
Abstract
Breast tumor cells are often resistant to tumor necrosis factor-related apoptosis-inducing ligand (tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)/APO-2 L). Here, we describe the sensitization by microtubule-interfering agents (MIAs) to TRAIL-induced apoptosis in breast tumor cells through a mitotic arrest and c-Jun N-terminal kinase (JNK)-dependent mechanism. MIA treatment resulted in BubR1-dependent mitotic arrest leading to the sustained activation of JNK and the proteasome-mediated downregulation of cellular FLICE-inhibitory protein (cFLIP) and myeloid cell leukemia-1 (Mcl-1) expression. The JNK inhibitor SP600125 abrogated MIA-induced mitotic arrest and downregulation of cFLIP and Mcl-1 and reduced the apoptosis caused by the combination of MIAs and TRAIL. Silencing of cFLIP and Mcl-1 expression by RNA interference resulted in a marked sensitization to TRAIL-induced apoptosis. Furthermore, in FLIP-overexpressing cells, MIA-induced sensitization to TRAIL-activated apoptosis was markedly reduced. In summary, our results show that mitotic arrest imposed by MIAs activates JNK and facilitates TRAIL-induced activation of an apoptotic pathway in breast tumor cells by promoting the proteasome-mediated degradation of cFLIP and Mcl-1.
Similar content being viewed by others


Main
Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)/APO-2 L, a member of the tumor necrosis factor (TNF) gene superfamily, induces apoptosis on binding to the death domain (DD)-containing receptors TRAIL-R1/DR4 and TRAIL-R2/DR5.1 As TRAIL induces apoptosis selectively in transformed cells of diverse origin but not in most normal cells in vitro, it is an attractive candidate for antitumor therapies.2, 3, 4 In the western world, breast cancer is the most common neoplasia among women, emphasizing the importance of developing effective treatments. Despite the fact that many cancer cells are sensitive to TRAIL-induced apoptosis, a number of them, and in particular breast cancer cells, are resistant to TRAIL.5 In these cases, a combination therapy using chemotherapeutic agents and TRAIL may be more suitable than using TRAIL as a single agent. Recently, we and others have reported that interferon-γ, DNA-damaging drugs, ionizing radiation and cyclin-dependent kinase inhibitors can sensitize breast cancer cells to TRAIL-induced apoptosis.6, 7, 8, 9
On binding to its proapoptotic receptors, TRAIL induces the formation of the death-inducing signaling complex (DISC), recruiting the dual adaptor Fas-associated death domain (FADD) molecule through its DD. In turn, this complex recruits the initiator caspase-8 through its death effector domain (DED),10 which is activated at the DISC by oligomerization. The processing and activation of caspase-8 at the DISC stimulates an apoptotic cascade that provokes cell death.11, 12 The apoptotic signal from the DISC may be inhibited by the cellular FLICE-inhibitory protein (FLIP).13 In most cells, two alternatively spliced isoforms of cellular FLICE-inhibitory protein (cFLIP) exist: a caspase-8 homolog cFLIPL that lacks the amino acids critical for proteolytic caspase activity; and cFLIPS, which consists of only the DEDs.13 Although the role of cFLIP in apoptotic signaling remains controversial, there is strong evidence that it displays antiapoptotic activity.14, 15, 16 The expression of cFLIP varies in a cell type-specific manner and fluctuates in response to various stimuli. Although it can be transcriptionally controlled by the nuclear factor-κB (NF-κB) pathway,17 altered rates of proteasomal degradation also regulate its protein activity,18 making it a versatile inhibitor of the apoptotic responses mediated by death receptors.
Microtubules are dynamic polymers composed of α/β tubulin dimers that have important roles in various cellular functions.19 Disruption of the spindle with microtubule-interfering agents (MIAs) such as nocodazole or taxol activates the spindle assembly checkpoint causing the arrest of cells in mitosis,20 which may eventually lead to apoptosis.21 Although several MIAs are successfully being used in cancer chemotherapy,22 the development of resistance against these drugs and their inherent toxicities call for either the development of new agents with improved efficacy or the use of combination regimes with other anticancer treatments.23
Here, we report that TRAIL-resistant human breast cancer cells can be sensitized to TRAIL-induced apoptosis by exposure to MIAs. The molecular mechanisms underlying the effects of MIAs involve the induction of a mitotic arrest and c-Jun N-terminal kinase (JNK)-dependent proteasome-mediated decrease in cFLIP and Mcl-1 levels that facilitates the activation by TRAIL of the apoptotic pathway.
Results
MIAs sensitize human breast tumor cell lines to TRAIL-induced apoptosis
Drugs that affect microtubules dynamics are one of the most important types of antitumor agents. To determine whether MIAs sensitize breast tumor cells to the apoptotic ligand TRAIL, several breast tumor cell lines were treated either with the microtubule-depolymerizing agent nocodazole or with the microtubule-stabilizing drug taxol before the addition of TRAIL. In MDA-MB231 and SKBr-3 cell lines, MIAs caused a significant arrest at the G2/M phase of the cell cycle (Figure 1a). Interestingly, in these cell lines, MIAs markedly sensitize the cells to TRAIL-induced apoptosis (Figure 1b). Similar results were obtained when apoptosis was determined by visualization of nuclear fragmentation by 4′6-diamidino-2-phenylindole (DAPI) staining (results not shown). In contrast, in the BT-474 cell line, MIAs only partially arrested cells at the G2/M phase and did not induce sensitization to TRAIL (Figures 1a,b). These data were further confirmed in dose–response experiments (Figure 1c). MDA-MB231 cells were markedly sensitized to TRAIL-induced apoptosis by MIAs and this correlated with G2/M arrest. In contrast, BT-474 cells were very resistant to G2/M arrest and apoptosis sensitization by nocodazole even at doses 100 times higher than those needed to sensitize MDA-MB231 cells to TRAIL. However, BT-474 cells can be sensitized to TRAIL by other agents such as flavopiridol and cycloheximide (Supplementary Figure S1a), which indicates that BT-474 cells have an intact TRAIL apoptosis pathway, as previously reported by our group using FLIP siRNA.8 Caspase activation was required for the sensitization observed as the pan-caspase inhibitor Z-VAD-fmk completely abrogated the cell death induced by the combination of MIA and TRAIL (Figure 1d). One of the consequences of MIA-induced G2/M arrest is the detachment of cells from the extracellular matrix. Furthermore, cell detachment from the extracellular matrix may increase the susceptibility of breast tumor cells to TRAIL.24 To further explore this issue, MDA-MB231 cells were seeded in poly-HEMA-coated tissue culture dishes to prevent cell attachment and apoptosis was assessed following incubation with TRAIL. Results shown in Figure 1e indicate that cell detachment is not sufficient to sensitize these cells to TRAIL-induced apoptosis.


