
Abstract
Sequential cleavage of the amyloid precursor protein (APP) by β- and then γ- secretase gives rise to Aβ1−40 (Aβ40), a major species of Aβ (β-amyloid) produced by neurons under physiological conditions. Aβ1−42 (Aβ42), a minor species of Aβ, is also produced by a similar but less understood mechanism of the γ-secretase. The physiological functions of these Aβ species remain to be defined. In this report, we demonstrate that freshly prepared soluble Aβ40 significantly promotes neurogenesis in primary neural progenitor cells (NPCs). First, Aβ40 increases neuronal markers as determined by NeuN expression and Tuj1 promoter activity, differing from Aβ42, which induces astrocyte markers in NPCs. Second, Aβ40 induces neuronal differentiation at the end of S-phase in the cell cycle. Third, Aβ40 promotes NPC entry into S-phase, playing a role in NPC self-renewal. Interestingly, Aβ40 does not significantly increase apoptotic indexes such as DNA condensation and DNA fragmentation. In addition, Aβ40 does not augment caspase-3 activation in NeuN+ or nestin+ cells. Collectively, this report provides strong evidence that Aβ40 is a neurogenic factor and suggests that the debilitated function of Aβ40 in neurogenesis may account for the shortage of neurons in Alzheimer's disease.
Similar content being viewed by others


Main
One of the hallmarks of Alzheimer's disease (AD) is the deposition of β-amyloid (Aβ) plaques in the brain. The major component and an initiating factor of the plaques is Aβ42.1 In contrast, Aβ40, a major species of Aβ produced by neurons under physiological conditions, appears to have a beneficial role in reducing the amyloid burden.2 Aβ40 levels are decreased by familial AD mutants, whereas Aβ42 levels remain unchanged.3, 4 The beneficial role of Aβ40 is also suggested by studies using PS1 knockout models, which undergo neurodegeneration in the absence of Aβ production.5 In the PS1 exon 10 deletion mice, there are very low levels of Aβ40 but accelerated amyloid plaque deposition.6 A very recent study shows that Aβ40 inhibits Aβ42-initiated amyloid deposition in vivo.2 Together, these findings suggest a hypothesis that decreases in Aβ40 may underlie the pathological process of AD, and that Aβ40, and even Aβ42, may have physiological functions before they aggregate and thus deserve more detailed investigation.7
Neural progenitor cells (NPCs) have the ability to self-renew and give rise to other CNS cell types in a process known as neurogenesis. Neurogenesis is altered in AD transgenic mouse models and in human postmortem brains. In an animal model with the Swedish and Indiana mutations of the amyloid precursor protein (APP) (APPswe/ind), a twofold increase in BrdU labeling is reported in 3-month-old mice before amyloid deposition and also in 1-year-old mice.8 However, a decrease in BrdU labeling is reported in adult mouse hippocampi expressing APPswe9 or APPind.10 Impaired neurogenesis is also reported in APPswe/PS1(P246L) knock-in mice.11 Similar impairment in neurogenesis was observed in several transgenic mouse models expressing presenilin-1 (PS1) familial AD mutants.12, 13, 14 The underlying mechanisms of altered neurogenesis are not clear in these animal models. It is anticipated that human studies using postmortem brains are even more complicated to interpret. In humans, neurogenic markers are shown to increase in the AD hippocampus15 but decrease in the subventricular zone.16 Another study finds evidence that increased proliferation reflects glial and vascular changes but not neurogenesis in the presenile AD hippocampus.17 Therefore, a consensus cannot be reached in explaining the controversial findings from both animal models and human postmortem brains.
Understanding stem cell development in neurodegenerative disease conditions may provide valuable information on potential stem cell therapies, involving either endogenous stem cells or stem cell transplantation.18, 19 We have developed an in vitro system to examine the effect of Aβ40 and Aβ42 in NPC growth and differentiation. The majority of the NPCs were proliferating and undergoing more symmetrical cell divisions in our cultures. With a sufficient number of such primary NPCs, we were able to use vigorous approaches such as western blotting and luciferase reporter assays to show that Aβ40 and Aβ42 played different roles in cell fate decisions with Aβ40 strongly promoting neurogenesis.
Results
Characteristics of NPCs isolated from rat E18 cerebral cortices
By day 10 in vitro after the initial plating (P0), NPCs had formed floating spheres in non-coated dishes or had grown into spindle-shaped cells on coated dishes (Figure 1A). After mechanical dissociation of the P0 cells and plating onto coated glass coverslips, up to 89% of the cells were stained positive for the proliferation marker Ki67 at 3 days after replating (Figure 1B).

