
Abstract
Apoptotic stimuli have been shown to trigger lysosomal membrane permeability (LMP), leading to the release of cathepsins, which activate death signaling pathways in the cytosol. However, it is unknown whether this process is an initiating or amplifying event in apoptosis. In this study, we used fibroblasts and monocytes exposed to etoposide, ultraviolet light, FasL or deprived of interleukin-3 (IL-3) to show that LMP and the cytosolic release of cathepsins B, L and D consistently depends on Bax/Bak and components of the apoptosome. Neither Bax nor Bak resided on the lysosomes, indicating that lysosomes were not directly perforated by Bax/Bak but by effectors downstream of the apoptosome. Detailed kinetic analysis of cells lacking cathepsin B or L or treated with the cysteine protease inhibitor, E64d, revealed a delay in these cells in etoposide- and IL-3 deprivation-induced caspase-3 activation and apoptosis induction but not clonogenic survival, indicating that cathepsins amplify rather than initiate apoptosis.
Similar content being viewed by others


Main
Lysosomes have long been implicated in necrotic or autophagic processes of eukaryotic cells. They belong to the acidic vacuolar apparatus, the main compartment for intracellular degradation of long-lived proteins and organelles, and contain a host of hydrolytic enzymes with pH optima between 4.5 and 6.5 for their catalysis. In 1969, de Duve and colleagues1 postulated that the release of these enzymes from the lysosomes would jeopardize cellular integrity. Although a series of subsequent studies clearly linked lysosomal rupture to necrotic cell death, Firestone and colleagues2, 3 reported that the induction of selective lysosomal rupture by lysosomotropic detergents induced apoptotic, rather than necrotic cell death. Further evidence showed that lysosomal perturbation is involved in apoptotic cell death induced by oxidative stress,4, 5 tumor necrosis factor alpha (TNFα) treatment,6, 7 lysosomotropic agents,8, 9 sphingosines10 or TRAIL.11 This suggests that the participation of lysosomal proteases in cellular processes goes beyond simple ‘garbage disposal’. The release of these enzymes into the cytosol during apoptosis is directly involved in the cleavage of proapoptotic substrates and/or activation of caspases.12
The most abundant lysosomal proteases are cathepsins B (CTB) and D (CTD) followed by cathepsin L (CTL).13, 14 CTL-deficient mice show a reduced rate of keratinocyte apoptosis during physiological regression of hair follicles resulting in a periodic hair loss phenotype.15, 16 CTB has a role in the processing of Bid and caspase-2 and the production of reactive oxygen species6, 17 and, together with other cathepsins, in the degradation of Bcl-2, Bcl-xL, Mcl-1, Bak and BimEL.18 All these events lead to mitochondrial membrane permeabilization and cytochrome c release. CTD seems to be implicated in apoptosis induced by staurosporine, p53, oxidative stress and TNFα.19, 20, 21 More recently, spontaneous neutrophil cell death was shown to be dependent on CTD-mediated activation of caspase-8.22 Although these findings suggest that cathepsins may contribute to apoptosis initiation, the precise mechanisms of lysosomal membrane permeabilization (LMP) and cathepsin release and the requirement of these enzymes for apoptosis induction/completion are still unclear.
Mitochondrial outer membrane permeabilization (MOMP) is considered a ‘point-of-no-return’ of apoptosis.23 The main effectors of MOMP are the proapoptotic members of the Bcl-2 family, Bax and Bak.24 In living cells, Bax is a cytosolic and Bak a mitochondrial protein. On apoptosis induction, Bax translocates to the mitochondria, and both Bax and Bak oligomerize, insert into the outer mitochondrial membrane and cause MOMP.25, 26 MOMP results in the release of cytochrome c from the intermembrane space of the mitochondria into the cytosol where it forms with Apaf-1 and pro-caspase-9 the so-called apoptosome.27 The apoptosome activates caspase-9, which in turn cleaves and activates the effector caspases-3 and -7 that are ultimately responsible for the apoptotic demolition of the cell.
More recently, it has been proposed that LMP might be due to the translocation of Bax to lysosomes independent of its movement to the mitochondria.11, 28, 29 If true, this process would represent another step before or concomitant with MOMP. The aim of this study was to investigate the requirements of Bax, Bak, the Apaf-1/caspase-9 apoptosome and caspases-3/-7 for LMP to identify whether cathepsins regulate early apoptotic events, such as MOMP, or rather serve the role of apoptosis amplifiers downstream or aside of the mitochondria.
Results
Lysosomal permeabilization and cathepsin release strictly depend on Bax/Bak in monocytes and fibroblasts
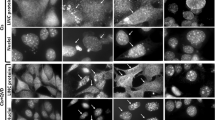
To investigate the role of lysosomes in apoptosis induction, we determined the degree of LMP in mouse monocytes (factor-dependent monocytes, FDMs) deprived of interleukin-3 (IL-3). We used the lysosomotropic probe acridine orange (AO), a metachromatic fluorophore, which mainly accumulates in acidic organelles and displays, at high concentration, a red to orange fluorescence when excited by blue light. On lysosomal rupture, the dye is released into the cytosol where its fluorescence spectrum changes from red to green.10, 30 Fluorescence-absorbent cell scanner (FACS) analysis of wild-type (wt) FDMs deprived of IL-3 for 24 h revealed a clear reduction of red fluorescence with a concomitant increase in green fluorescence (Figure 1a). A similar decrease in red fluorescence was observed using the lysosomotropic probe LysoTracker Red DND-99 (Figure 1a). Astonishingly, the IL-3 deprivation-induced loss of red fluorescence of both AO and LysoTracker Red was completely prevented in FDMs deficient for both Bax and Bak (Bax/Bak double knockout (DKO)) (Figures 1a and b). These data indicate that LMP in response to IL-3 deprivation is absolutely dependent on Bax, Bak or both. To define the time course of LMP, and whether LMP was differentially affected by Bax or Bak, we performed AO FACS analysis of wt, Bax−/−, Bak−/− and Bax/Bak DKO FDMs deprived of IL-3 for 0–48 h. As shown in Figure 1b, the loss of lysosomal membrane integrity was time dependent and effectively prevented in Bax/Bak DKO cells, even at 48 h. Bax and Bak single-deficient cells exhibited a similar LMP kinetic as wt cells. This suggests that either Bax or Bak alone are sufficient to result in IL-3 deprivation-induced LMP.