MIAs sensitize human breast tumor cells to TRAIL-induced apoptosis. (a, b) Different breast tumor cells were incubated for 15 h in complete medium without or with nocodazole (0.4 μg/ml) or taxol (10 μM) before the addition of TRAIL. Apoptosis was measured 6 h after the addition of TRAIL (500 ng/ml) as the percentage of cells with sub-G1 DNA content, as described in Materials and Methods. Error bars represent S.D. from three independent experiments. **P<0.01. (c) Cells were treated for 15 h with a range of concentrations of either nocodazole or taxol before incubation with or without TRAIL for 6 h. Quantitative analysis of cell cycle and apoptosis was determined as described in Materials and Methods. Error bars represent S.D. from three independent experiments. (d) MDA-MB231 cells incubated in the presence or absence of nocodazole (0.4 μg/ml) for 15 h were treated with or without Z-VAD-fmk (50 μM) for 1 h and subsequently exposed to TRAIL (500 ng/ml) for 6 h in the same culture media. Apoptosis was measured as described in Materials and Methods. Error bars represent S.D. from three independent experiments. (e) MDA-MB231 cells were seeded on regular culture plates and treated with nocodazole for 15 h (nocodazole) or on poly-HEMA-coated dishes for 24 h (poly-HEMA). Following these incubations, cells were treated without or with TRAIL (500 ng/ml) for 6 h and apoptosis was assessed as the percentage of cells with sub-G1 DNA content. Error bars represent S.D. from three independent experiments
To get further insight into the mechanism of sensitization to TRAIL-induced apoptosis promoted by MIA in breast tumor cells, we examined different biochemical events that occur on TRAIL binding to its receptors at the cell surface. TRAIL initiates apoptosis by inducing the recruitment of the adapter molecule FADD to the apoptotic TRAIL receptors and the subsequent engagement and activation of procaspase-8.11, 12 We determined whether caspase-8 was activated in MDA-MB231 cells by analyzing the processing of the pro-caspase to the 43/41 kDa intermediate proteolytic fragments, and through the generation of the mature p18 caspase-8 subunits on TRAIL stimulation. Accordingly, TRAIL-induced activation of caspase-8 was clearly enhanced by pretreatment with nocodazole or taxol (Figure 2a and Supplementary Figure S1b). Activation of caspase-8 leads to the processing of its substrate BID, generating a 15-kDa fragment that translocates to the mitochondria.25 Data shown in Figure 2a show that only in nocodazole-pretreated cells was TRAIL capable of inducing a decrease in the level of intact Bid and the generation of truncated BID (tBID). Moreover, on death receptor activation, the cytoplasmic protein Bax migrates to the mitochondria, where it cooperates with tBID in the release of cytochrome c (Cyt c).26 Thus, we examined the activation of the mitochondria-controlled apoptotic pathway by TRAIL in nocodazole-treated MDA-MB-231 cells by determining the release of Cyt c from the mitochondria. Results shown in Figure 2a indicate the presence of Cyt c in cytosolic extracts from MDA-MB-231 cells on pretreatment with nocodazole and TRAIL receptor ligation. This effect was associated with the loss of Cyt c levels in the mitochondria-containing membrane fraction of digitonin-lysed MDA-MB-231 cells. Furthermore, processing of procaspase-9 and complete activation of caspase-3 were only observed in cells treated with MIAs and TRAIL (Figure 2a and Supplementary Figure S1b).

MIAs facilitate the activation by TRAIL of a mitochondria-operated apoptotic pathway. (a) MDA-MB231 cells were incubated with nocodazole (0.4 μg/ml) for 15 h before treatment with TRAIL (500 ng/ml) for 6 h. Bid cleavage, caspase-9 processing and caspase-8/caspase-3 activation were assessed by immunoblotting. Asterisk indicates an unspecific band detected with caspase-9 antibody. Tubulin was used as a protein loading control. Release of cytochrome c from mitochondria was assessed by western blotting as described in Materials and Methods. (b) Cells were incubated with nocodazole for 15 h and cell surface expression of TRAIL receptors was determined by flow cytometry with antibodies against TRAIL receptors (solid line) or no antibody (dotted line) as described in Materials and Methods. Results are representative of at least two independent experiments
Several treatments have been shown to upregulate the expression of the TRAIL death receptors, resulting in enhanced TRAIL-induced apoptosis.27 We therefore examined the effect of nocodazole on the surface expression of TRAIL death and decoy receptors by flow cytometry (Figure 2b). MDA-MB231 cells only express TRAIL-R2 and -R4 on the cell surface and the expression of these TRAIL receptors at the cell membrane was not significantly altered by exposure to nocodazole (Figure 2b). Hence, nocodazole does not appear to sensitize MDA-MB231 cells to TRAIL-induced apoptosis by modulating the surface expression of TRAIL receptors.
Treatment of breast tumor cells with MIAs alters the cellular levels of different apoptosis-related proteins
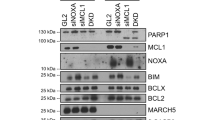
To elucidate the molecular mechanisms underlying the sensitization to TRAIL-induced apoptosis by MIAs in breast tumor cells, we determined the expression levels of different pro- and antiapoptotic proteins, which are known to be relevant to mechanisms of TRAIL signaling, in MDA-MB231 cells treated either with nocodazole or with taxol. After treatment with MIAs for times up to 24 h, no changes in the levels of Bid, Bax, procaspase-8, procaspase-9, procaspase-3, procaspase-2 and XIAP were observed (data not shown). Interestingly, we observed a marked decrease in the cellular levels of the antiapoptotic Bcl-2 family member Mcl-1 in cells treated with nocodazole (Figure 3a) or taxol (Figure 3b), with barely detectable levels observed after 24 h. Likewise, treatment with nocodazole or taxol caused an important decline in the levels of the cFLIP isoforms FLIPL and FLIPS, inhibitors of caspase-8 activation at the DISC in death receptor-mediated apoptosis (Figures 3a and b). In addition, treatment of MDA-MB231 cells with MIAs resulted in changes in expression levels and/or the electrophoretic mobility of the apoptosis-related proteins Bim, FADD and Bcl-2 (data not shown), which is indicative of alterations in their phosphorylation pattern.28, 29, 30 Interestingly, none of these effects of MIAs were observed in the BT-474 cell line (Figure 3c), refractory to MIA-induced sensitization to TRAIL-activated apoptosis (Figures 1b and c).