NPC cultures and monolayer NPC differentiation into major CNS cell types. (A) NPCs grown in vitro. Left, NPC grown in a non-coated dish; Right, NPCs grown in a poly-ornithine-coated dish. (B) Monolayer NPCs were stained for Ki67 (green) and counter-stained with DAPI. (C) Monolayer NPCs at 10 day in vitro were replated onto glass coverslips. After 2 days in NPC growth medium, cells were fixed for nestin staining (a) or switched to a lineage-specific defined medium (B–D) for 4 days before immunostaining for MAP2 (b), O4 (c), or S100B (d). The percentage of cells labeled with a specific marker was calculated against the total number of cells stained with DAPI (blue or purple), n=2
The monolayer cultures contained mostly bipolar or monopolar nestin+ cells at the first plating (P1) from the P0 cells (Figure 1C, a), and resembled cortical progenitor cells in vivo.20, 21 These monolayer cells were multipotent and could differentiate into major brain cell types after switching to a defined medium for a specific cell type according to the protocols described before22 (Figure 1C, b–d). After 4 days in the respective defined medium, about 80% of the cells were MAP2+ neurons, 70% were S100B+ astrocytes, and 40% were O4+ oligodendrocytes. These differentiated cells also appeared to have cell type-specific morphologies. These experiments suggest that our NPC cultures underwent more symmetrical divisions and possessed multipotencies. NPCs in the experiments described below were grown as monolayer cultures and used at P1 or second plating (P2) after P0 plating.
Aβ40 and Aβ42 elevated lineage-specific protein markers
To determine the effect of Aβ40 or Aβ42 in NPC differentiation in vitro, we undertook three independent approaches: immunofluorescence labeling and cell counting, western blotting, and cell type-specific promoter activity assays. The latter two strategies rely on a population of cells and are more suggestive albeit probably less sensitive. In the case of immunofluorescence labeling and cell counting, samples were stained and coded by one person, and the data were collected blindly by another person to minimize the observer's bias. In these experiments, NPCs were treated with freshly prepared, soluble Aβ40 or Aβ42 at 1.5 μM or as specified for 25 h. Unless noted as aggregated, freshly prepared (soluble) Aβ species were used to treat NPCs.
The very first set of experiments was performed by western blotting. After 25 h of treatment, NPCs were lysed and equal amount of total protein was resolved on SDS-PAGE gels and analyzed for NeuN and S100B expressions using γ-tubulin as the loading control. These original experiments revealed that Aβ40 increased the neuronal marker NeuN but not the astrocyte marker S100B in NPCs (Figure 2a). In contrast, Aβ42 increased the astrocyte marker S100B expression in these cells. The concentration effect of Aβ40 on the expression of NeuN was further analyzed (Figure 2b & c). NPCs were treated with freshly prepared Aβ40 or Aβ42 for 25 h in a range of concentrations. Cells were lysed and analyzed for NeuN expression using γ-tubulin as the loading control. Figure 2b shows that NeuN levels were increased in an Aβ40 dose-dependent manner. NeuN was significantly induced by 1, 1.5, and 5 μM compared to that in the control (Figure 2c). In contrast, freshly prepared Aβ42, but not Aβ40, consistently increased the astrocyte marker S100B expression in a dose-dependent manner (Figure 2a and data not shown).

Aβ40 and Aβ42 increased lineage-specific markers. (a) A representative blot shows that Aβ40 increased NeuN expression, whereas Aβ42 increased S100B expression (n>3). MW are marked on the right. Intervening lanes were cut out. (b) A representative blot shows that Aβ40 induced NeuN expression in a dose-dependent manner. (c) Western blot analyses of the concentration effect of Aβ40 on NeuN expression. The average NeuN expression in NPCs was based on three independent experiments. Two-tailed t-tests: vehicle versus 1 pM, P=0.60; vehicle versus 1 nM, P=0.18; vehicle versus 1 μM, P<0.001; vehicle versus 1.5 μM=0.01; vehicle versus 5 μM, P=0.03. The 48 kDa NeuN was presented and analyzed in (b, c)
Aβ40 promoted neurogenesis in NPCs exiting cell cycle S-phase
It has been shown that NPC assumes a neuronal fate by responding to unknown environmental cues at approximately S-phase of the cell cycle during cortical development.23, 24 We noticed that in our experimental conditions in which NPCs were treated with Aβ40 for 25 h in the presence of BrdU, the NeuN+ cells were rarely BrdU+ (Figure 3a). To determine if these NeuN+ cells were newborn neurons and if it took NPCs more than 25 h to develop a neuronal fate, we examined S-phase NPCs in response to Aβ40 treatment (Figure 3b). In this experiment, NPCs were pulse-labeled for 3 h with BrdU to mark the S-phase cells. The medium was then replaced with fresh medium containing Aβ40 without BrdU. Cells were fixed at 25 h after treatment and immunostained for BrdU and NeuN. As shown in Figure 3b, almost all NeuN+ cells were now BrdU+, suggesting that NPCs responded to Aβ40 to differentiate into neurons only if they were already in S-phase. A closer examination revealed that two populations of BrdU-labeled cells were present: cells were either brightly or faintly labeled. NeuN+ cells almost exclusively overlapped with those faintly labeled with BrdU. Cells that incorporated less BrdU and were thus faintly labeled might be at the end of S-phase in the cell cycle,25 or they might have just entered the S-phase shortly before BrdU withdrawal. The later prediction was unlikely because concomitant Aβ40 and BrdU in the medium did not result in NeuN+ and BrdU+ double-labeling (Figure 3a). These data suggest that only those NPCs traversing the later stage of the S-phase, that is, exiting the S-phase, assumed a neuronal cell fate in response to Aβ40. These data also suggest that in our primary NPC cultures, neuronal differentiation took longer than 25 h.

