
Abstract
The mechanism by which the cell death mediator Bax becomes activated to cause mitochondrial damage, a key step for the intrinsic pathway to apoptosis, remain highly contentious. Although some data support a role for certain BH3-only proteins, such as Bim or tBid, to directly activate Bax, others have led to the conclusion that BH3-only proteins act indirectly by antagonizing the prosurvival Bcl-2 proteins, thereby allowing Bax activation to proceed. A recent paper in Nature by Gavathiotis et al. provides the first biophysical evidence for a direct interaction between a BH3 domain, that of Bim, with Bax. Here, we review these intriguing observations and discuss their implications for our understanding of how the BH3-only proteins initiate apoptosis.
Similar content being viewed by others


Main
The Bcl-2 protein family lies at the heart of the intrinsic pathway to apoptosis, the interactions between them being central to determining if a cell lives or dies.1 Members of this family can either promote or block cell survival. Bcl-2 itself and closely related proteins such as Bcl-xL, Bcl-w, Mcl-1 and A1 impose cell survival, their action countered by the related proapoptotic proteins. These family members share regions of primary sequence and structural similarity. The prosurvival proteins and the cell death mediators Bax and Bak display four conserved Bcl-2 homology (BH) domains,2 in addition to a C-terminal membrane anchor. Remarkably, the prosurvival proteins such as Bcl-xL3 and proapoptotic Bax,4 or Bak,5 have a common structural fold (Figure 1).

Similar structural folds of the multidomain Bcl-2 family proteins. (a) A well-characterized hydrophobic groove on surface of Bcl-2 prosurvival proteins is the site of interaction for BH3 domains, as displayed in the Mcl-1/BimBH3 complex (PDB ID: 2NL9; BimBH3 coloured magenta).31 (b) In the case of the soluble, inactive cell death mediator Bax, this groove is occupied by its putative transmembrane anchor (coloured orange) (PDB ID: 1F16)4
The other subclass of proapoptotic proteins are the BH3-only proteins, which includes Bim, Bid, Puma, Noxa and Bad. As the name suggests, these proteins possess only the BH3 domain, and, unlike the multidomain proteins Bax and Bak that mediate mitochondrial damage,6, 7, 8 they act at an apical step to initiate apoptosis.9, 10 The BH3-only proteins are intrinsically unstructured proteins,11 but their BH3 domains form amphipathic α-helices on binding to the hydrophobic groove on the surface of the prosurvival proteins (Figure 1a). Among the BH3-only proteins, Bid is unique in that it adopts the same structure as multidomain Bcl-2 family proteins12, 13 and contains the BH4 motif within the α1 helix which is common to proteins with the Bcl-2 protein fold 4. Whether inactivating the prosurvival proteins is the only, or critical, action of the BH3-only proteins is the subject of much debate.
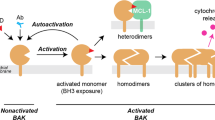
During apoptosis Bax and/or Bak assemble into large homo-oligomers that damage the outer mitochondrial membrane, consequently becoming permeable to proteins normally constrained within the intermembrane space of the mitochondria. The release of factors, such as cytochrome c, trigger activation of the initiator caspase, caspase-9, setting off a chain of events leading to the eventual destruction of the cell. Precisely, how the BH3-only proteins trigger Bax and/or Bak activation is uncertain. It has been proposed that Bax and Bak are held in check by prosurvival proteins.14, 15 On receipt of an apoptotic stimulus, activated BH3-only proteins remove Bax and Bak from the prosurvival constraints to promote their activation indirectly. However, this model (Figure 2a) appears to be in conflict with the absence of significant amounts of Bax and Bak in complex with the prosurvival proteins. Indeed, in most cell types, Bax is located in the cytosol,16, 17 away from the intracellular membranes where Bcl-2 and its close relatives are found.