Protein changes in MIAs-treated breast tumor cells. MDA-MB231 cells were incubated with nocodazole (a) or taxol (b) for the indicated times. Expression of apoptosis-related proteins was determined by western blotting with specific antibodies. In (c) BT-474 cells were incubated with nocodazole for the indicated times and protein expression determined by western blotting. GAPDH expression was used as a protein loading control. Results are representative of at least three independent experiments
MIA treatment causes the proteasome-mediated degradation of apoptosis-related proteins
Mcl-1 and cFLIP are short half-life proteins that are subject to constitutive polyubiquitination and subsequent degradation by the proteasome.31, 32 To further explore the mechanism by which MIAs promoted the sensitization to TRAIL in breast tumor cells, we first examined the role of the proteasome in the loss of Mcl-1 and cFLIP proteins observed on MIAs treatment. As shown in Figure 4a, pretreatment of MDA-MB231 cells with the proteasome inhibitor MG-132 completely prevented the disappearance of both Mcl-1 and cFLIP proteins induced following 24 h exposure to either nocodazole or taxol, suggesting a role for the ubiquitin–proteasome pathway in the disappearance of these antiapoptotic proteins on MIA treatment. It was recently reported that the BH3-only E3 ubiquitin ligase Mule/HectH9/ARF-BP1/HUWE1 is responsible for the polyubiquitination of Mcl-1 in different cell types.33 To examine the role of Mule in the degradation of Mcl-1 in breast tumor cells treated with MIAs, we eliminated Mule expression by an RNA interference approach and examined the effect of nocodazole treatment on the cellular levels of Mcl-1 protein. Data shown in Figure 4b show that Mule silencing prevented the loss of Mcl-1 by nocodazole treatment in MDA-MB231 cells. In contrast, downregulation of cFLIP (Figure 4b) and Bim (data not shown) levels by nocodazole was not affected by Mule siRNA. The E3 ubiquitin ligase Itch has recently been reported to mediate the ubiquitination and proteasomal degradation of cFLIP in TNF α-induced apoptosis.34 To determine whether Itch was involved in the proteasomal degradation of both FLIPL and FLIPS in breast tumor cells treated with nocodazole, we performed RNA interference experiments with an siRNA specific for Itch. Downregulation of the Itch protein by siRNA did not prevent the proteasomal degradation of FLIPL and FLIPS in MDA-MB231 cells treated with nocodazole (Figure 4c). We are currently characterizing the E3 ubiquitin ligase responsible for the proteasomal degradation of cFLIP in MIA-induced sensitization to TRAIL-activated apoptosis in breast tumor cells.

MIAs treatment causes the proteasome-mediated degradation of cFLIP and Mcl-1. (a) MDA-MB231 cells were treated without or with MG-132 (0.5 μM) for 30 min before incubation in the presence or absence of nocodazole (top panel) or taxol (bottom panel) for 24 h. Protein expression was determined by western blotting. GAPDH expression was used as a protein loading control. Results are representative of at least three independent experiments. (b) MDA-MB231 cells were transfected with Mule siRNA or control siRNA and Mule mRNA levels were determined by RT-PCR as described in Materials and Methods. The RT-PCR product of β-actin was used as a control for mRNA input. After 48 h, transfection medium was replaced with regular medium and cultures were treated without or with nocodazole (0.4 μg/ml) for 24 h. Expression levels of Mcl-1 and cFLIP were determined by western blotting. GAPDH expression was used as a protein loading control. Results are representative of at least three independent experiments. (c) MDA-MB231 cells were transfected for 48 h with Itch siRNA or control siRNA. After this incubation, transfection medium was replaced with regular medium and cultures were treated without or with nocodazole (0.4 μg/ml) for 24 h. Expression levels of Itch and cFLIP were determined by western blotting. GAPDH expression was used as a protein loading control. Results are representative of at least three independent experiments
cFLIP and Mcl-1 downregulation cooperate in the sensitization of breast tumor cells to TRAIL
Cellular levels of both cFLIP and Mcl-1 have been reported to have an important role in the resistance of tumor cells to TRAIL.8, 35 To ascertain whether downregulation of these antiapoptotic proteins by nocodazole was an important event in the sensitization of breast tumor cells to TRAIL, we performed RNA interference experiments with siRNA oligonucleotides specific for cFLIP or Mcl-1. Thus, we tested whether the specific silencing of cFLIP expression by siRNA has a similar effect on TRAIL-induced apoptosis. In MDA-MB231 cells, the levels of both cFLIPL and cFLIPS were substantially reduced by a specific siRNA (Figure 5a), whereas a similar concentration of a scrambled control siRNA did not modify cFLIP protein expression. Most importantly, silencing of cFLIP expression resulted in a clear sensitization to TRAIL-induced apoptosis (Figure 5b) as previously reported.8 These data correlated well with the effects of nocodazole and support the hypothesis that downregulation of cFLIP expression by MIAs is critical for the sensitization to TRAIL-induced apoptosis in breast tumor cells. In contrast, silencing Mcl-1 expression by specific siRNA (Figure 5a) has a very weak effect on TRAIL-induced apoptosis (Figure 5b). Interestingly, simultaneous knockdown of both cFLIP and Mcl-1 by siRNA oligonucleotides significantly augmented the sensitization caused by either siRNA alone (Figure 5b).

cFLIP and Mcl-1 down-regulation co-operate in the sensitization of breast tumor cells to TRAIL. (a) Cell lysates from MDA-MB231 cells transfected with siRNAs (50 nM) for 48 h were analyzed for protein expression as described in Materials and Methods. (b) MDA-MB231 cells were transfected with siRNAs for 48 h and treated with TRAIL (500 ng/ml) for 6 h. Apoptosis was assessed as the percentage of cells with sub-G1 DNA content. Error bars represent S.D. from three independent experiments. *P<0.05. (c) Mock-transfected and MDA-MB231 cells transfected with the pCR3.V64-Met-Flag-FLIPL vector were treated with or without nocodazole (0.4 μg/ml) for 15 h before the addition of TRAIL (500 ng/ml). Apoptosis was measured 6 h after the addition of TRAIL as the percentage of cells with sub-G1 DNA content, as described in Materials and Methods
To further substantiate the role of cFLIP in the sensitization observed, we used MDA-MB231 cells overexpressing cFLIPL and determined the effect of nocodazole treatment on apoptosis by TRAIL. Results shown in Figure 5c show that cells overexpressing cFLIPL were clearly more resistant to nocodazole-induced sensitization to TRAIL apoptosis than cells expressing normal cFLIP levels.