Removal of IL-3 triggers a Bax/Bak-dependent LMP and cytosolic release of cathepsin D and B proteins and B/L activity in FDMs. (a) Quantitative FACS analysis of wt and Bax/Bak DKO FDMs stained with 10 μM acridine orange (AO) (10 min) or 75 nM LysoTracker Red DND-99 (45 min) before (+IL-3, gray lines and dots) or after removing IL-3 for 24 h (−IL-3, black lines and dots). Dot plots, histograms and fluorescence microscopy (Zeiss Axiovert, magnification: × 630) show a reduction of red fluorescence in the lysosomes (FL-3 or red dots by microscopy) concomitant with an increase in green fluorescence in the cytosol (FL-1). (b) Time-resolved FACS analysis of AO-stained wt (circle), Bax−/− (triangle), Bak−/− (square) and Bax/Bak DKO FDMs (rhomboid) at 0–48 h following IL-3 deprivation. The graph depicts percentage of red stained cells (+IL-3: 100%). The data represent the means from three independent experiments±S.E.M., P<0.02. (c) Anti-cathepsin D (CTD) and B (CTB) western blot analysis showing the processed forms of the proteases (ca. 25 kD) in the membrane and cytosolic fractions of FDM cells deprived of IL-3 for 0–20 h. (d) Anti-CTD and CTB western blot analysis as described in (c), comparing the abundance of both cathepsins in the membrane and cytosolic fractions of healthy and IL-3-deprived (24 h) wt and Bax/Bak DKO FDMs. In both (c) and (d), Cox V is shown for equal membrane and actin for equal cytosolic protein loading. (e) Cathepsin B/L cysteine protease activity in the cytosol of IL-3-treated or -deprived wt and Bax/Bak DKO FDMs, measured with the fluorogenic substrate z-FR-AMC in the absence or presence of the cathepsin B/L inhibitors Z-FA.fmk (10 μM), CA074 (25 μM), JPM-OEt (50 μM), the cysteine protease inhibitor E-64d (50 μM) or the cathepsin D inhibitor pepstatin A (Pep A, 50 μM). The data are depicted as means of relative fluorescence units (RFU) from three independent experiments±S.E.M., P<0.03. The color reproduction of the figure is available on the html full text version of the paper
Lysosomal permeability is accompanied by the release of cathepsins into the cytosol where they may trigger cellular responses, including apoptosis. In particular, CTB and CTD were described to have a role in apoptotic signaling.19, 22, 31, 32 We therefore monitored the appearance of cathepsins B, L and D in the cytosol of IL-3-deprived FDMs by western blotting and CTB /CTL activity assay using the substrate z-Phe-Arg-7-amino-coumarin (z-FR-AMC). The processed forms of both CTD and B were gradually lost from the membrane fraction (which also contained lysosomes) and appeared in the cytosol after 8–12 h of IL-3 deprivation (Figure 1c). Although this occurred at different kinetics for the two proteases, it nicely corresponded with the kinetics of LMP (Figure 1c). A similar cytosolic release was observed for CTL (Supplementary Figure S1). Concomitantly, we measured increased cytosolic CTB/CTL activity even at neutral pH. This activity was specific because it was not only prevented by z-Phe-Ala-fluoromethylketone (z-FA.fmk), but also by three other CTB/CTL inhibitors, CA074, E64d and JPM-OEt, but not by the CTD inhibitor pepstatin (Figure 1e). Importantly, as for LMP, the lysosomal loss and concomitant cytosolic appearance of CTB and CTD proteins (Figure 1d) and CTB/CTL activity (Figure 1e) were effectively prevented in Bax/Bak DKO cells.
Given this surprising finding, it was important to show that LMP and cathepsin release were also Bax/Bak dependent in other cell types and in response to other apoptotic stimuli. We therefore exposed wt, Bax−/−, Bak−/− and Bax/Bax DKO FDMs as well as spontaneously transformed, early-passage mouse embryo fibroblasts (3T9 mouse embryonic fibroblasts (MEFs)) to 20–50 μM etoposide or 1200 J/m2 ultraviolet (UV) light for 24 and 48 h (Figure 2). We also treated the MEF cell lines with 50 ng/ml FasL for 2–6 h, as these cells were previously shown to require Bax/Bak for FasL-induced apoptosis.33 Quantitative Annexin-V/PI and AO FACS analysis revealed a dose- and time-dependent increase in apoptosis (Supplementary Figures S2a and b) and decrease in the lysosomal membrane integrity in etoposide and UV-treated FDMs and MEFs, as well as in FasL-treated MEFs (Figures 2a and b). Neither apoptosis nor LMP were majorly detected in Bax/Bak DKO cells, even at high drug/UV doses after 48 h. As shown in IL-3-deprived FDMs (Figure 1), UV-, etoposide- and FasL-treated MEFs exhibited a Bax/Bak- and time-dependent decrease of CTB and CTD expression in the membrane (lysosomal) fraction and a concomitant appearance of both proteins in the cytosol (Figures 3a and b and Supplementary Figure S3). This correlated with increased cytosolic CTB /CTL (z-FRase) activity that was not seen in the absence of Bax/Bak and was blocked with the CTB/CTL inhibitor z-FA.fmk (Figure 3c). Importantly, the apoptotic cells did not seem to lose all CTB and CTD from lysosomes (Figures 3a and b), indicating that LMP was most likely due to a discrete membrane perforation rather than a total rupture of the lysosomes. Thus, apoptotic monocytes and fibroblasts show a discrete LMP and the cytosolic release of active cathepsin enzymes in response to several apoptotic stimuli, but both processes consistently depend on Bax/Bak.