BH3-only proteins can trigger the activation of Bax and Bak, either (a) indirectly, by inactivating the prosurvival Bcl-2 proteins, or (b) directly
Alternatively, it has been suggested that a subset of the BH3-only proteins, the so-called activators, such as Bim, Bid and Puma, are capable of directly activating Bax and Bak unless kept in check by the prosurvival proteins.18, 19, 20, 21, 22 This threshold can be breached by the action of sensitizer BH3-only proteins, such as Bad and Noxa, which displace the activators to promote Bax and Bak activation. Furthermore, binding of the BH3-only proteins, potentially into the hydrophobic groove of Bax, akin to binding the prosurvival proteins, may promote translocation of Bax to the mitochondrial membranes because the putative transmembrane domain normally accommodated in the groove would be displaced (Figure 1b).
Although the interaction of the BH3-only proteins with the prosurvival Bcl-2-like proteins can be readily demonstrated, evidence for a direct interaction with Bax or Bak has so far been lacking. In an important advance, Gavathiotis et al. have been able to use NMR to derive a model for the interaction between a stapled BimBH3 peptide and Bax.23 The in vitro action of the stapled BimBH3 peptide has provoked further discussions of how BH3-only proteins, in particular Bim, can drive Bax activation through direct physical contact.24
Stapled BH3 Peptides
As most BH3 domain polypeptides are unstructured,11 Walensky and colleagues developed a neat approach to lock these domains in a helical conformation, as seen in their complexes with the prosurvival proteins. They have synthesized hydrocarbon-stapled BH3 peptides that display enhanced helicity in the absence of a binding partner.25, 26 To construct such peptides, two residues predicted to be on the same face of the helix, are replaced with (S)-pentenyl alanine derivatives and then joined by a metathesis reaction. Such peptides, dubbed ‘stabilized α-helix of Bcl-2 domains’ (SAHB), display enhanced binding affinities for prosurvival proteins. 26 In particular, the threefold increase in affinity of the stapled BimBH3 over its parent for Bcl-xL (16 nM compared with 52 nM, respectively, in fluorescence polarization assays) accords with the reported fourfold increase in helicity of the peptide on stapling. Stapled BimBH3 peptides also displayed binding activity for Bax (24 nM) in fluorescence polarization assays but, curiously, no binding to Bax with the unstapled Bim peptide was detected. One explanation for these in vitro data is that the artificial staple actively participates in the interaction and substantially contributes to the interaction with Bax, other than by promoting helical propensity.
Regardless, the stapled BimBH3 peptide induces oligomerization and a conformational change in recombinant Bax, consistent with that seen in cells undergoing apoptosis. Bax-mediated membrane permeabilization is also enhanced by stapled BimBH3 as demonstrated with release of labelled dextran from liposomes or cytochrome c from mitochondria. These observations are consistent with, and extend, previous observations using stapled BidBH3 peptides.
A Model for the Stapled BimBH3/Bax Interaction
This group has now utilized 1H-15N heteronuclear single quantum coherence (HSQC) experiments to map an interaction site for stapled BimBH3 on Bax. NMR peaks in fast exchange are restricted to residues on the α1, α1–α2 loop and α6 of Bax, albeit with small chemical shifts. This places the interaction surface on Bax at a site distinct from the canonical BH3 interaction groove on prosurvival proteins (Figures 1a and 3). We note that fast exchange reported here is indicative of considerably weaker affinity than the value previously reported by these same authors 26 for the stapled BimBH3/Bax interaction, 24 nM.