Mitotic arrest imposed by MIAs activates JNK and facilitates TRAIL-induced apoptosis
Accumulated evidence indicates that altering microtubule dynamics by treatment with MIAs activates the JNK/SAPK pathway in a variety of cells.36 On the other hand, the activity of the JNK/SAPK pathway may regulate the sensitivity of human tumor cells to TRAIL.37 To further investigate the mechanism of MIA-induced sensitization to TRAIL-activated apoptosis in breast tumor cells, we analyzed the activation of the JNK/SAPK pathway in MDA-MB231 cells treated with nocodazole or taxol. We first examined the effect of MIAs on the activation of JNK/SAPK by measuring the dual phosphorylation of JNK/SAPK (Thr183/Tyr185) as a function of time. Interestingly, incubation of cells with nocodazole or taxol caused a sustained activation of JNK/SAPK that persisted up to 24 h, the latest time examined in our studies (Figure 6a). Moreover, incubation of cells with MIAs in the presence of the JNK/SAPK inhibitor SP60012538 completely abrogated the phosphorylation observed. To find out the role of JNK/SAPK activation by MIAs in the sensitization to TRAIL, we next determined the effect of SP600125 on the changes in pro- and antiapoptotic proteins observed in cells treated with MIAs. Most importantly, the presence of SP600125 in the incubation medium completely prevented the loss of FLIPL, FLIPS and Mcl-1 proteins induced by MIA treatment in MDA-MB231 cells (Figures 6b and c). Furthermore, the changes in the electrophoretic mobility of the apoptosis-related proteins FADD, Bim and Bcl-2 observed in cells treated with MIAs were markedly inhibited in the presence of the JNK/SAPK inhibitor (results not shown).

Activation of JNK/SAPK by MIAs is required for the proteasomal degradation of cFLIP and Mcl-1 and the sensitization to TRAIL-induced apoptosis. (a) MDA-MB231 cells were incubated with or without nocodazole (0.4 μg/ml, top panel) or taxol (10 μM, bottom panel) in the presence or absence of SP600125 (25 μM) for the indicated times. JNK activation was determined by western blotting with an antibody against p-JNK. GAPDH expression was used as a protein loading control. Results are representative of at least three independent experiments. (b, c) MDA-MB231 cells were treated as described in Figure 6a. Expression levels of the various apoptosis-related proteins were determined by western blotting with specific antibodies. GAPDH and tubulin expression were used as protein loading controls. Results are representative of at least three independent experiments. (d, e) MDA-MB231 cells were incubated for 15 h in the presence or absence of SP600125 and nocodazole (d) or taxol (e) before treatment with TRAIL for 4 h. Apoptosis was assessed as described in Materials and Methods. Error bars represent S.D. from three independent experiments. (f) MDA-MB231 cells were incubated for 15 h in the presence or absence of nocodazole (0.4 μg/ml), flavopiridol (100 nM) or cycloheximide (1 μg/ml), with or without SP600125 (25 μM). Following these incubations, cells were treated without or with TRAIL (500 ng/ml) for 6 h and apoptosis was assessed as the percentage of cells with sub-G1 DNA content. Results are the average and range of two independent experiments in duplicate
We next examined the effect of SP600125 on apoptosis induced by incubation of cells with either nocodazole or taxol and subsequently treated with TRAIL. Results in Figures 6d and e show that SP600125 drastically reduced the apoptosis elicited by the combination of MIA and TRAIL, consistent with a role of JNK/SAPK activation in the regulation of cFLIP and Mcl-1 degradation by the proteasome- and TRAIL-induced apoptosis. Interestingly, the inhibitor of JNK/SAPK specifically inhibited the apoptosis induced by MIAs and TRAIL but not by other combination treatments (flavopiridol, cycloheximide) with TRAIL, which do not cause a sustained activation of the JNK/SAPK pathway (Figure 6f and results not shown).
To further investigate the mechanism of MIA-induced sensitization to TRAIL, we examined the importance of the mitotic checkpoint induction in the sensitization process. Mitotic checkpoint activation was assessed by monitoring the levels of BubR1 and histone H3 phosphorylation in cells treated with nocodazole.39 As shown in Figure 7a, nocodazole treatment resulted in a shift of BubR1 to its hyperphosphorylated form and a marked phosphorylation of histone H3 in MDA-MB231 cells. Other treatments such as flavopiridol or cycloheximide, which cause SP600125-insensitive sensitization to TRAIL, did not induce mitotic arrest (Supplementary Figure S2). Interestingly, BT-474 cells were clearly refractory to nocodazole-induced activation of these mitotic checkpoint-related events, thus confirming the cell cycle data shown in Figure 1a that indicated inefficient G2/M arrest in these cells following MIAs treatment. It has been reported that the JNK inhibitor SP600125 could abrogate spindle assembly checkpoint function in human cells by inhibiting the activity of the mitotic checkpoint kinase monopolar spindle 1 (Mps1).40 We have investigated this issue by monitoring histone H3 phosphorylation in MDA-MB231 cells treated with nocodazole. Histone H3 phosphorylation induced by nocodazole was markedly reduced in cells treated with SP600125 (Figure 7b), thus indicating that cells did not enter mitotic arrest under these conditions. It has been described that siRNA knockdown of BubR1 results in mitosis acceleration and spindle assembly checkpoint abrogation,41 thus indicating that the control of BubR1 levels might be an essential regulatory step in checkpoint activity. To further study the role of the mitotic arrest imposed by MIAs in the activation of JNK and the regulation of TRAIL-induced apoptosis, we performed siRNA experiments to knock down BubR1 expression levels in MDA-MB231 cells. Silencing of BubR1 expression drastically reduced histone H3 phosphorylation (Figure 7c) and rounding up of cells (not shown) induced by nocodazole, indicating an efficient bypass of the mitotic arrest imposed by MIAs. Interestingly, BubR1 knockdown clearly inhibited the activation of JNK and the downregulation of Mcl-1 and FLIP proteins by nocodazole. Furthermore, silencing of BubR1 expression significantly reduced the apoptosis elicited by the combination of MIA and TRAIL (Figure 7d), consistent with a role of mitotic arrest in TRAIL-induced apoptosis.