Etoposide, UV and FasL trigger a dose- and time-dependent LMP in FDMs and MEFs in a Bak/Bax-dependent manner. Quantitative FACS analysis of red fluorescent (AO)-stained wt, Bax−/−, Bak−/− or Bax/Bak DKO FDMs exposed to 50 μM etoposide or 1200 J/m2 UV light for 0–48 h (a) and MEFs exposed to the indicated doses of etoposide or UV for 24 h, or to 50 ng/ml FasL-Fc for 0–6 h (b). The data represent the means of three independent experiments±S.E.M., P<0.02

Cathepsin D and B proteins and B/L activity appear in the cytosol of etoposide-, UV- and FasL-treated MEFs in a Bak/Bax-dependent manner. Anti-cathepsin D (CTD) and anti-cathepsin B (CTB) western blot analysis of the membrane and cytosolic fractions of wt or Bax/Bak DKO MEFs exposed to either 50 μM etoposide (a) or 1200 J/m2 UV light (b) for the times indicated. Cox V is shown for equal membrane and actin for equal cytosolic protein loading. In panel (a) the membrane fraction of cathepsin D−/− cells was used as a control for the specificity of the cathepsin D antibody. (c) Cathepsin B/L activities (z-FR-AMC) in the cytosol of wt and Bax/Bak DKO MEFs exposed to 50 μM etoposide, 1200 J/m2 UV light or 50 ng/ml FasL for the times indicated. The specificity of the cathepsin B/L activity was checked by the addition of the cathepsin B/L inhibitor 10 μM z-FA.fmk. The data are the means of three independent experiments±S.E.M., P<0.03. Note that all apoptotic stimuli trigger the appearance of the processed forms of cathepsin B and D as well as cathepsin B/L activity in the cytosol in a Bax/Bak-dependent manner, although the release is not complete
LMP is not due to a direct localization/action of Bax and Bak on the lysosomes
Bax and Bak are required for increased mitochondrial membrane permeability to provoke caspase-dependent and -independent apoptotic signaling in the cytosol.24 Our data show that Bax and Bak are also required for LMP. Recent evidence suggested that Bax may localize to the lysosomes in addition to the mitochondria.11, 28, 29 We therefore envisaged the possibility that Bax and Bak resided and acted on lysosomes to directly trigger LMP during apoptosis. We purified lysosomes from untreated and etopsoide-treated MEFs by a novel gradient centrifugation technique and tested the fractions for the co-presence of lysosomal acid phosphatase activity, the lysosomal membrane protein lysosomal-associated membrane protein-1 (Lamp-1), the mitochondrial protein cytochrome c oxidase IV (COX IV) and Bax and Bak. As shown in Figures 4a and b, the first three fractions of the purification contained most of the Lamp-1 protein and acid phosphatase activity but no Bax protein. The majority of Bax and Bak were present in fractions expressing high amounts of mitochondrial COX IV (fractions 5–8). Here both Bax and Bak were activated in response to etoposide (but not in control cells), as they could be immunoprecipitated with conformation-specific antibodies selectively detecting the active forms (Figure 4b, lanes 5–7, IP). Low amounts of Bak were found in the first three Lamp-1-positive fractions (Figure 4b, lanes 1–3, WB) but this form of Bak was not immunoprecipitated with active-site antibodies from control or etoposide-treated cell extracts (Figure 4b, lanes 1–3, WB). In addition, these fractions were slightly contaminated with the mitochondria as they were positive for COX IV (Figure 4b). Finally, immunofluorescence analysis of the Bak protein in control, etoposide- and UV-treated MEFs showed that Bak poorly colocalized with Lamp-1 (Figure 4c). Taken together, these data show that even though Bax and Bak are required for LMP, this probably does not occur by a direct action of Bax or Bak at the lysosome.

Bax and Bak do not localize to the lysosomes in healthy or etoposide-treated cells. Differential centrifugation of lysates of FDMs untreated (control) or treated with 50 μM etoposide for 16 h (ca. 50% cell death), using the lysosomal purification kit from Sigma. Each fraction was assayed for protein concentration and acid phosphatase activity, (a) as well as anti-Lamp-1, anti-Bax, anti-Bak and anti-COX IV (mitochondrial marker) western blotting (b, WB). In addition, the fractions were immunoprecipitated with conformation-specific anti-Bax (6A7) and anti-Bak (NT) antibodies and the immunoprecipitates were tested for active Bax and Bak by anti-Bax and anti-Bak western blotting, respectively (b, IP). Note that fractions 1–3 with the highest lysosomal acid phosphatase activity are positive for Lamp-1, but not for Bax. Although Bak is present in these fractions, it is not in an active form, and the fractions appear to be slightly contaminated with mitochondria based on anti-COX IV immunoreactivity. (c) Anti-Bak (green) and anti-Lamp-1 (red) immunofluorescence analysis of MEFs exposed to 50 μM etoposide or 600 J/m2 UV for 16 h and viewed under a Zeiss Axiovert fluorescence microscope at magnifications × 400 (Ctrl) and × 630 (etoposide, UV). Note that there is only little, if any, overlap (yellow) of anti-Bak and anti-Lamp-1
LMP occurs downstream of the apoptosome by a caspase-dependent mechanism
The finding that the Bax/Bak dependence of LMP was not because of the lysosomal localization/action of the two proapoptotic proteins raised the possibility that LMP occurred as a result of apoptosome formation and caspase activation. To determine this, we measured apoptosis and LMP in Apaf-1−/− FDMs deprived of IL-3 or treated with various doses of etoposide or UV for 24 h. In addition, we treated Apaf-1−/− and caspase-9−/− MEFs with etoposide and UV. In all cases, LMP (Figures 5a and b) and apoptosis (Supplementary Figures S4a and b) were strongly delayed in caspase-9−/− and Apaf-1−/− cells. Next, we investigated the effector caspase requirement for apoptosis and LMP by exposing MEFs deficient in caspases-3 and -7 (caspase-3/-7 DKO) to 50 and 100 μM etoposide or 600–1200 J/m2 UV for 24–48 h. As shown in Supplementary Figure S4c, apoptosis was majorly caspase dependent for both stimuli because caspase-3/-7 DKO MEFs displayed a slower apoptosis kinetic than wt cells. With regard to LMP this caspase requirement depended on the stimulus and the duration of the apoptotic stress. While in etoposide-treated caspase-3/-7 DKO cells, lysosomal membrane integrity was largely maintained for up to 48 h (Figure 5c), this was only the case for the first 24 h of UV exposure. These data show that LMP requires the assembly of the apoptosome and, dependent on the apoptotic stimulus and time of exposure, the activation of effector caspase-3/-7.

LMP induced by etoposide, UV or IL-3 deprivation requires components of the Apaf-1/caspase-9 apoptosome and caspase-3/-7. Quantitative FACS analysis of AO-stained wt, Apaf-1−/− and caspase-9−/− MEFs treated with 50 or 100 μM etoposide or exposed to 1200 J/m2 UV light for 24 h (a), of AO-stained wt and Apaf-1−/− FDMs treated with the indicated doses of etoposide or UV, or deprived of IL-3 for 24 h (b), or of AO-stained wt and caspase-3/-7 DKO MEFs treated with the indicated doses of etoposide or UV for 24 or 48 h (c). The data are the means of three independent experiments±S.E.M., P<0.03
CTB and CTL knockout cells are partially resistant to apoptosis induced by etoposide and IL-3 deprivation, but cannot be rescued from cell death
We reasoned that if LMP was important for the initiation of apoptosis, deletion of cathepsins should result in protection against apoptotic stimuli. Alternatively, if LMP was largely a phenomenon resulting from the activation of apoptosis, cathepsin deficiencies would not permit cells to survive apoptotic stimuli in the long-term. To test these predictions, we generated IL-3-dependent FDMs from CTB, CTL and CTD knockout mice as previously described,34 and tested the cells for their sensitivity to apoptosis induced by IL-3 deprivation, etoposide or UV treatment. All the knockout cells exhibited similar kinetics of LMP in response to apoptotic stimuli, indicating that the lysosomal membrane integrity was not controlled by particular cathepsins (data not shown). However, CTB−/− and CTL−/− FDMs showed a strong delay of apoptosis induction in response to etoposide (Figures 6a and b) or IL-3 deprivation (Figures 6a and c) as compared with wt cells. No major difference in apoptosis sensitivity between wt and knockout cells was noted in CTD−/− FDMs (Figure 6) or when the cells were exposed to UV (Figures 6a and d), indicating that the lysosomal contribution to apoptosis is restricted to certain cathepsin isoforms and apoptotic stimuli. Detailed kinetic analysis revealed that the slower apoptosis rate of CTB−/− and CTL−/− FDMs nicely correlated with delayed caspase-3 activation. Although in wt or CTD−/− FDMs maximal caspase-3 activity peaked at 24 h after IL-3 deprivation or etoposide treatment and declined thereafter, CTB−/− and CTL−/− FDMs exhibited maximal caspase-3 activity only after 36–48 h (Figures 6b and c).