Aβ40 increased the number of NeuN+ cells by affecting NPCs exiting S-phase of the cell cycle. (a) BrdU was added along with each treatment and stained for NeuN and BrdU after fixation. (b) NPCs were prelabeled with BrdU before Aβ40 treatment. Arrows point to the cells with apparent BrdU and NeuN double-labeling. BrdU+ staining was less bright in the double-labeled cells. Representative images are shown in (a, b)
Aβ42 induced NPCs to become astrocytes
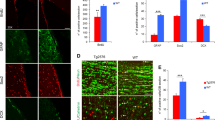
Aβ is implicated in promoting glia cell fate.17 Here we determined whether Aβ40 or Aβ42 affected astrocyte lineage determination in NPCs. After Aβ40 or Aβ42 treatment for 25 h, NPCs were fixed and stained for S100B, a marker for astrocyte development.26 As shown in Figure 4, Aβ42 significantly increased the number of S100B+ astrocytes compared to samples treated with Aβ40 or the control sample treated with the vehicle DMSO.

Aβ42 dramatically increased the percentage of S100B+ cells. Top, bar graphs show the analyses of 10 random fields of cells immunostained with rabbit anti-S100B. A representative experiment is presented. Two tail t-test: Aβ42 versus DMSO, P=0.0004; Aβ40 versus DMSO, P=0.1; Aβ40 versus Aβ42, P=0.03. Bottom, representative images of S100B-stained cells are shown (S100B, green, DAPI, red). The number of * indicates the degree of significance compared to the control
Aβ40 and Aβ42 increased lineage-specific promoter activity
To further quantify the lineage-specific effects of Aβ40 and Aβ42, NPCs were transfected with a lineage-specific promoter-luciferase reporter construct before Aβ40 or Aβ42 treatment. After transfection, cells were treated with Aβ40 or Aβ42 for 25 h and luciferase activities were measured in the cell lysate. A SOX2 promoter-driven luciferase construct was used as an undifferentiated NPC reporter construct. A βIII-tubulin (Tuj1) promoter-driven luciferase construct was used as a neuron-specific reporter construct. The GFAP and MBP promoter-driven constructs were used as astrocyte and oligodendrocyte markers, respectively. As shown in Figure 5a, Aβ40 significantly increased Tuj1 promoter activity, whereas Aβ42 had the opposite effect on the Tuj1 promoter. A reversed pattern was observed in the GFAP promoter activity: Aβ40 significantly decreased, whereas Aβ42 increased the GFAP promoter activity (Figure 5b). In contrast, no consistent effect was observed on the activities of the MBP promoter or on the SOX2 promoter by Aβ40 or Aβ42 treatment (data not shown).