Mitotic arrest induced by MIAs activates JNK and facilitates TRAIL-induced apoptosis. (a) Breast tumor cells were incubated in complete medium with or without nocodazole (0.4 μg/ml) for 24 h. After this incubation, BubR1 electrophoretic mobility shift and histone H3 phosphorylation were determined by western blotting. (b) MDA-MB231 cells were incubated with or without nocodazole (0.4 μg/ml) in the presence or absence of SP600125 (25 μM) for 24 h. JNK activation and histone H3 phosphorylation were determined by western blotting with specific antibodies. GAPDH expression was used as a protein loading control. (c) MDA-MB231 cells were transfected for 48 h with BubR1 siRNA (50 nM) or control siRNA. After this incubation, transfection medium was replaced with regular medium and cultures were treated without or with nocodazole for 24 h. Expression levels of proteins were determined by western blotting. GAPDH expression was used as a protein loading control. Results are representative of three independent experiments. (d) MDA-MB231 transfected with BubR1 siRNA as described in (c) were incubated in complete medium in the presence or absence of nocodazole for 15 h before treatment with or without TRAIL for 6 h. Apoptosis was assessed as described in Materials and Methods. Error bars represent S.D. from three independent experiments, *P<0.05
Discussion
TRAIL induces selective cell death in human tumor cells, sparing most untransformed cells. On the basis of these findings, phase 1 and 2 clinical trials of TRAIL and agonistic TRAIL receptor antibodies for the treatment of cancer are currently under way.42 However, resistance to TRAIL is not uncommon in primary tumors and tumor cell lines, similar to those of the breast. Hence, sensitization of cells to TRAIL-induced apoptosis through different strategies would augment the therapeutic potential of TRAIL and combination treatments have been implemented to facilitate TRAIL apoptotic signaling.43 Microtubules are a major component of the cytoskeleton and have important roles in many cellular functions. They are a crucial component of the cell division machinery, and as such are highly attractive targets for anticancer drug design. In this work, we have examined how altering microtubules dynamics may increase the sensitivity of breast tumor cells to the death ligand TRAIL. Although an increase in the total expression of proapoptotic TRAIL receptor proteins by taxol and other chemotherapeutic agents has been reported,44, 45 we do not observe an upregulation of TRAIL-R1 or TRAIL-R2 at the cell surface in breast tumor cells treated with MIAs. Hence, the step(s) involved in the sensitization to TRAIL should reside downstream of ligand binding to the receptor.
We have been studying the resistance of human breast tumor cells to TRAIL and previously reported that the formation of DISC is a common target for different sensitizing regimes.8, 46 Furthermore, resistance of tumor cells to TRAIL can be overcome by treatment with metabolic inhibitors such as cycloheximide or actinomycin D.47 These compounds induce a strong downregulation of cFLIP expression.48 In this regard, a potential mechanism involved in the regulation of TRAIL sensitivity is the modulation of the expression of the caspase-8 inhibitors cFLIPL and cFLIPS,13 proteins with very short half-lives18 that are expressed at high levels in breast tumors.49 Although enhancement of TRAIL-induced caspase-8 activation and apoptosis does not always correlate with cFLIP levels,46 it has been shown that different treatments can induce downregulation of cFLIP and subsequent sensitization to TRAIL-induced apoptosis.50, 51 In our study, treatment of breast tumor cells with MIAs induced a marked downregulation of both cFLIPL and cFLIPS proteins. It is well documented that cFLIP can be transcriptionally regulated through the NF-κB pathway.17 However, we did not observe a decrease in cFLIP mRNA in breast tumor cells treated with MIAs at concentrations that caused maximal sensitization to TRAIL (results not shown), thus excluding the transcriptional regulation of cFLIP as the major target for the inhibitory action of MIAs on cFLIP levels. cFLIP protein levels can be downregulated by proteasomal degradation18 and this modulation may lead to TRAIL sensitization.52 Our results show that in breast tumor cells MIA treatment leads to the proteasome-mediated degradation of both cFLIP isoforms through a mitotic arrest and JNK-dependent mechanism. Inhibition of cytoskeletal dynamics results in the activation of signal transduction pathways. MIAs have been shown to induce a sustained activation of JNK in a variety of human cells through both Ras and apoptosis signal-regulating kinase (ASK1)-dependent pathways.36 MEK1 kinase (MEKK1) is also required for the activation of JNK following exposure of cells to MIAs.53 In this study, we have shown that inhibiting microtubule dynamics causes a sustained activation of the JNK and markedly sensitizes breast tumor cells to TRAIL-induced apoptosis. We have also shown that JNK activation by MIAs requires the spindle checkpoint machinery as it is markedly abrogated in cells depleted of the mitotic checkpoint kinase BubR1. Downregulation of cFLIP and sensitization to TRAIL-induced apoptosis correlated well with the activation of JNK by MIAs in the various breast tumor cell lines examined. Regarding the mechanism of cFLIP degradation, it has been shown that JNK activation by TNF-α reduces cFLIPL stability by a mechanism involving JNK-mediated phosphorylation and activation of the E3 ubiquitin ligase Itch, which ubiquitinates cFLIPL and induces its proteasomal degradation.34 Furthermore, in cisplatin-treated tumor cells, Itch forms a complex with cFLIP and p53 to promote cFLIP ubiquitination and degradation through the proteasome.54 However, our siRNA data suggest that Itch is not responsible for the proteasomal degradation of cFLIP in breast tumor cells treated with MIAs, indicating that other E3 ubiquitin ligases must be involved in the degradation of cFLIP proteins following inhibition of microtubule dynamics.
The role of Mcl-1 in the resistance of tumor cells to TRAIL-induced apoptosis is a controversial issue. It has been reported that downregulation of Mcl-1 expression either by RNA interference or by the multikinase inhibitor sorafenib sensitizes tumor cells to TRAIL-induced apoptosis.55, 56, 57 On the contrary, our results indicate that siRNA to Mcl-1 only weakly facilitates activation of apoptosis by TRAIL in breast tumor cells. These conflicting results suggest that Mcl-1 involvement in the resistance of cells to TRAIL might be cell type-dependent. Interestingly, simultaneous silencing of Mcl-1 and cFLIP by siRNA significantly enhances sensitization to apoptosis induced by siRNA alone, suggesting the cooperation between Mcl-1 and cFLIP in the mechanism of resistance to TRAIL in breast tumor cells. Hence, treatments that target cFLIP and Mcl-1 for proteasomal degradation, such as antimicrotubule agents, may be therapeutically relevant tools to fight cancer in combination with agonist TRAIL receptor antibodies or recombinant TRAIL. Once activated by TRAIL, caspases may further contribute to tumor cell death by MIAs through the degradation of protein components of the checkpoint machinery.58
We show for the first time that MIAs induce the proteasomal degradation of both apoptosis inhibitors by a mechanism involving the spindle checkpoint machinery and the sustained activation of JNK. Proteasome-mediated degradation of Mcl-1 is regulated by phosphorylation.59 In growth factor-dependent cells, Mcl-1 protein levels are regulated by growth factor signaling at the level of protein half-life through phosphorylation by GSK-3.60 It has been suggested that JNK could be responsible for the priming phosphorylation of Mcl-161 necessary for the docking of GSK-3 to Mcl-1. It remains to be determined whether in breast tumor cells treated with MIAs, GSK-3 is also involved in the proteasome-mediated degradation of Mcl-1. Mcl-1 is a target for the BH3-containing E3 ubiquitin ligase Mule (also known as Huwe1, UreB1, ARF-BP1, Lasu1 and HectH9).33 Our results indicate that Mule is required for the proteasome-mediated degradation of Mcl-1 in breast tumor cells treated with MIAs. It will be important to determine whether phosphorylation of Mcl-1 by JNK and possibly GSK-3 may regulate the direct recognition of Mcl-1 by the HECT-family member Mule.