Delay of etoposide and IL-3 deprivation-induced, but not UV-induced apoptosis and caspase activation of cells deficient of cathepsin L and/or B. (a) FACS dot plots and quantitation of Annexin-V/PI-negative wt, cathepsin L−/− (CTL−/−), cathepsin B−/− (CTB−/−) or cathepsin D−/− (CTD−/−) FDMs deprived of IL-3 (dark gray bars) or exposed to 50 μM etoposide (light gray bars) or 1200 J/m2 UV light (black bars) for 24 h. (b–e) Quantitation of cytosolic caspase activity (fluorogenic DEVDase assay) (bars) and Annexin-V/PI negativity (lower left quadrant of FACS analysis) (graphs) of wt, cathepsin L−/− (CTL−/−), cathepsin B−/− (CTB−/−) or cathepsin D−/− (CTD−/−) FDMs exposed to 50 μM etoposide (b), deprived of IL-3 (c) or exposed to 1200 J/m2 UV light (d), as well as of wt, cathepsin L−/− (CTL−/−), cathepsin B−/− (CTB−/−) or cathepsin B−/−, L−/− (CTB/L DKO) MEFs treated with 50 μM etoposide for 0–48 h (e). In panel (e) wt MEFs were also co-treated with etoposide and E64d (30 μM). The data are the means of three independent experiments±S.E.M., P<0.02. Note that apoptosis and caspase-3 activation of CTL−/− and CTB−/−, but not CTD−/− FDMs, was delayed for etoposide treatment and IL-3 deprivation, but not for UV exposure. For etoposide-treated MEFs, apoptosis and caspase-3 activation/processing was delayed in CTB−/− and CTB/L DKO as well as in wt MEFs treated with E64d, but not in CTL−/− cells
We also isolated MEFs from mice deficient of CTB, CTL or both (CTB/L DKO) and exposed them to etoposide and UV. As shown in Figure 6e, CTB−/− and B/L DKO MEFs exhibited a delay in etoposide-induced Annexin-V/PI staining and caspase-3 activation. This was also the case when we inhibited CTB with the cysteine protease inhibitor, E64d, in etoposide-treated wt MEFs, indicating that the apoptosis-enhancing effect of CTB was due to its proteolytic activity rather than another yet undescribed function of the enzyme. In contrast, CTL −/− MEFs died with similar time kinetics as wt cells, as did all the cathepsin knockout cell lines exposed to UV (data not shown). These data show that, as previously reported, cathepsins can influence apoptosis sensitivity but this depends on the cell type, the apoptotic stimulus and the kind of cathepsin.
We have previously shown that a delay in the appearance of the morphological characteristics of apoptosis does not necessarily mean that cells have not committed to apoptosis.34 To determine whether CTB or CTL were crucial for cell commitment to apoptosis, we performed clonogenicity assays. In this assay, monocytes were tested for their potential to form colonies in soft agar after they had been exposed to etoposide or deprived of IL-3, but then allowed to recover in fresh medium with survival factors (IL-3). As shown in Figure 7, Bax/Bak DKO FDMs were fully rescued from apoptosis induced by IL-3 deprivation or etoposide treatment. By contrast, none of the cathepsin knockout cells could form colonies after IL-3 re-addition (Figure 7a) or removing etoposide (Figure 7b). This was also true for cells deficient in both cathepsins B and L and for wt FDMs treated with E64d, indicating that even the lack or blockage of two cathepsins did not fully prevent apoptosis (Figures 7a and b). This finding clearly shows that cathepsins B and L do not contribute to the commitment step of apoptosis (as Bax and Bak do), but enhance the efficiency of apoptosis through an amplification loop.

Lack of clonogenic growth of IL-3-deprived or etoposide-treated wt, cathepsin L−/−, B−/− or L/B DKO FDMs after re-adding IL-3 or removing etoposide. Relative clonogenicity (logarithmic scale) of wt (open square), cathepsin L−/− (gray triangle), B−/− (gray circle), L/B DKO (black triangle) or Bax/Bak DKO (black square) FDMs deprived of IL-3 (a) or exposed to 50 μM etoposide (b) for 0–60 h followed by plating the cells on agar in the absence of etoposide and/or in the presence of IL-3 for 21 days. The analysis was also carried out for etoposide and UV-treated wt FDMs in the presence of 30 μM E64d (black circle). 100% represents the number of colonies generated per 1000 cells at day 0 (before removing IL-3 or adding etoposide). The data are the means of three independent experiments±S.E.M., P<0.02
Discussion
Our findings resolve a crucial aspect of apoptosis regulation that has been highly debated in the past; namely, at what step of the signaling cascade LMP and the cytosolic release of cathepsins contribute to the death fate of mammalian cells. These processes have been thought to occur at early time points of apoptosis, leading to the cleavage of Bid,17, 18, 35, 36 the activation of caspase-217 or -8,22 or the translocation of Bax,21 all events triggering the Bax/Bak-mediated perforation of the mitochondrial outer membrane (MOMP), a crucial commitment step for apoptosis induction. However, in this study, we show, using four different apoptotic stimuli (UV, etoposide, FasL, IL-3 deprivation) on two different cell lines (MEFs and FDMs), that LMP and cathepsin release are late events of apoptosis signaling occurring downstream of MOMP in a manner dependent on Bax/Bak, the apoptosome and effector caspases-3 and -7. Indeed, in all cases tested, the kinetics of apoptosis (Supplementary Figures S2 and S4) were slightly faster than those of LMP (Figures 2 and 5), indicating that LMP is more likely to occur after than before apoptosis induction.
Why has this interdependence between LMP and MOMP been dismissed? First, numerous studies used lysosomotropic agents such as LeuLeuOMe12 to force cathepsins release into the cytosol. Such experiments placed the action of cathepsins automatically upstream of MOMP, but failed to show where exactly these proteases act in response to apoptotic stimuli. Second, apoptosis induced by TNFα, H2O2 or other stimuli was considered to be ‘inhibited’ by genetically deleting or pharmacologically inhibiting CTB, CTL or CTD.6, 11, 19, 20, 21, 31 However, in the absence of clonogenic assays, it is impossible to discriminate whether such an ‘inhibition’ is only a delay or a real blockage of cell death. Third, although LMP and cathepsin release was sometimes shown to precede Bax/Bak-mediated cytochrome c release,21, 22, 37 the causal relationship between these two events has not yet been tested by genetically deleting apoptosis signaling components between LMP and MOMP. All three aspects were addressed in our study. We found that cathepsins were released downstream, rather than upstream of MOMP, and the lack of clonogenic survival of cathepsin KO or even DKO cells indicated that these proteases did not have an impact on the point of life-or-death decisions.