Aβ40 and Aβ42 increased lineage-specific promoter activities. (a) Aβ40 significantly increased Tuj1 promoter activity, whereas Aβ42 decreased it. Two tail t-test: Aβ40 versus DMSO, P=6 × 10−4; Aβ42 versus DMSO, P=0.01; Aβ40 versus Aβ42, P=6 × 10−4. (b) Aβ42 dramatically increased GFAP promoter activity, whereas Aβ40 did not. Two tail t-test: Aβ42 versus DMSO, P=7 × 10−5; Aβ40 versus DMSO, P=0.014; Aβ40 versus Aβ42, P=8 × 10−4. The number of * indicates the degree of significance compared to the control, n=3
Both Aβ40 and Aβ42 increased NPC proliferation during 25 h of treatment
So far we have shown that Aβ40 and Aβ42 had distinct roles in cell fate determination based on cell type-specific markers (Figure 2, 3, 4 and 5). Evidence suggests that these Aβ species may also alter NPC proliferation depending on the time and type of animal models used.8, 12, 13, 14 We hypothesized that the discrepancy in animal models may reflect the existence of different levels of soluble Aβ40 and/or Aβ42 in each animal model. Another less-likely factor might be that a relatively small number of stem cells were normally examined in vivo. In our cell culture model, both of these factors could be controlled. In this experiment, NPC cultures were treated for 25 h with fresh Aβ40 and Aβ42 based on the report that NPCs have a cell cycle of about 25 h in adult rat dentate gyrus.25 BrdU and Aβ40 or Aβ42 were concomitantly present for 25 h before fixation and immunofluorescence staining. In mock-treated cultures, more than 70% of the cells incorporated BrdU, that is, in the S-phase, during the 25 h of incubation (Figure 6a), demonstrating that the majority of the cells were proliferating and similar to the results by Ki67 staining (Figure 1B). The percentage of cells that underwent S-phase and became BrdU+ during the 25 h interval was similar to that as predicted in the dentate gyrus of adult rats.25 Treatment with Aβ40 or Aβ42 significantly increased the number of BrdU+ cells with Aβ42 having a stronger effect (Figure 6a). Representative images are shown in Figure 6b.

Both Aβ40 and Aβ42 significantly increased the number of NPCs in S-phase of the cell cycle. (a) Analyses of 10 random fields of cells immunostained with rabbit anti-BrdU and counter-stained with DAPI. Two tail t-test: Aβ40 versus DMSO, P=0.01; Aβ42 versus DMSO, P=8 × 10−8; Aβ40 versus Aβ42, P=0.05. The number of * indicates the degree of significance compared to the control. (b) Representative images of BrdU-stained cells are shown (BrdU, pink; DAPI, blue). (c) Flow cytometric analyses of NPC cell cycle. Left, histograms show the DNA content distribution (PI stain intensity) of cells on the x-axis for each treatment condition. Right, the percentage of the S-phase cells is shown in the bar graph. A representative experiment is shown, n=3
So far we have shown that a high percentage of NPCs were proliferating as measured by Ki67 staining and BrdU uptake, and that Aβ40 and Aβ42 increased the number of NPCs in S-phase by BrdU labeling. To validate these results, the DNA content distribution of NPCs was analyzed by flow cytometry. In this experiment, cells were stained with propidium iodide for determining DNA content after treatment with each Aβ peptide or the vehicle DMSO. As shown in Figure 6c (left) histograms, NPC exhibited a DNA content profile similar to proliferating human embryonic stem cell lines with an abbreviated G1 phase.27 Cell cycle-associated DNA distribution analyses suggested that the percentage of cells in the S-phase was also increased by Aβ40 and Aβ42 treatment (Figure 6c, right). Together, these data demonstrate that Aβ40 and Aβ42 accelerates NPC cell cycle.
Aβ40 and Aβ42 did not induce NPC DNA fragmentation or DNA condensation
Aβ42 is known to induce neuronal death.28, 29, 30 In addition, amyloid induces or predisposes neurons to DNA damage.31 However, it does not appear to affect NPC death.16, 17, 32 In these NPCs, no increase in cell death was obvious after 25 h of Aβ40 or Aβ42 treatment as judged by Trypan blue exclusion (data not shown). A hallmark of apoptosis is DNA fragmentation. In the next experiment, DNA fragmentation was determined by the Comet assay after Aβ40 or Aβ42 treatment. Fragmented DNA should appear like a comet after propidium iodide staining and electrophoresis. The positive control showed that primary neurons underwent DNA fragmentation after prolonged Aβ42 treatment (see comets in Figure 7a, control). However, NPCs did not show DNA fragmentation as no comets were observed in the Aβ40- or Aβ42-treated samples (Figure 7a).

Aβ40 did not significantly induce NPC DNA fragmentation or DNA condensation. (a) Aβ40 or Aβ42 treatment of NPCs did not induce DNA fragmentation as shown by comet assays. The positive control with ‘comets,’ that is, fragmented DNA, was primary neurons treated with Aβ42. Representative images are shown, n=3. (b) Condensed nuclei were counted in NPCs after treatment with freshly prepared Aβ species or preaggregated species (-agg) for 25 h before fixation and DAPI staining. Condensed nuclei were counted and presented as the percentage of total nuclei in the bar graph. Two-tailed t-tests did not reveal any P-values < 0.05 between the vehicle and an Aβ species, n=3
Cells often undergo DNA condensation during apoptosis.33 In this experiment, the percentage of cells that had condensed nuclei was determined by DAPI staining in NPCs. Cells were treated with freshly prepared soluble Aβ or preaggregated Aβ for 25 h before fixation and staining. Condensed nuclei were much brighter than normal nuclei, and these condensed nuclei were gated for counting by the MetaVue software. No significant differences were observed among these treatment conditions (Figure 7b).