JNK activation by MIAs also posttranslationally affects other apoptosis-related proteins such as Bim, Bcl-2 and the death receptor adaptor FADD. Although the role of these modifications on TRAIL-induced apoptosis is not clear, we cannot exclude that they may contribute to the sensitization observed. JNK-mediated phosphorylation of Bim has been proposed to mediate sensitization to TRAIL in other tumor cells.62 Furthermore, it has been shown that under nonapoptotic conditions, Bim is sequestered by Mcl-1 and loss in Mcl-1 expression significantly enhances the mitochondrial apoptotic response to TRAIL mediated by freed Bim.63 On the other hand, it has been reported that Bcl-2 phosphorylation by JNK is functionally linked to apoptosis induced by taxol.64 Similarly, FADD phosphorylation through a JNK-dependent mechanism is observed in tumor cells treated with taxol and is closely associated with chemosensitivity.65 However, the role of Bcl-2 and FADD phosphorylation by JNK in the regulation of TRAIL-induced apoptosis remains to be elucidated.
In conclusion, it is likely that inhibition of microtubule function by MIAs sensitizes tumor cells to TRAIL by reducing the cellular levels of apoptosis inhibitors, which leads to the complete activation of the apoptotic machinery as we have shown in this work. The present findings provide further support for the general strategy of combining TRAIL and agents that affect the mitotic checkpoint in anticancer strategies.
Materials and Methods
Reagents and antibodies
MG-132, nocodazole and taxol were obtained from Sigma Chemical Corp. (St. Louis, MO, USA). Soluble human His-tagged recombinant TRAIL was generated in our laboratory as described.66 Antihuman TRAIL Receptor R1, R2, R3 and R4 antibodies and anti-cFLIP monoclonal antibody (NF6) were from Alexis Corp. (San Diego, CA, USA). Cyt c, BubR1, Itch and FADD antibodies were from BD Bioscience (Erembodegem, Belgium). The monoclonal antibody to α-tubulin was purchased from Sigma Chemical Corp. Antibodies against GAPDH, H3 histone, phospho-H3 histone and Mcl-1 (S-19) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Mouse anti-Bcl-2 mAb was purchased from DAKO (Glostrup, Denmark). Antibodies to Bim and phospho-JNK were from Calbiochem (San Diego, CA, USA) and Cell Signaling (Denvers, MA, USA), respectively. Anti-caspase 8 was a gift from Dr. Gerald Cohen (Leicester University, Leicester, UK). Rabbit anti-Bid polyclonal antibody was generously provided by Dr. X. Wang (Howard Hughes Medical Institute, Dallas, TX, USA). Horseradish peroxidase or FITC-conjugated, goat anti-mouse and goat anti-rabbit secondary antibodies were obtained from DAKO (Cambridge, UK). Z-VAD-fmk was from Bachem AG (Bachem, Bubendorf, Switzerland). Cleaved caspase-3 polyclonal antibody was from Cell Signaling (Danvers, MA, USA). Caspase 9 monoclonal antibody was purchased from MBL International (Woburn, MA, USA).
Cell culture
The human tumor cell line MDA-MB231 was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 40 μg/ml gentamycin. SKBr-3 and BT-474 were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 40 μg/ml gentamycin. The cells were maintained at 37°C in a humidified 5% CO2, 95% air incubator. A stable cell line overexpressing cFLIPL was generated on transfection of MDA-MB231 cells with pCR3.V64-Met-Flag-FLIPL (a kind donation from Dr. J. Tschopp, University of Lausanne, Lausanne, Switzerland) by electroporation. Mock-transfected cells and cells overexpressing FLIPL were selected in culture medium with 1 mg/ml G418 (Sigma Chemical Co., St. Louis, MO, USA) and analyzed for the expression cFLIPL by western blot. For cultures of MDA-MB231 cells in suspension, 6-well plates were coated with a film of poly-HEMA following published procedures.67
Analysis of apoptosis
Cells (3 × 105 per well) were treated in six-well plates as indicated in the figure legends. After treatment, hypodiploid apoptotic cells were detected by flow cytometry according to published procedures.9 Basically, cells were washed with phosphate-buffered saline (PBS), fixed in 70% cold ethanol and then stained with propidium iodide while treating with RNAse. Quantitative analysis of the cell cycle and sub-G1 cells was carried out in a FACSCalibur cytometer using the Cell Quest software (Becton Dickinson, Mountain View, CA, USA).
RT-PCR
RT-PCR analyses were performed according to standard protocols. Total RNA was isolated from cells with Trizol reagent (Life Technologies, Inc., Grand Island, NY, USA) as recommended by the supplier. cDNA was synthesized from 2 μg of total RNA using an RNA PCR kit (Perkin-Elmer, Indianapolis, IN, USA) with the supplied Random Hexamers under the conditions described by the manufacturer. PCR reactions were performed using specific primers for Mule (forward: 5′-GGGGTTATGACCCAAGAGGT-3′ and reverse: 5′-CCCATCTCGAGACTCCTCTG-3′) and actin.
Immunoblot analysis of proteins
After detachment with trypsin, cells (3 × 105) were washed with PBS, and protein content was measured following cell lysis using the Bradford reagent (Bio-Rad Laboratories, Hercules, CA, USA) before adding Laemmli sample buffer. Samples were sonicated, and proteins were resolved on SDS-polyacrylamide minigels and detected as described previously.9
Measurements of Cyt c release
Cells (3 × 105 per well) were treated in six-well plates as indicated in the figure legends. After treatment, cells were detached from the plate with RPMI/EDTA and trypsin, washed with PBS and lysed in 30 μl ice-cold lysis buffer (80 mM KCl, 250 mM sucrose, 500 μg/ml digitonin and protease inhibitors in PBS). For measurements of Cyt c release from mitochondria, cell lysates were centrifuged for 5 min at 10 000 g to separate the supernatant (cytosolic fraction) and pellet (mitochondria-containing fraction). The amount of protein in each fraction was determined by the Bradford protein assay (BIO-RAD, Hertfordshire, UK). Proteins from the supernatant and pellet were mixed with Laemmli buffer and resolved on SDS-12% PAGE minigels. Cyt c was determined by western blot analysis.