Putative Bim interaction site on Bax (PDB ID: 2K7W).23 The BimBH3, and by extension Bim, interaction interface on Bax is shown in blue. The C-terminal transmembrane region of Bax and its interaction interface are shown in orange
Further characterization of the stapled BimBH3/Bax interaction by standard NMR methods was confounded by the aggregation of Bax on addition of stapled BimBH3. To overcome this limitation, paramagnetic relaxation enhancement (PRE) was used to further investigate the stapled BimBH3/Bax complex. This method has recently been shown to be useful for the characterization of low affinity interactions in a variety of systems.28, 29
Use of PRE allowed for residues on Bax in the vicinity of the paramagnetic label on the peptide to be identified. Placing the label at the N terminus of the stapled BimBH3 peptide identified residues within α6, α1, α1–α2 loop and α4 of Bax. Alternatively, placement of the label at the C terminus of the peptide identifies residues mostly within α1 and also within α6. From such data, the authors calculated a model for the stapled BimBH3/Bax complex using distance restraints from the PRE measurements and published structures for Bax4 and BimBH3 in complex with Mcl-1 (PDB ID: 2PQK). Readers not familiar with these techniques may assume that Gavathiotis et al. have determined a de novo structure of a complex between Bax and BimBH3.
The NMR data support the conclusion that on binding the stapled BimBH3 peptide, the Bax structure is only mildly perturbed from the published structure of free Bax as presented in the Bax/BimBH3 model. The Bax aggregation observed in the presence of stapled BimBH3 suggests that this interaction probably induces larger scale conformational changes in Bax. Regardless, no data are provided to support the conclusion that the stapled peptide remains helical beyond the bounds of the staple when it binds to Bax as presented in the model. The label used for the PRE experiments is attached in two different places in the Bim peptide, residue 147 or 164 (numbering based on human BimEL). A staple placed between residues 154 and 158 would suggest that the central portion of the peptide is helical. If the connecting sequences N- or C terminal to the PRE labels are indeed helical, then the distances from the labels to the staple are each of the order of 10Å. If, on the other hand, these sequences fold as extended strands when the peptide binds to Bax, these distances could approach 20Å, with concomitant consequences on modelling the interaction between the peptide and Bax.
The calculated model of the stapled BimBH3/Bax complex places the peptide, unexpectedly, within a groove on the α1/α6 surface of the protein. The α1–α2 loop of Bax, which displays some flexibility in the original Bax NMR structure, is displaced in the model to accommodate the peptide. Mutating residues on the stapled BimBH3 predicted by the model to lie at the interface with Bax disrupted the interaction, as measured by Bax oligomerization and cytochrome c release assays. Further, a staple across residues in the BimBH3 predicted to lie at the interface disrupts the activity of the stapled BimBH3 peptides, whereas staples across residues predicted to lie on the solvent exposed surface of the BimBH3 do not. Finally, consistent with previous observations, a stapled BadBH3 peptide does not display any activity on Bax,26 though a mutated BadBH3 domain (containing a norleucine for methionine) was used in this control experiment.
The authors also tested their model by undertaking mutagenesis on Bax. They chose to mutate Lys21 as this produced the most pronounced chemical shift in HSQC binding experiments. Bax (K21E) displayed a decrease in propensity to oligomerize and in its ability to release cytochrome c from mitochondria when incubated with stapled BimBH3. Further, the stapled peptide has a reduced killing activity in cells expressing only Bax (K21E) compared to those expressing wild-type Bax. Intriguingly, in the proposed model, Lys21 of Bax interacts with Glu158 on the BimBH3. However, in the stapled BimBH3 peptide Glu158 has been replaced with the staple (Figure 4a). That this Bax mutation (K21E) affects the binding of stapled BimBH3 further implicates the artificial staple in the interaction of the stapled peptide with Bax. It would be interesting to test other stapled BimBH3 peptides against the Bax (K21E) mutant to investigate this possibility.