Aβ40 and Aβ42 in lineage-specific caspase-3 activation
So far we have shown that Aβ40 or Aβ42 did not significantly induce NPC DNA fragmentation or DNA condensation. Apoptotic cell death is often preceded by caspase-3 activation in many other cell types. Using a well-characterized and widely used antibody that recognizes activated caspase-3, we determined if caspase-3 was activated by Aβ40 or Aβ42 in a cell-type specific manner (Figure 8). In these experiments, NPCs were treated with either freshly prepared or preaggregated Aβ for 25 h before fixation. Cells were double-stained with the rabbit anti-activated caspase-3 antibody and the mouse anti-NeuN or the mouse anti-nestin antibodies followed by appropriate Alexa Fluor-conjugated secondary antibodies. Figure 8a confirmed our previous observation that Aβ40 significantly increased the number of NeuN+ cells. Aggregated Aβ40 or soluble Aβ42 also increased the number of NeuN+ cells. However, Aβ40 did not significantly increase caspase-3 activation in nestin+ cells (Figure 8c). Aβ40 actually significantly reduced caspase-3 activation in NeuN+ cells (Figure 8b). These data suggest that S-phase entry promoted by Aβ40 does not induce apoptosis in NPCs and may be supportive for newborn neuron survival.

Effect of Aβ species on caspase-3 activation in NeuN+ and nestin+ cells in NPC cultures. (a) Freshly prepared Aβ40 significantly induced NeuN expression in NPCs. Aggregated Aβ40 (Aβ40-agg) or freshly prepared Aβ42 also showed moderate effect on NeuN expression. Two tail t-test: vehicle versus Aβ40, P=0.008; vehicle versus Aβ40-agg, P=0.05; vehicle versus Aβ42, P=0.03; vehicle versus Aβ42-agg, P=0.78, n=3. (b) Percentage of NeuN+ cells with activated caspase-3. One-tail t-test: Aβ40 versus vehicle, P=0.036; Aβ42 versus vehicle, P=0.194, n=3. (c) Percentage of nestin+ cells with activated caspase-3. Two-tailed t-tests: vehicle versus Aβ42, P=0.04, n=3. (d) Migration patterns of Aβ species on protein gels as probed by 4G8. Left, freshly prepared soluble Aβ40 or Aβ42 (0.25 μg per lane) migrated as monomers. Right, the migration patterns of aggregated Aβ40 or Aβ42 (0.5 μg per lane). The MW standard (BioRad) is shown on the left side of the blot. The same blot was duplicated with the top of the blot reversed in color (separated by a white line) (far right). We are not sure why the band around 66 kDa behaved differently in color using our system. The migration position of monomers and oligomers are shown on the right
The cell counting method appeared to be more sensitive than western blotting by showing a significant effect of soluble Aβ42 (although much less than Aβ40) at increasing the percentage of NeuN+ cells (Figure 8a). This demonstrates that there is some overlapping function between these two peptides. However, soluble Aβ42 might not be beneficial in the long run due to its higher aggregation tendency. It was less certain whether freshly prepared Aβ42 was toxic to NPCs, although it reached a statistical significance compared with the control (Figure 8c). Aβ42 augmented caspase-3 activation in nestin+ cells, but a large variation existed among the microscopic cell counts as indicated by the large error bars (Figure 8c). Figure 8d shows that freshly prepared Aβ40 migrated as monomers, whereas the aggregation protocol produced a mixture of monomers and oligomers. In contrast, although the majority of freshly prepared Aβ42 migrated as monomers, some existed as a high molecular weight (MW) species. Over the course of aggregation, all Aβ42 appeared as a single high MW species. Together, these experiments suggest that soluble Aβ40 is the major effective species in neurogenesis and that aggregation ablates such a function.
Discussion
In this report, we employed primary, multipotent, proliferating NPC cultures (Figure 1 and Figure 6). Using such NPCs, we demonstrated that freshly prepared Aβ40 significantly stimulated neurogenesis, whereas Aβ42 induced astrocyte lineage in these cells (Figures 2, 3, 4 and 5, 8a). The neurogenic effect is greatly reduced when Aβ40 was preaggregated, corresponding to reduced levels of monomers (Figure 8 a and d). We further showed that Aβ40 induced neuronal cell fate in NPCs that were exiting S-phase of the cell cycle (Figure 3). Besides their differential effects on NPC differentiation, we provided evidence that both Aβ40 and Aβ42 accelerated progenitor cell proliferation (Figure 6). Aβ40 and Aβ42 did not significantly induce NPC DNA fragmentation or DNA condensation (Figure 7). We further showed that Aβ40 did not augment caspase-3 activation in NeuN+ or nestin+ cells (Figure 8b and c), but Aβ40 might be protective to neurons as it significantly decreased the number of caspase-3+NeuN+-cells (Figure 8b). Taken together, these data suggest a dynamic disease process in AD, where an increase in neurogenesis is followed by an overall decline, as Aβ40 aggregates and loses the function. Accordingly, diminution of neurons as observed in postmortem AD brains may originate from reduced neurogenesis later in the disease process. These findings may also partially explain the discrepancies in neurogenesis reported in animal models. In such AD animal models, neurogenesis may vary according to the levels of soluble Aβ40 and the time point when the data are collected.