The simplest explanation for the dependence of LMP on Bax/Bak would be that these two proapoptotic proteins directly perforate the lysosomal membrane, in a manner analogous to the function of Bax and Bak at the mitochondrial membrane. Some experimental evidence does support such a model.11, 28 However, in this study we could not convincingly immunolocalize Bak on lysosomes in response to etoposide or UV, and acid phosphatase-positive lysosomal fractions were entirely negative for inactive or active Bax. Although some Bak contaminated these fractions, it was not in an active conformation. We therefore contend that LMP does not result from a direct perforation of lysosomes by Bax and Bak. Rather, our data point toward the role of the apoptosome and caspase-3/-7 in triggering LMP. The discrepancy between previous reports and our study may be explained by the possibility that Bax does not translocate to the lysosomes from the cytosol but ends up in this compartment after autophagic sequestration of damaged mitochondria during apoptosis. Similarly, the BH3-only protein Bim could first activate Bax/Bak on the mitochondria and then move to the lysosomes for degradation.11 This would be in agreement with the slight contamination of lysosomal fractions with mitochondrial proteins. Preliminary immunoelectron microscopy data suggest that Bax localizes to lysosomes exclusively in multivesicular structures reminiscent of autophagosomes (data not shown). In addition, treatment with the autophagy inhibitor 3-MA diminished the association of Bax with these structures, indicating that Bax arrived at the lysosomes by autophagy and not by direct translocation from the cytosol.
What might be the signaling pathway linking the apoptosome and/or activated effector caspases-3/-7 to LMP? Gyrd-Hansen et al.38 implicated caspase-9, but not Apaf-1, in TNFα-induced LMP, suggesting that caspase-9 had an Apaf-1–, and hence apoptosomal/mitochondrial independent function to cause LMP. We however propose an apoptosome- and effector caspases-3/-7-dependent mechanism, which may result in the cleavage of particular substrates mediating LMP. Candidate substrates could include regulators of oxidative stress, sphingolipid, arachidonic acid and fatty acid metabolism, components of ion channels or pumps (for example, calcium), hsp70, a protein shown to inhibit LMP, or the caspase-3 substrate NDUFS1, the p75 subunit of mitochondrial respiratory complex I, leading to the production of reactive oxygen species.10, 12, 39, 40, 41, 42 Recently, a global mapping of the topography and magnitude of proteolytic events in apoptosis has unveiled a surge of new caspase substrates, which can now be tested for their involvement in mediating LMP.43
Our study shows that CTB and CTL amplify death signaling induced by IL-3 deprivation or etoposide. Thus, these proteases still contribute to apoptosis after MOMP has occurred, although their involvement may vary in different cell types and with the death stimulus. Although immortalized MEFs treated with TNFα/CHX were shown to require CTB and CTL, but not CTD for apoptosis amplification, the opposite was true when the cells were treated with staurosporine.44 Similarly, primary neutrophils mainly rely on CTD, but not CTB or CTL, for efficiently executing spontaneous apoptosis.22 We have shown that in response to UV irradiation, the mitochondrial signaling pathway leading to caspase-3/-7 activation is sufficient for apoptosis, despite the fact that LMP and cathepsin release occur later on. In contrast, our results show that caspase-3 activation is delayed in CTB−/−, CTL−/− and B/L DKO myeloid progenitors treated with etoposide or deprived of IL-3. In this case, cytosolic CTB and CTL may stimulate a positive feedback loop enhancing caspase-3 processing and activity. Alternatively, these proteases may further stimulate caspase-independent processes such as necrosis.45 To resolve this issue, we need to identify cytosolic cathepsin substrates mediating alternative death signaling pathways.
What role might cathepsin-mediated amplification of apoptosis signaling have in vivo? Under normal circumstances, apoptosis and phagocytosis of dead cells is rapid. A delay in this process, as observed in the prolonged survival of CTD-deficient neutrophils, may exaggerate innate immune responses in experimental bacterial infection and in septic shock.22 Moreover, reduced keratinocyte apoptosis in CTL-deficient mice was found to result in periodic hair loss,15, 16 and CTB-deficient animals showed diminished liver damaged on TNFα challenge.6 Our study here reveals that, in order to fully understand a particular biological process such as the role of LMP and cathepsin in apoptosis, we need robust and physiologically relevant, experimental tools and an accurate kinetic analysis of the sequence of events involved.
Materials and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), penicillin, streptomycin and fetal calf serum (FCS) were purchased from Gibco/Life Technologies (Rockville, MD, USA). Z-FR-AMC and Z-FA.fmk were from Calbiochem (Calbiochem-Merck, Damstadt, Germany), pepstatin A from Fluka (Steinheim, Germany), E-64d from Biomol GmbH (Biomol GmbH, Hamburg, Germany), and CA074, etoposide and the LYSISO1 lysosomal isolation kit from Sigma-Aldrich (Steinheim, Germany). Polyclonal goat antibodies against CTB, CTD and CTL were obtained from R&D Systems (Wiesbaden, Germany), rat monoclonal to Lamp-1 (clone 1D4B) from Abcam (Cambridge, UK), rabbit polyclonal anti-Bax NT and the conformation-specific anti-Bak (NT) antibody from Upstate, Millipore GmbH (Schwalbach, Germany), polyclonal rabbit anti-caspase-3 (9661 and 9662) from Cell Signaling (Frankfurt am Main, Germany), monoclonal mouse anti-β actin (C4) from MP Biomedical (Eschwege, Germany), mitochondrial anti-COX IV and anti-COX V from Molecular Probes (Karlsruhe, Germany) and the conformation-specific anti-Bax antibody (6A7) from BD Biosciences (Heidelberg, Germany). The secondary AlexaFluor546-conjugated goat anti-rat and the AlexaFluor488-conjugated goat anti-rabbit secondary IgG antibodies were obtained from Invitrogen (Karlsruhe, Germany), the horseradish peroxidase (HRP)-conjugated anti-goat from Sigma-Aldrich, and the HRP-conjugated anti-rabbit and anti-mouse IgG antibodes from Jackson ImmunoReserach (Suffolk, UK). AO was bought from Invitrogen and tetramethylrhodamine ethyl ester perchlorate, LysoTracker Green and Red DND 99 from Molecular Probes. Recombinant murine FasL-Fc was kindly provided by Pascal Schneider, Lausanne. JMP-OEt was a gift from Matt Bogyo, Stanford University, CA, USA. Recombinant mouse IL-3 was obtained from the supernatant of WEHI3B cells as described34 and recombinant His-GFP-Annexin-V was produced according to Egger et al.46