Analysis of TRAIL receptors by flow cytometry
MDA-MB231 cells were detached with RPMI 1640/EDTA, washed in ice-cold PBS and resuspended in PBS. Cells were then labeled with anti-TRAIL receptor antibodies (5 μg/ml) or no antibody (negative control), and then incubated with goat anti-mouse FITC-conjugated antibody F(ab′)2 fragment. Labeled cells were analyzed by flow cytometry using the CellQuest software (Becton Dickinson, Mountain View, CA, USA).
RNA interference
siRNAs against cFLIP (5′-GGGACCUUCUGGAUAUUUUtt-3′), Mcl-1 (5′-GAAACGCGGUAAUCGGACUtt-3′), Mule (5′-GAGUUUGGAGUUUGUGAAGtt-3′), BubR1 (5′-CUUCACUUGCGGAGAACAUtt-3′), Itch (5′-AAGUGCUUCUCAGAAUGAUGAtt-3′) and nontargeting scrambled siRNA were synthesized by Sigma Proligo (St. Louis, MO, USA). Cells were transfected with siRNAs using DharmaFECT-1 (Dharmacon, Lafayette, CO, USA) as described by the manufacturer. After 48 h, the transfection medium was replaced with regular medium before further analysis.
Statistical analysis
Data are presented as mean±S.E.M. of at least three independent experiments. Differences between treatment groups were determined by using Student's t-test. Statistical significance was evaluated by calculating P-values. P-values below 0.05 were considered significant.
Abbreviations
- cFLIP:
-
cellular FLICE-inhibitory protein
- Mcl-1:
-
myeloid cell leukemia-1
- JNK:
-
c-Jun N-terminal kinase
- TRAIL:
-
tumor necrosis factor-related apoptosis-inducing ligand
- TNF:
-
tumor necrosis factor
- DISC:
-
death-inducing signaling complex
- FADD:
-
Fas-associated death domain
- Cyt c:
-
cytochrome c
- DD:
-
death domain
- DED:
-
death effector domain
- DAPI:
-
4′6-diamidino-2-phenylindole
References
Ashkenazi A, Dixit VM . Death receptors: signaling and modulation. Science 1998; 281: 1305–1308.
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 1999; 104: 155–162.
Reed JC . Drug insight: cancer therapy strategies based on restoration of endogenous cell death mechanisms. Nat Clin Pract Oncol 2006; 3: 388–398.
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 1999; 5: 157–163.
Keane MM, Ettenberg SA, Nau MM, Russell EK, Lipkowitz S . Chemotherapy augments TRAIL-induced apoptosis in breast cell lines. Cancer Res 1999; 59: 734–741.
Chinnaiyan AM, Prasad U, Shankar S, Hamstra DA, Shanaiah M, Chenevert TL et al. Combined effect of tumor necrosis factor-related apoptosis-inducing ligand and ionizing radiation in breast cancer therapy. Proc Natl Acad Sci USA 2000; 97: 1754–1759.
Ohtsuka T, Buchsbaum D, Oliver P, Makhija S, Kimberly R, Zhou T . Synergistic induction of tumor cell apoptosis by death receptor antibody and chemotherapy agent through JNK/p38 and mitochondrial death pathway. Oncogene 2003; 22: 2034–2044.
Palacios C, Yerbes R, Lopez-Rivas A . Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res 2006; 66: 8858–8869.
Ruiz-Ruiz C, Lopez-Rivas A . Mitochondria-dependent and -independent mechanisms in tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis are both regulated by interferon-gamma in human breast tumour cells. Biochem J 2002; 365: 825–832.
Sprick MR, Weigand MA, Rieser E, Rauch CT, Juo P, Blenis J et al. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000; 12: 599–609.
Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P et al. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat Cell Biol 2000; 2: 241–253.
Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A . Apo2 L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000; 12: 611–620.
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190–195.
Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S . Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem 2001; 276: 20633–20640.
Sharp DA, Lawrence DA, Ashkenazi A . Selective knockdown of the long variant of cellular FLICE inhibitory protein augments death receptor-mediated caspase-8 activation and apoptosis. J Biol Chem 2005; 280: 19401–19409.
Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 2000; 12: 633–642.
Kreuz S, Siegmund D, Scheurich P, Wajant H . NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol 2001; 21: 3964–3973.
Fukazawa T, Fujiwara T, Uno F, Teraishi F, Kadowaki Y, Itoshima T et al. Accelerated degradation of cellular FLIP protein through the ubiquitin–proteasome pathway in p53-mediated apoptosis of human cancer cells. Oncogene 2001; 20: 5225–5231.
Gelfand VI, Bershadsky AD . Microtubule dynamics: mechanism, regulation, and function. Annu Rev Cell Biol 1991; 7: 93–116.
Rudner AD, Murray AW . The spindle assembly checkpoint. Curr Opin Cell Biol 1996; 8: 773–780.
Sorger PK, Dobles M, Tournebize R, Hyman AA . Coupling cell division and cell death to microtubule dynamics. Curr Opin Cell Biol 1997; 9: 807–814.
Mollinedo F, Gajate C . Microtubules, microtubule-interfering agents and apoptosis. Apoptosis 2003; 8: 413–450.
Drukman S, Kavallaris M . Microtubule alterations and resistance to tubulin-binding agents (review). Int J Oncol 2002; 21: 621–628.
Goldberg GS, Jin Z, Ichikawa H, Naito A, Ohki M, El-Deiry WS et al. Global effects of anchorage on gene expression during mammary carcinoma cell growth reveal role of tumor necrosis factor-related apoptosis-inducing ligand in anoikis. Cancer Res 2001; 61: 1334–1337.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 1999; 144: 891–901.
Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL . Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL. Mol Cell Biol 2000; 20: 205–212.
Alappat EC, Feig C, Boyerinas B, Volkland J, Samuels M, Murmann AE et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Mol Cell 2005; 19: 321–332.
Haldar S, Basu A, Croce CM . Bcl2 is the guardian of microtubule integrity. Cancer Res 1997; 57: 229–233.
Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ . Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 2003; 278: 18811–18816.
Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev 2003; 17: 1475–1486.
Poukkula M, Kaunisto A, Hietakangas V, Denessiouk K, Katajamaki T, Johnson MS et al. Rapid turnover of c-FLIP short is determined by its unique C-terminal tail. J Biol Chem 2005; 280: 27345–27355.
Zhong Q, Gao W, Du F, Wang X . Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121: 1085–1095.
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006; 124: 601–613.
Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res 2004; 64: 3517–3524.
Wang TH, Wang HS, Ichijo H, Giannakakou P, Foster JS, Fojo T et al. Microtubule-interfering agents activate c-Jun N-terminal kinase/stress-activated protein kinase through both Ras and apoptosis signal-regulating kinase pathways. J Biol Chem 1998; 273: 4928–4936.