The major difference of our in vitro studies from many others is that we used early stage, proliferating NPCs with very few other cell types. Under such cultural conditions, Aβ40 had a predominant effect on neurogenesis, differing from a report showing that soluble Aβ42 has the major effect.34 We showed that soluble Aβ42 increased the number of NeuN+ cells, but Aβ40 is more potent in neurogenesis in our culture conditions (Figure 8a). Another study shows that soluble Aβ40 impairs neurogenesis at a very high concentration,35 which probably contains few monomers. We decided to start with a much lower concentration of Aβ40 due to the concern that higher concentrations may result in a nonspecific, toxic effect which would mask potential physiological functions. Higher concentrations of Aβ40 may also result in faster aggregation and loss of function. We showed that Aβ40 significantly induced NeuN expression between 1 and 5 μM (Figure 5b). The less effectiveness of Aβ40 in neurogenesis at 5 μM may be due to aggregation. These data indicate that Aβ40 may be a promising tool for neurogenesis, but at high concentrations, Aβ40 may not be beneficial unless aggregation can be effectively controlled. We are currently investigating better strategies of using Aβ40 for neurogenesis.
Aβ42 is strongly implicated in AD pathogenesis, but it did not induce apoptotic indexes such as DNA fragmentation and condensation in NPCs (Figure 7). In a closer look, we showed that Aβ42 significantly induced caspase-3 activation in nestin+ cells, but there were large variations between the microscopic fields as indicated by the large error bars (Figure 8c). Freshly prepared Aβ42 or aggregated Aβ42 did not significantly increase caspase-3 activation in NeuN+ cells (Figure 8b); this suggests that neither the monomers nor the 66 kDa species are toxic to neurons. Maybe the smaller MW Aβ42 oligomers are toxic but our protocol did not appear to produce detectible amount of oligomers from Aβ42 (Figure 8d). Aβ42 is known to aggregate faster. The absence of lower MW Aβ42 oligomer suggests that most of the monomers had evolved into the higher MW species (about 66 kDa) over 3 days of prior incubation. In contrast, the same aggregation protocol produced a whole range of Aβ40 species including low MW oligomers (Figure 8d). This is associated with reduced neurogenesis, but not with significant increases in cell death indexes (Figure 7b and Figure 8).
We showed that Aβ42 induced astrocyte differentiation in NPCs in addition to NPC proliferation (Figures 2a, 4, and 6). Activated astrocytes may secrete cytokines toxic to neurons and inhibit axonal growth. Thus, Aβ42 may serve as a double-edged sword depending on the situation. In contrast, Aβ40 was predominantly neurogenic. It cannot be excluded that these Aβ species may differ in their affinity for potential receptors and thus result in mainly different functions. Detailed mechanistic studies of Aβ40 or Aβ42 should provide valuable information on neuroregeneration, as well as understanding AD pathogenesis.
Similar to the findings from several reports, we found that more NPCs incorporated BrdU after Aβ40 or Aβ42 treatment (Figure 6a and b). We also found that Aβ40 promoted neuronal cell fate during the later stage of S-phase in the cell cycle (Figure 3). Therefore, Aβ40 positively affects two stages of the cell cycle: one is at S-phase entry and the other at S-phase exit. The cumulative effect of Aβ40 appears to be beneficial as it not only promotes NPC self-renewal but also induces neuronal differentiation. Furthermore, the effect of Aβ40 on NPC differentiation suggests that it might be protective to neurons that are induced to re-enter the cell cycle; in response to Aβ40, such neurons may exit the S-phase as with NPCs, retain partial function, and survive for a long time.36 These observations also open new avenues for future investigations such as how Aβ40 functions at the molecular level at these two stages of the cell cycle during NPC development.