(a) In the proposed model of Bax/BimBH3 interaction, Lys21 of Bax interacts with Glu158 of the BimBH3. However, assays reporting the role of Bax Lys21 are performed using a stapled BimBH3 peptide in which Glu158 and Arg154 (yellow side chains) are replaced with the hydrocarbon staple. (b) A previous study19 has identified an interaction between Asp33 of Bax (magenta) and Arg84 of Bid, the equivalent residue in Bim is Ile146 (green). These residues on the BH3 domains and Bax are distant on the proposed model for interaction between ‘activator’ BH3 domains and Bax. The numbering of the residues is based on human BimEL and human Bid
As noted by Gavathiotis et al., a previous study has proposed the α1 of Bax as an interaction site for ‘activator’ BH3 domains.19 However, in that work, mutagenesis studies identified Arg84 of Bid (numbering base on human Bid) as directly interacting with Asp33 of Bax. This interaction is inconsistent with the proposed BimBH3/Bax model as Bax Asp33 is on a different face to that proposed for BimBH3 interaction, far removed from Ile146 of Bim, the equivalent residue to Arg84 of Bid (Figure 4b).
Implications
Gavathiotis et al. provides intriguing new insights into the elusive nature of the physical interaction between the BH3-only proteins and Bax. They showed that in the context of a stapled BimBH3 peptide, this peptide could bind weakly and transiently to a novel face for binding BH3 peptides on the Bcl-2 protein fold. They also show that this interaction can instigate changes (conformation, oligomerization) in recombinant Bax similar to that seen with endogenous Bax in dying cells and trigger permeabilization in vitro of both artificial and mitochondrial membranes.
The results and conclusions from the Walensky group raise many questions, most of which are experimentally tractable. Do the studies with the stapled peptide faithfully reflect the physiological role of Bim? Is the BimBH3/Bax interaction necessary and sufficient to drive Bax activation? Is the key event initiated by the stapled BimBH3 peptide to promote the translocation of Bax from the cytoplasm to the mitochondrial membrane? If the Bim/Bax interaction is the key step initiating Bax translocation, what is the sequence of events that culminate in cytochrome c release from the mitochondria? Once at the membrane, will Bax be inhibited by the presence of prosurvival Bcl-2 proteins as proposed in the ‘indirect’ model? Although the experiments with stapled BimBH3 suggest that its interaction with Bax is sufficient to fully drive Bax activation, they do not resolve this conundrum because these peptides retain high affinity for prosurvival proteins.26 In this regard it is intriguing that the stapled BimBH3 peptide only causes killing of some cell lines, suggesting that other mechanisms need to be activated or suppressed before full Bax activation occurs. In the context of cancer therapeutics, the idea that a Bax activator could have utility is an interesting one provided there is sufficient therapeutic index so that normal tissues are spared.
It will also be pertinent to determine if Bim acts in a similar manner on the other cell death mediator Bak, which, unlike Bax, is predominantly at the mitochondria and does not require the same translocation step. Do other BH3-only proteins, such as tBid, which has also been proposed to be an activator of Bax and interacts with Bax in a lipid environment,30 trigger Bax and/or Bak in a similar manner?
Concluding Remarks
Gavathiotis and co-workers provide biophysical evidence for an interaction between a derivatized BH3 domain and Bax. As discussed, there are some caveats about the conclusions drawn from these experiments, but the work provides a model to test the role of these newly described weak interactions. In particular, the physiological significance of these interactions in the biological context of cells undergoing apoptosis remains to be fully established.
Abbreviations
- BH:
-
Bcl-2 homology domain
- HSQC:
-
heteronuclear single quantum coherence
- NMR:
-
nuclear magnetic resonance
- PRE:
-
paramagnetic relaxation enhancement
- PDB:
-
protein data bank
- SAHB:
-
stabilized α-helix of Bcl-2 domains
References
Youle RJ, Strasser A . The Bcl-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Kvansakul M, Yang H, Fairlie WD, Czabotar PE, Fischer SF, Perugini MA et al. Vaccinia virus anti-apoptotic F1 L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ 2008; 15: 1564–1571.
Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996; 381: 335–341.
Suzuki M, Youle RJ, Tjandra N . Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 2000; 103: 645–654.
Moldoveanu T, Liu Q, Tocilj A, Watson MH, Shore G, Gehring K . The X-ray structure of a Bak homodimer reveals an inhibitory zinc binding site. Mol Cell 2006; 24: 677–688.