Herr I, Wilhelm D, Meyer E, Jeremias I, Angel P, Debatin KM . JNK/SAPK activity contributes to TRAIL-induced apoptosis. Cell Death Differ 1999; 6: 130–135.
Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 2001; 98: 13681–13686.
Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ . Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol 1999; 146: 941–954.
Schmidt M, Budirahardja Y, Klompmaker R, Medema RH . Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep 2005; 6: 866–872.
Meraldi P, Draviam VM, Sorger PK . Timing and checkpoints in the regulation of mitotic progression. Dev Cell 2004; 7: 45–60.
Ashkenazi A . Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug Discov 2008; 7: 1001–1012.
Johnstone RW, Frew AJ, Smyth MJ . The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer 2008; 8: 782–798.
Nimmanapalli R, Perkins CL, Orlando M, O’Bryan E, Nguyen D, Bhalla KN . Pretreatment with paclitaxel enhances apo-2 ligand/tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of prostate cancer cells by inducing death receptors 4 and 5 protein levels. Cancer Res 2001; 61: 759–763.
Singh TR, Shankar S, Chen X, Asim M, Srivastava RK . Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res 2003; 63: 5390–5400.
Munoz-Pinedo C, Ruiz-Ruiz C, Ruiz de Almodovar C, Palacios C, Lopez-Rivas A . Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspase-8 processing. J Biol Chem 2003; 278: 12759–12768.
Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G et al. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J Biol Chem 2000; 275: 24357–24366.
Fulda S, Meyer E, Debatin KM . Metabolic inhibitors sensitize for CD95 (APO-1/Fas)-induced apoptosis by down-regulating Fas-associated death domain-like interleukin 1-converting enzyme inhibitory protein expression. Cancer Res 2000; 60: 3947–3956.
Conticello C, Pedini F, Zeuner A, Patti M, Zerilli M, Stassi G et al. IL-4 protects tumor cells from anti-CD95 and chemotherapeutic agents via up-regulation of antiapoptotic proteins. J Immunol 2004; 172: 5467–5477.
Hietakangas V, Poukkula M, Heiskanen KM, Karvinen JT, Sistonen L, Eriksson JE . Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol Cell Biol 2003; 23: 1278–1291.
Kim Y, Suh N, Sporn M, Reed JC . An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J Biol Chem 2002; 277: 22320–22329.
Zhang S, Shen HM, Ong CN . Down-regulation of c-FLIP contributes to the sensitization effect of 3,3′-diindolylmethane on TRAIL-induced apoptosis in cancer cells. Mol Cancer Ther 2005; 4: 1972–1981.
Yujiri T, Fanger GR, Garrington TP, Schlesinger TK, Gibson S, Johnson GL . MEK kinase 1 (MEKK1) transduces c-Jun NH2-terminal kinase activation in response to changes in the microtubule cytoskeleton. J Biol Chem 1999; 274: 12605–12610.
Abedini MR, Muller EJ, Brun J, Bergeron R, Gray DA, Tsang BK . Cisplatin induces p53-dependent FLICE-like inhibitory protein ubiquitination in ovarian cancer cells. Cancer Res 2008; 68: 4511–4517.
Meng XW, Lee SH, Dai H, Loegering D, Yu C, Flatten K et al. Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43–9006)-induced TRAIL sensitization. J Biol Chem 2007; 282: 29831–29846.
Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007; 12: 66–80.
Rosato RR, Almenara JA, Coe S, Grant S . The multikinase inhibitor sorafenib potentiates TRAIL lethality in human leukemia cells in association with Mcl-1 and cFLIPL down-regulation. Cancer Res 2007; 67: 9490–9500.
Kim M, Liao J, Dowling ML, Voong KR, Parker SE, Wang S et al. TRAIL inactivates the mitotic checkpoint and potentiates death induced by microtubule-targeting agents in human cancer cells. Cancer Res 2008; 68: 3440–3449.
Opferman JT . Unraveling MCL-1 degradation. Cell Death Differ 2006; 13: 1260–1262.
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR . Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21: 749–760.
Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J et al. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem 2002; 277: 43730–43734.
Wang X, Chen W, Zeng W, Bai L, Tesfaigzi Y, Belinsky SA et al. Akt-mediated eminent expression of c-FLIP and Mcl-1 confers acquired resistance to TRAIL-induced cytotoxicity to lung cancer cells. Mol Cancer Ther 2008; 7: 1156–1163.
Han J, Goldstein LA, Gastman BR, Rabinowich H . Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem 2006; 281: 10153–10163.
Srivastava RK, Mi QS, Hardwick JM, Longo DL . Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci USA 1999; 96: 3775–3780.
Shimada K, Matsuyoshi S, Nakamura M, Ishida E, Kishi M, Konishi N . Phosphorylation of FADD is critical for sensitivity to anticancer drug-induced apoptosis. Carcinogenesis 2004; 25: 1089–1097.
Harper N, Farrow SN, Kaptein A, Cohen GM, MacFarlane M . Modulation of tumor necrosis factor apoptosis-inducing ligand-induced NF-kappa B activation by inhibition of apical caspases. J Biol Chem 2001; 276: 34743–34752.
Folkman J, Moscona A . Role of cell shape in growth control. Nature 1978; 273: 345–349.
Acknowledgements
This work was supported by grants from Ministerio de Educación y Ciencia (SAF2006-00633), Red Temática de Investigación Cooperativa en Cáncer (RTICC) (RD06/0020/0068) and Junta de Andalucía (CTS-211) to ALR. GO-F and TS-P were supported by a contract and fellowship from Ministerio de Ciencia e Innovación, respectively. We thank Ana Isabel López-Pérez for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Ashkenazi
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Sánchez-Pérez, T., Ortiz-Ferrón, G. & López-Rivas, A. Mitotic arrest and JNK-induced proteasomal degradation of FLIP and Mcl-1 are key events in the sensitization of breast tumor cells to TRAIL by antimicrotubule agents. Cell Death Differ 17, 883–894 (2010). https://doi.org/10.1038/cdd.2009.176
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.176
Keywords
This article is cited by
-
Mechanism of action of the third generation benzopyrans and evaluation of their broad anti-cancer activity in vitro and in vivo
Scientific Reports (2018)
-
Release of c-FLIP brake selectively sensitizes human cancer cells to TLR3-mediated apoptosis
Cell Death & Disease (2018)
-
UMI-77 primes glioma cells for TRAIL-induced apoptosis by unsequestering Bim and Bak from Mcl-1
Molecular and Cellular Biochemistry (2017)
-
Delaying mitotic exit downregulates FLIP expression and strongly sensitizes tumor cells to TRAIL
Oncogene (2015)
-
The NOXA–MCL1–BIM axis defines lifespan on extended mitotic arrest
Nature Communications (2015)