Cell lines
Interleukin-3-dependent (factor-dependent myeloid (FDM) cells were generated by co-culturing E14.5 fetal liver single cell suspensions with fibroblasts expressing a HoxB8 retrovirus in the presence of high levels of IL-3.34 The cells were expanded in DMEM supplemented with 10% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin and 10% IL-3 from WEHI3B supernatant. FDMs bearing gene deletions for Bax, Bak or both (Bax/Bak DKO) have been described.47 In addition, FDMs were generated from C57Bl6 mice bearing deletions for Apaf-1 (kindly provided by A Strasser) and from FVB mice that were either wt or deficient for CTB, CTL or CTD. 3T9 mouse embryo fibroblasts (MEFs) from wt C57BL/6 mice or mice deficient of Apaf-1, caspase-9, Bax, Bak or both (Bax/Bak DKO) were obtained from A Strasser, WEHI, Melbourne. SV40 T antigen transformed MEFs deficient of both caspase-3 and -7 (caspase-3/-7 DKO) were kindly provided by R Flavell, Yale, USA. MEFs deficient of CTL, B or both were isolated from the respective FVB knockout mice. All MEFs were 3T9 immortalized and cultured in DMEM including 10% FCS, 100 units/ml penicillin and 100 μg/ml streptomycin as previously described.46
Apoptosis quantitation
Apoptosis was induced by etoposide (Sigma), UV irradiation (Stratalinker UV Crosslinker 2400 (Stratagene Europe, Amsterdam, The Netherlands), 254 nm UV), FasL-Fc or IL-3 withdrawal (for FDMs). Quantitation of apoptotic cells was performed by FACS analysis of His-GFP-Annexin-V/PI stained cells as previously described.46
Measurement of LMP
To measure lysosomal membrane integrity, healthy or apoptotic cells were stained with either 10 μM AO for 10–15 min or 75 nM LysoTracker Red DND 99 for 45 min at 37 °C, and washed several times in phosphate-buffered saline (PBS) to reduce background. Both dyes accumulate in intact acidic compartments. For qualitative immunofluorescence analysis, the cells were cytospun and viewed on glass coverslips under a Axiovert Zeiss fluorescence microscope (Flo Jo, Tree Star, Ashland, OR, USA). LMP was quantified by measuring the reduction of red or green fluorescence using a FACS Calibur from BD Biosciences. The FlowJo software (Flo Jo, Tree Star, Ashland, OR, USA) was used to analyze the flow cytometry measurements.
Immunofluorescence analysis
The 3T9 MEFs were grown on glass coverslips and fixed with 4% paraformaldehyde in PBS containing 0.1 M PIPES (pH 6.8) for 30 min. Cells were permeabilized with 0.1% Triton-X100 in PBS for 20 min and blocked with PBS containing 3% bovine serum albumin at room temperature for 1 h. After incubation with rat monoclonal anti-Lamp-1 (1 : 200) and rabbit polyclonal anti-Bak (1 : 150) at room temperature for 90 min, the cells were washed three times with PBS and treated with the AlexaFluor546-conjugated goat anti-rat and the AlexaFluor488-conjugated goat anti-rabbit secondary antibody at dilutions 1 : 200 for 90 min. Cells were washed three times in PBS and in H2O, mounted onto slides using Mowiol (6 g glycerine, 2.4 g Mowiol, 6 ml H2O, 12 ml 0.2 M Tris, pH 8.5, 0.1% DABCO) and stored in the dark at 4 °C until observation.
Clonogenic assay
Clonogenic assays were carried out as previously described.34 Briefly, 1 × 105 cells/ml were grown with or without IL-3 or in the absence or presence of 50 μM etoposide. At the various time points after apoptosis induction, known dilutions of culture were plated in 36 mm Petri dishes in DMEM, 20% FCS, 10% IL-3 supernatant and 0.3% agar. After 21 days the number of colonies were counted and expressed as a percentage relative to the number of colonies generated per 1000 cells plated immediately before culture without IL-3 or with etoposide, that is, after washing and determining of cell number. For example, if a clone generated 200 colonies per 1000 cells plated at day 0 and 50 colonies per 1000 cells after culture without IL-3 or with etoposide for 1 day, the relative clonogenicity at days 0 and 1 were 100 and 25%, respectively, at day 1. In each independent experiment, at least three independent clones of each genotype were used.
Cellular fractionation and immunoblot analysis
For subcellular fractionation into the cytosol and mitochondria, the cells were washed two times with PBS and incubated with twice the volume of MSH-Buffer (210 mM mannitol, 70 mM sucrose, 20 mM HEPES pH 7.5, 1 mM EDTA, 300 μM Pefabloc, 100 μM PMSF) for 45 min on ice. Cells were lysed using a 25-G needle until 50% of the cells were trypan blue positive. Cells were centrifuged for 5 min at 350 × g to pellet cellular debris and the nuclei. The supernatant was centrifuged for 20 min at 16 000 × g to obtain the mitochondria. The resulting supernatant was subjected to 100 000 × g centrifugation for 45 min to obtain the cytosol. To obtain a crude membrane/lysosomal fraction for cathepsin western blotting (containing all other organelles expect the nuclei), the postnuclear supernatant was directly spun at 100 000 × g and the pellet resuspended in MSH buffer +1% Triton. Equal amount of protein (100 μg) was resolved by 12–15% SDS-PAGE, transferred to PVDF membrane and blocked with 5% bovine serum albumin in PBS containing 0.05% Tween 20 (v/v) for 1 h. After washing, the membranes were incubated overnight with goat anti-CTB, CTD and CTL (each 1 : 5000) diluted in PBS-Tween containing 5% bovine serum albumin. After further washings the PVDF membrane was incubated with the corresponding HRP-conjugated rabbit anti-goat secondary antibody (1/5000) and visualized by enhanced chemiluminescence (ECL, PIERCE, Themo Scientific, Schwerte, Germany).
Immunoprecipitations
The fractions from the LYSISO1 purification were lysed in MSH buffer containing 1% CHAPS and subjected to immunoprecipitations using the conformation-specific anti-Bax 6A7 and anti-Bak NT antibodies specifically detecting active Bax and Bak in their native states, respectively. A total of 100 μg of protein was incubated with 1 μg of anti-Bax 6A7 or anti-Bak NT on ice for 1 h, followed by the addition of 50 μl of 50% (w/v) Protein A Sepharose. The antibody–antigen complexes were captured on Sepharose beads on a rotating wheel at 4°C for 90 min. The beads were centrifuged and washed three times in 20 volumes (1 ml) of MSH and then dissolved in hot SDS-sample buffer under nonreducing conditions (to keep the immunoglobulins intact). The immunoprecipitates were analyzed by anti-Bax NT and anti-Bak NT western blotting using ECL detection as described above.