In this report, we relied on embryonic instead of adult cortical NPCs due to the ease of isolation and culturing. Surprisingly, these NPCs have a similar DNA profile as in human embryonic stem cell lines.27 Disturbed neurogenesis during early development can result in neurodegeneration in adulthood as exemplified by the spinocerebellar ataxia type 1 mouse model.37 Thus, the effect of Aβ40 and Aβ42 on prenatal cerebral NPCs may reflect their physiological functions during development. The possibility that a perturbation of early neural development could contribute to adult onset of AD may be investigated in animal models in vivo. Further, the effects of Aβ40 and Aβ42 should be scrutinized in adult NPCs, especially in hippocampal NPCs. These future investigations will provide valuable information on the potential of a stem cell therapy against neurodegeneration including in AD.
Materials and Methods
Antibodies
The following antibodies were used: rabbit anti-BrdU antibody (Megabase Research Products), mouse anti-NeuN (Chemicon), rabbit anti-S100B (gift from Dr. Linda J Van Eldik), mouse anti-MAP2 (HM-2 clone) and mouse anti-γ-tubulin (both from Sigma), mouse anti-nestin (BD Bioscience), mouse anti-O4 (gift from Dr. Steven Pfeiffer),38 rabbit anti-Ki67 (ABCam), mouse anti-Aβ (4G8 clone, Signet), rabbit anti-activated caspase-3 (Cell Signaling), and Alexa Fluor 594 goat anti-mouse and Alexa Fluor 488 goat anti-rabbit (Molecular Probes).
Neural progenitor cells
The animal protocol was preapproved by the Institutional Animal Care Committee. Briefly, cortices from embryonic day 18 (E18) rat were dissected, mechanically dissociated, then first centrifuged through a sucrose cushion at 200 × g for 7 min, and later through a 4% BSA/DMEM solution at 700 × g for 10 min. The cell pellet containing progenitor cells were washed twice with growth medium containing neurobasal medium supplemented with 2% of B-27 supplement without vitamin A and 1x penicillin/streptomycin (all from Invitrogen) and with growth factors (EGF and FGF at 20 ng ml−1 each, Sigma) and L-glutamine at 0.5 mM (Invitrogen). Single cells were initially plated in the growth medium in a 60 mm dish at a density of 3 × 106 per dish either on poly-ornithine-coated dishes for monolayer cultures or non-coated dishes for floating spheres. These initial plating was designated P0 cells. Half of the culture medium was replaced every 2 days. Cells were maintained at 37 °C with 5% CO2 in an incubator with high humidity. Cells were mechanically dissociated before replating. Experiments were performed using P0 cells at first (P1) or second (P2) plating in coated dishes. Most of the experiments were performed in NPCs initially plated as a monolayer and repeated using NPCs initially plated as spherical cultures.
Primary neurons
Primary neurons were grown as described in the following cited report.33 Cells were treated at 7 or 8 day in vitro with 1.5 μ M of Aβ42 for 72 h.
Fixation, immunofluorescence labeling, and microscopy
Cells were fixed in 4% paraformaldehyde for 40 min at room temperature (RT). Cells were washed 4 × with PBS and incubated in blocking buffer (1% normal goat serum/0.5% Triton X-100 in PBS) for 15 min. Primary antibodies were diluted in the blocking buffer and incubated for 2 h at RT. Cells were then washed with the blocking buffer 2 × 5 min before incubation with Alexa Fluor-conjugated secondary antibodies (1 : 400) for 1 h. Cells were then stained with 20 ng ml−1 of DAPI (Sigma) for 2 min. Coverslips were washed with PBS (4 × ) and mounted in 90% glycerol. Negative controls were cells incubated without the primary antibody.
For staining with mouse anti-NeuN, rabbit anti-BrdU, and rabbit anti-Ki67, cells were treated with 2 N HCL for 15 min at RT before blocking. For mouse anti-O4 staining, live cells were stained following a protocol described previously.38
Images were collected with × 20 or × 40 objectives on a Nikon Eclipse 600 fluorescence microscope. The microscope was equipped with a CoolSnap camera (Photometrics). Images were collected using MetaView (version 6.2r2, Universal Imaging Corporation). Composite images were arranged in Adobe Photoshop (version 6.0). Data were analyzed by ANOVA and t-test assuming unequal variance using the Excel data analysis tool (Microsoft). Error bars represent the standard error.
NPC differentiation
NPC differentiation was carried out in cells plated onto coated glass coverslips following a protocol described.22 Half of the medium was replaced after 48 h of incubation in the respective defined medium. Cells were then fixed and stained for nestin, MAP2, S100B, or O4, as in the staining protocol described above.