Lindsten T, Ross AJ, King A, Zong W, Rathmell JC, Shiels HA et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–1399.
Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T et al. Bcl-2, Bcl-xL sequester BH3 domain-only molecules preventing Bax- and Bak-mediated mitochondrial apoptosis. Mol Cell 2001; 8: 705–711.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic Bax and Bak: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Bouillet P, Metcalf D, Huang DCS, Tarlinton DM, Kay TWH, Köntgen F et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 1999; 286: 1735–1738.
Huang DCS, Strasser A . BH3-only proteins – essential initiators of apoptotic cell death. Cell 2000; 103: 839–842.
Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC et al. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ 2007; 14: 128–136.
Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G . Solution structure of Bid, an intracellular amplifier of apoptotic signaling. Cell 1999; 96: 615–624.
McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Cowburn D . Solution structure of the proapoptotic molecule Bid: a structural basis for apoptotic agonists and antagonists. Cell 1999; 96: 625–634.
Oltvai ZN, Milliman CL, Korsmeyer SJ . Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993; 74: 609–619.
Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007; 315: 856–859.
Hsu Y-T, Wolter KG, Youle RJ . Cytosol-to-membrane redistribution of Bax and Bcl-xL during apoptosis. Proc Natl Acad Sci USA 1997; 94: 3668–3672.
Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ . Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 1997; 139: 1281–1292.
Letai A, Bassik M, Walensky L, Sorcinelli M, Weiler S, Korsmeyer S . Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002; 2: 183–192.
Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P et al. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and Puma. Mol Cell 2004; 16: 807–818.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 2005; 17: 525–535.
Certo M, Moore Vdel G, Nishino M, Wei G, Korsmeyer S, Armstrong SA et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic Bcl-2 family members. Cancer Cell 2006; 9: 351–365.
Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A . BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007; 12: 171–185.
Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG et al. Bax activation is initiated at a novel interaction site. Nature 2008; 455: 1076–1081.
Green DR, Chipuk JE . Apoptosis: stabbed in the Bax. Nature 2008; 455: 1047–1049.
Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004; 305: 1466–1470.
Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J et al. A stapled BID BH3 helix directly binds and activates Bax. Mol Cell 2006; 24: 199–210.
Chipuk JE, Green DR . How do Bcl-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 2008; 18: 157–164.
Battiste JL, Wagner G . Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear Overhauser effect data. Biochemistry 2000; 39: 5355–5365.
Clore GM, Tang C, Iwahara J . Elucidating transient macromolecular interactions using paramagnetic relaxation enhancement. Curr Opin Struct Biol 2007; 17: 603–616.
Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B et al. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 2008; 135: 1074–1084.
Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DCS et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci USA 2007; 104: 6217–6222.
Acknowledgements
Work in our laboratories is supported by grants and fellowships from the Australian NHMRC, the US NCI, the Leukemia and Lymphoma Society, the Cancer Council of Victoria and the Victorian State Government. We thank our colleagues for many stimulating discussions.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by G Nunez
Rights and permissions
About this article
Cite this article
Czabotar, P., Colman, P. & Huang, D. Bax activation by Bim?. Cell Death Differ 16, 1187–1191 (2009). https://doi.org/10.1038/cdd.2009.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.83
Keywords
This article is cited by
-
Protective effect of Cordyceps sinensis against diabetic kidney disease through promoting proliferation and inhibiting apoptosis of renal proximal tubular cells
BMC Complementary Medicine and Therapies (2023)
-
Artesunate improves venetoclax plus cytarabine AML cell targeting by regulating the Noxa/Bim/Mcl-1/p-Chk1 axis
Cell Death & Disease (2022)
-
The pro-apoptotic domain of BIM protein forms toxic amyloid fibrils
Cellular and Molecular Life Sciences (2021)
-
An epigenetic screening determines BET proteins as targets to suppress self-renewal and tumorigenicity in canine mammary cancer cells
Scientific Reports (2019)
-
Targeting BAX to drug death directly
Nature Chemical Biology (2019)