Caspase and cathepsin assays
Caspase-3/-7 activity was measured in the cytosol of healthy and apoptotic cells using the fluorogenic substrate Ac-DEVD-AMC (7-amino-4-methylcoumarin, Alexis Biochemicals, Enzo Life Sciences GmbH, Lörrach, Germany), as described.46 CTB-/CTL-specific activity was measured in the cytosol of healthy and apoptotic cells. In all, 10 μl of the cytosol was mixed with 85 μl of assay buffer (200 mM sodium acetate, 1 mM EDTA, 0.05% Brij, pH 5.0) in the presence or absence of inhibitors for CTB/CTL (z-FA.fmk, CA074, JMP-OEt, E-64d) or CTD (pepstatin A). Altogether, 5 μl of the CTB/CTL substrate z-FR-AMC (25 μM) was added and incubated at 37°C for 30 min before the fluorescence was measured in a microtiter fluorescence reader (Ex: 380 nm, Em: 460 nm).
Lysosomal purification
In all, 3 × 108 cells per sample were grown to 90% confluency, treated with 50 μM etoposide and harvested by trypsinization. All subsequent steps of the lysosomal isolation were performed according to manufacturer's description (LYSISO1). In brief, cells were centrifuged at 600 × g for 5 min, resuspended in 2.7 packed cell volume of 1 × extraction buffer and homogenized in a glass Dounce homogenizer. The degree of breakage was checked under the microscope using trypan blue until 80–85% of the cells were broken. The nuclei were removed by centrifugation at 1.000 × g for 10 min. The postnuclear supernatant was centrifuged at 20.000 × g for 20 min and the resulting pellet, containing the crude lysosomal fraction, was resuspended in a minimal volume of 1 × extraction buffer (0.4 ml per 108 cells). To enrich the lysosomes, the suspension was further purified by density gradient centrifugation at 150.000 × g for 4 h on a multistep OptiPrep (Sigma-Aldrich, Steinheim, Germany) gradient according to the manufacturer's description. Altogether, 0.5 ml fractions were collected starting from the top of the gradient. Each fraction was assayed for protein concentration, acid phosphatase activity and amount of Lamp-1, Bax, Bak and Cox IV proteins by western blotting.
Acid phosphatase enzymatic assay
Phosphatase activity was assayed with the help of the acid phosphatase assay kit from Sigma, according to the manufacturer's instruction. In brief, prewarmed (37 °C) substrate solution and reaction components (sample and citrate buffer) were mixed in a 96-well plate and incubated at 37 °C for 10 min. The reaction was stopped by adding 0.2 ml of stop solution (0.5 N NaOH) to each well and the absorption was measured at 405 nm. Acid phosphatase activity was measured in triplicates (a blank reaction as well as standard solution was run in parallel) and calculated according to the following equation:

DF: dilution factor of the original sample; time: time of incubation at 37 °C in minutes; Venz: volume of enzyme sample added to assay in ml; 0.05: concentration (μM/ml) of 4-nitrophenol in standard solution; 0.3: total assay volume including stop solution (ml).
Statistical analysis
Data were expressed as mean values±S.E.M. Statistical evaluation was determined by two-tailed Student's t-test. A value of P<0.05 was considered to be statistically significant. All statistical analysis was carried out with the program SigmaPlot for Windows, Version 10.0 (Systat Software Inc., Chicago, IL, USA).
Abbreviations
- AO:
-
acridine orange
- FDMs:
-
factor-dependent monocytes
- IL-3:
-
interleukin-3
- DKO:
-
double knockout
- Lamp-1:
-
lysosomal-associated membrane protein-1
- LMP:
-
lysosomal membrane permeabilization
- MEFs:
-
mouse embryonic fibroblasts
- PI:
-
propidium iodide
- MOMP:
-
mitochondrial outer membrane permeabilization
- TMRE:
-
tetramethylrhodamine ethyl ester perchlorate
- ROS:
-
reactive oxygen species
- z-FR-AMC:
-
z-Phe-Arg-7-amino-coumarin
- z-FA.fmk:
-
z-Phe-Ala-fluoromethylketone
- DEVD-AMC:
-
Asp-Glu-Val-Asp-7-amino-coumarin
- CTD:
-
cathepsin D
- CTL:
-
cathepsin L
- CTB:
-
cathepsin B
- COX:
-
cytochrome c oxidase
- UV:
-
ultraviolet light
- TNFα:
-
tumor necrosis factor alpha
- FACS:
-
fluorescence-absorbent cell scanner
- GFP:
-
green fluorescent protein
- BSA:
-
bovine serum albumin
- PBS:
-
phosphate-buffered saline
- DMEM:
-
Dulbecco's modified Eagle's medium
References
Baudhuin F, Hertoghe-Lefevre D, de Duve C . Differential release of alkaline deoxyribonuclease and of glutamate dehydrogenase from rat-liver mitochondrial fractions. Biochem Biophys Res Commun 1969; 35: 548–555.
Firestone RA, Pisano JM, Bonney RJ . Lysosomotropic agents. 1. Synthesis and cytotoxic action of lysosomotropic detergents. J Med Chem 1979; 22: 1130–1133.
Wilson PD, Firestone RA, Lenard J . The role of lysosomal enzymes in killing of mammalian cells by the lysosomotropic detergent N-dodecylimidazole. J Cell Biol 1987; 104: 1223–1229.
Zdolsek J, Zhang H, Roberg K, Brunk U . H2O2-mediated damage to lysosomal membranes of J-774 cells. Free Radic Res Commun 1993; 18: 71–85.
Brunk UT, Zhang H, Dalen H, Ollinger K . Exposure of cells to nonlethal concentrations of hydrogen peroxide induces degeneration-repair mechanisms involving lysosomal destabilization. Free Radic Biol Med 1995; 19: 813–822.
Guicciardi ME, Deussing JD, Miyoshi H, Bronk SF, Svingen PA, Peters C et al. Cathepsin B contributes to TNFα-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest 2000; 106: 1127–1137.
Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ . Tumor necrosis factor-alpha-associated lysosomal permeabilization is cathepsin B dependent. Am J Physiol Gastrointest Liver Physiol 2002; 283: G947–G956.
Li W, Yuan X, Nordgren G, Dalen H, Dubowchik GM, Firestone RA et al. Induction of cell death by the lysosomotropic detergent MSDH. FEBS Lett 2000; 470: 35–39.
Boya P, Andreau K, Poncet D, Zamzami N, Perfettini J-L, Metivier D et al. Lysosomal membrane permeabilization induces cell death in a mitochondria-dependent fashion. J Exp Med 2003; 197: 1323–1334.
Kagedal K, Zhao M, Svensson I, Brunk UT . Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem J 2001; 359: 335–343.
Werneburg NW, Guicciardi ME, Bronk SF, Kaufmann SH, Gores GJ . Tumor necrosis factor-related apoptosis-inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl-2 proteins. J Biol Chem 2007; 282: 28960–28970.
Boya P, Kroemer G . Lysosomal membrane permeabilization in cell death. Oncogene 2008; 27: 6434–6451.
Turk B, Stoka V, Rozman-Pungercar J, Cirman T, Droga-Mazovec G, Oresic K et al. Apoptotic pathways: involvement of lysosomal proteases. Biol Chem 2002; 383: 1035–1044.
