-
room temperature
References
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC . Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110.
Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU . Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 1985; 4: 110–125.
Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress-mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007; 56: 2016–2027.
Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007; 50: 2486–2494.
Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ et al. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol 2010; 176: 861–869.
Mulder H, Ahren B, Stridsberg M, Sundler F . Non-parallelism of islet amyloid polypeptide (amylin) and insulin gene expression in rats islets following dexamethasone treatment. Diabetologia 1995; 38: 395–402.
Butler PC, Chou J, Carter WB, Wang YN, Bu BH, Chang D et al. Effects of meal ingestion on plasma amylin concentration in NIDDM and nondiabetic humans. Diabetes 1990; 39: 752–756.
Silvestre RA, Peiro E, Degano P, Miralles P, Marco J . Inhibitory effect of rat amylin on the insulin responses to glucose and arginine in the perfused rat pancreas. Regul Pept 1990; 31: 23–31.
Haataja L, Gurlo T, Huang CJ, Butler PC . Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev 2008; 29: 303–316.
Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC . Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP rat) - A new model for type 2 diabetes. Diabetes 2004; 53: 1509–1516.
Hoppener JWM, Oosterwijk C, Nieuwenhuis MG, Posthuma G, Thijssen JHH, Vroom TM et al. Extensive islet amyloid formation is induced by development of type II diabetes mellitus and contributes to its progression: pathogenesis of diabetes in a mouse model. Diabetologia 1999; 42: 427–434.
Huang CJ, Haataja L, Gurlo T, Butler AE, Wu XJ, Soeller WC et al. Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab 2007; 293: E1656–E1662.
Meusser B, Hirsch C, Jarosch E, Sommer T . ERAD: the long road to destruction. Nat Cell Biol 2005; 7: 766–772.
Kirkin V, McEwan DG, Novak I, Dikic I . A role for ubiquitin in selective autophagy. Mol Cell 2009; 34: 259–269.
Mizushima N . Autophagy: process and function. Genes Dev 2007; 21: 2861–2873.
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T . LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117: 2805–2812.
Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG et al. Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62′s role in neurodegenerative disease. J Biomed Biotechnol 2006; 2006: 62079.
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131: 1149–1163.
Waguri S, Komatsu M . Biochemical and morphological detection of inclusion bodies in autophagy-deficient mice. Methods Enzymol 2009; 453: 181–196.
Huang CJ, Gurlo T, Haataja L, Costes S, Daval M, Ryazantsev S et al. Calcium-activated calpain-2 is a mediator of beta cell dysfunction and apoptosis in type 2 diabetes. J Biol Chem 2010; 285: 339–348.
Komatsu M, Ichimura Y . Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010.
Kuusisto E, Salminen A, Alafuzoff I . Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. NeuroReport 2001; 12: 2085–2090.
Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002; 160: 255–263.
Seidel K, Brunt ER, de Vos RA, Dijk F, van der Want HJ, Rub U et al. The p62 antibody reveals various cytoplasmic protein aggregates in spinocerebellar ataxia type 6. Clin Neuropathol 2009; 28: 344–349.
Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171: 603–614.
Moscat J, Diaz-Meco MT . p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009; 137: 1001–1004.
Babu JR, Seibenhener ML, Peng JM, Strom AL, Kemppainen R, Cox N et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 2008; 106: 107–120.
Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC . Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33: 517–527.
Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 2008; 8: 325–332.
Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 2008; 8: 318–324.
Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009; 52: 1083–1086.
Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC . The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999; 48: 491–498.
Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE et al. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 2004; 279: 46363–46366.
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH . Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 2009; 35: 385–398.
Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S . LAMP proteins are required for fusion of lysosomes with phagosomes. EMBOJ 2007; 26: 313–324.
Kimura S, Noda T, Yoshimori T . Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3: 452–460.
Munafo DB, Colombo MI . Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic 2002; 3: 472–482.
Butler AE, Janson J, Soeller WC, Butler PC . Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 2003; 52: 2304–2314.
Soeller WC, Janson J, Hart SE, Parker JC, Carty MD, Stevenson RW et al. Islet amyloid-associated diabetes in obese A(vy)/a mice expressing human islet amyloid polypeptide. Diabetes 1998; 47: 743–750.
Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab 2006; 3: 211–222.
Naldini L, Blomer U, Gage FH, Trono D, Verma IM . Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA 1996; 93: 11382–11388.
Hohmeier HE, Mulder H, Chen GX, Henkel-Rieger R, Prentki M, Newgard CB . Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 2000; 49: 424–430.
Acknowledgements
These studies were supported by grants from the National Institutes of Health (DK059579) and the Larry L Hillblom Foundation (2007-D-003-NET and 2008-D-027-FEL to SC). We thank Chang Liu and Heather Cox for their excellent technical help. We thank UCLA CURE Vector Core (NIH: 2P30DK04301)/JCCC Vector shared resource facility (NIH: CA-016042) for the generation of shRNA lentivirus. We thank Dr. S Kimura for LC3-RFP plasmid.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by S Kumar
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
Rivera, J., Gurlo, T., Daval, M. et al. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: protective role of p62-positive cytoplasmic inclusions. Cell Death Differ 18, 415–426 (2011). https://doi.org/10.1038/cdd.2010.111
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.111
Keywords
This article is cited by
-
The nuclear receptor REV-ERBα is implicated in the alteration of β-cell autophagy and survival under diabetogenic conditions
Cell Death & Disease (2022)
-
Optimal allogeneic islet dose for transplantation in insulin-dependent diabetic Macaca fascicularis monkeys
Scientific Reports (2021)
-
IAPP toxicity activates HIF1α/PFKFB3 signaling delaying β-cell loss at the expense of β-cell function
Nature Communications (2019)
-
Pancreatic β cells overexpressing hIAPP impaired mitophagy and unbalanced mitochondrial dynamics
Cell Death & Disease (2018)
-
Proteasomal degradation of the histone acetyl transferase p300 contributes to beta-cell injury in a diabetes environment
Cell Death & Disease (2018)

{kind=link}
{kind=link}
{kind=link}