Guicciardi ME, Leist M, Gores GJ . Lysosomes in cell death. Oncogene 2004; 23: 2881–2890.
Tobin DJ, Foitzik K, Reinheckel T, Mecklenburg L, Botchkarev VA, Peters C et al. The lysosomal protease cathepsin L is an important regulator of keratinocyte and melanocyte differentiation during hair follicle morphogenesis and cycling. Am J Pathol 2002; 160: 1807–1821.
Reinheckel T, Hagemann S, Dollwet-Mack S, Martinez E, Lohmüller T, Zlatkovic DJ et al. The lysosomal cysteine protease cathepsin L regulates keratinocyte proliferation by control of growth factor recycling. J Cell Sci 2005; 118: 3387–3395.
Guicciardi ME, Bronk SF, Werneburg NW, Yin X-M, Gores GJ . Bid is upstream of lysosome-mediated caspase 2 activation in tumor necrosis factor α-induced hepatocyte apoptosis. Gastroenterology 2005; 129: 269–284.
Droga-Mazovec G, Bojic L, Petelin A, Ivanova S, Romih R, Repnik U et al. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of Bid and anti-apoptotic Bcl-2 homologues. J Biol Chem 2008; 283: 19140–19150.
Deiss LP, Galinka H, Berissi H, Cohen O, Kimchi A . Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J 1996; 15: 3861–3870.
Kagedal K, Johansson U, Ollinger K . The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. FASEB J 2001; 15: 1592–1594.
Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumon C et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem 2003; 278: 31401–31411.
Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S et al. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med 2008; 205: 685–698.
Kroemer G, Galluzzi L, Brenner C . Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA et al. The combined functions of proapoptotic Bcl-2 family members bax and bak are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–1399.
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ . Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 1997; 139: 1281–1292.
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002; 111: 331–342.
Zou H, Li Y, Liu X, Wang X . An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 1999; 274: 11549–11556.
Kagedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS et al. Lysosomal membrane permeabilization during apoptosis – involvement of Bax? Int J Exp Pathol 2005; 8: 309–321.
Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ . Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol 2006; 290: G1339–G1346.
Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L et al. Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci USA 2002; 99: 6286–6291.
Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M et al. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol 2001; 153: 999–1010.
Canbay A, Guicciardi ME, Higuchi H, Feldstein A, Bronk SF, Rydzewski R et al. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Clin Invest 2003; 112: 152–159.
Walter D, Schmich K, Vogel A, Pick R, Kaufmann T, Hochmuth FC et al. Switch from type II to I Fas/CD95 death signaling upon in vitro culturing of primary hepatocytes. Hepatology 2008; 48: 1942–1953.
Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ et al. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J Cell Biol 2004; 165: 835–842.
Stoka V, Turk B, Schendel SL, Kim T-H, Cirman SJ, Snipas SJ et al. Lysosomal protease pathways to apoptosis. J Biol Chem 2001; 276: 3149–3157.
Cirman T, Oresic K, Mazovec GD, Turk V, Reed JC, Myers RM et al. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem 2004; 279: 3578–3587.
Roberg K . Relocalization of cathepsin D cytochrome c early in apoptosis by immuno-electron microscopy. Lab Inves 2001; 81: 149–158.
Gyrd-Hansen M, Farkas T, Fehrenbacher N, Bastholm L, Hoyer-Hansen M, Elling F et al. Apoptosome-independent activation of the lysosomal cell death pathway by caspase-9. Mol Cell Biol 2006; 26: 7880–7891.
Zhao M, Brunk UT, Eaton JW . Delayed oxidant-induced cell death involves activation of phospholipase A2. FEBS Lett 2001; 509: 399–404.
Foghsgaard L, Lademann U, Wissing D, Poulsen B, Jäättelä M . Cathepsin B mediates tumor necrosis factor-induced arachidonic acid release in tumor cells. J Biol Chem 2001; 277: 39499–39506.
Nylandsted J, Hansen MG, Danielewich A, Fehrenbacher N, Lademann U, Hoyer-Hansen M et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 2004; 200: 425–435.
Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N et al. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 2001; 117: 773–786.
Dix MM, Simon GM, Cravatt BF . Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 2008; 134: 679–691.
Fehrenbacher N, Gyrd-Hansen M, Poulsen B, Felbor U, Kallunki T, Boes M et al. Sensitization of the lysosomal cell death pathway upon immortalization and transformation. Cancer Res 2004; 64: 5301–5310.
Golstein P, Kroemer G . Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 2007; 32: 37–43.
Egger L, Schneider J, Rheme C, Tapernoux M, Häcki J, Borner C . Serine proteases mediate apoptosis-like cell death and phagocytosis under caspase inhibiting conditions. Cell Death Differ 2003; 10: 1188–1203.
Ekert PG, Jabbour AM, Manoharan A, Heraud JE, Yu J, Pakusch M et al. Cell death provoked by loss of interleukin-3 signalling is independent of Bad, Bim and PI3 kinase/AKT, but depends in part on Puma. Blood 2006; 108: 1461–1468.
Acknowledgements
We thank A Strasser, WEHI, for the Bax−/−, Bak−/−, Bax/Bak DKO, Apaf-1−/− and caspase-9−/− mice and 3T9 MEFs; R Flavell, Yale, for the SV40 transformed caspase-3/-7 DKO cells; P Schneider, Lausanne, for the FasL-Fc; and M Bogyo, Stanford, for the cathepsin B/L inhibitor JMP-OEt. This work was supported by the Deutsche Forschungsgemeinschaft (BO-1933), as well as by the Spemann Graduate School of Biology and Medicine (SGBM, GSC-4), and the Cluster BIOSS funded by the Excellence Initiative of the German Federal and State Governments (EXC 294) (to CO, JB and CB). TR and MT were funded by the Deutsche Krebshilfe (Re106977) and by the European Union FP-7, Grant 201279 Microenvimet. PGE was supported by National Health and Medical Research Council (project Grants 436936 and 384404) and the Sylvia and Charles Senior Medical Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Oberle, C., Huai, J., Reinheckel, T. et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 17, 1167–1178 (2010). https://doi.org/10.1038/cdd.2009.214
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.214
Keywords
This article is cited by
-
A fluorogenic, peptide-based probe for the detection of Cathepsin D in macrophages
Communications Chemistry (2023)
-
The Achilles’ heel of cancer: targeting tumors via lysosome-induced immunogenic cell death
Cell Death & Disease (2022)
-
Overexpression of V-ATPase B2 attenuates lung injury/fibrosis by stabilizing lysosomal membrane permeabilization and increasing collagen degradation
Experimental & Molecular Medicine (2022)
-
S-Nitrosylation of cathepsin B affects autophagic flux and accumulation of protein aggregates in neurodegenerative disorders
Cell Death & Differentiation (2022)
-
Autophagy as a modulator of cell death machinery
Cell Death & Disease (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}