Amyloid preparation and treatment
Aβ40 and Aβ42 peptides (American peptide) were prepared essentially as described by Dahlgren et al.39 Briefly, the peptides were dissolved in hexafluoro-2-propanol, divided into smaller aliquots, dried in a SpeedVac, and stored in −84°C. Soluble peptides were prepared by first diluting a dry stock in DMSO to 5 mM and then further diluting to 100 μM in neurobasal medium (Invitrogen) immediately before adding to the cells. Control culture NPCs were treated with similarly diluted DMSO. Soluble peptides were boiled in sample loading buffer with β-mercaptoethanol and resolved by electrophoresis on a 16% bicine-tris-acrylamide gel with urea. Soluble peptides migrated as monomers (Figure 8d, left). To prepare for aggregated Aβ, the peptides were dissolved as described above to 100 μM and left at 4°C for 3 days before further dilution and cell treatment. These aggregated Aβ peptides were boiled in sample loading buffer with β-mercaptoethanol and then resolved on a 4–20% precise protein gel (Pierce). Both of the blots were probed with 4G8 (Figure 8d). Unless noted otherwise, freshly diluted soluble Aβ peptides were used to treat the cells. Aβ40 and Aβ42 were at a final concentration of 1.5 μM unless noted otherwise. The final concentration of the vehicle DMSO present in the experimental samples was 0.0003 μl ml−1 for 1.5 μM of the peptides and 0.001 μl ml−1 for 5 μM of the peptides. No significant effect of DMSO on NPC differentiation was found within this range compared to no-DMSO controls (medium only).
Western blotting analyses of experimental samples
Cells were washed with cold PBS and scraped in a lysis buffer containing 150 mM NaCl, 50 mM Tris-HCL, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS plus a protease and phosphatase inhibitor cocktail (Sigma). After 30 min on ice, the lysate was briefly sonicated and precleared by centrifugation. Protein concentrations were determined by the bicinchromic acid method using a kit from Pierce. Equal amount of protein was boiled in SDS sample buffer and resolved on 7.5% or 12% SDS-PAGE gels. Proteins were transferred onto nitrocellulose, blocked for 1 h with 3% BSA in TBS-Tween and incubated with primary antibodies overnight. Chemiluminescence reactions were carried out using ChemiGlow (Alpha Innotech) and images were collected on Chemilmager (Alpha Innotech). At least three independent experiments were performed in each western blot analysis.
Dual luciferase assay
The luciferase reporter plasmids were as described.22 Two days after plating into 35 mm dishes, cells were transfected with a cell lineage-specific reporter construct (1 μg per dish) and the Renilla luciferase reporter as internal control for transfection efficiency (1 ng per dish) (CMV-PRL, Promega) using lipofectamine and plus reagents (Invitrogen). One day after transfection, cells were treated with amyloid in fresh NPC growth medium for 25 h before harvest. Lysates were prepared in passive lysis buffer, and dual luciferase assays were performed according to the manufacturer's protocols (Promega). Luciferase activity was measured by a dual-injector microplate luminometer (Turner Biosystems) using Veritas program as arbitrary units. The luminescence signal of a specific luciferase reporter plasmid was normalized to the internal control Renilla luciferase reporter intensity. The normalized luminescence signals representing specific luciferase reporter activities was shown in the graphs. Each experiment was performed in duplicates or triplicates.
Analyses of DNA synthesis phase in NPCs by BrdU incorporation and fluorescence microscopy
In these assays, P0 progenitor cells were mechanically dissociated and plated into poly-ornithine-coated 24-well plates with glass coverslips at 3 × 105 per well 2 days before the treatment. Cells were fixed and stained for BrdU, NeuN, S100B, and DAPI as described above. BrdU incorporation was visualized by immunofluorescence labeling using a rabbit anti-BrdU antibody. Images were collected with a Nikon Eclipse 600 fluorescence microscope.
To determine the effect of Aβ40 or Aβ42 on NPC proliferation during the entire treatment period of 25 h, BrdU (Sigma, 10 μM ml−1) was present in the culture medium concomitantly with Aβ40 or Aβ42.
To determine the effect of Aβ40 or Aβ42 on cells that were exiting S-phase before Aβ treatment, cells were preincubated with BrdU (20 μM ml−1) for 3 h before they were switched to fresh medium with Aβ40 or Aβ42 for another 25 h before fixation.
Flow cytometric analyses
The percentage of cells in the S-phase of cell cycle was evaluated by flow cytometry. Briefly, cells were trypsinized, washed with PBS, and fixed in 70% ethanol at 4°C overnight. Cells were then stained with propidium iodide for DNA contents. The data were collected using FACSCalibur with the CellQuest software (Beckman Dickinson). The samples were analyzed for cell cycle distribution using the software ModFit LT (Version 3.0, Verity).
Detecting DNA fragmentation by the comet assay
The Comet assay was performed essentially as described.40 Briefly, cells were dissociated in PBS from plates after fixation. Approximately 104 cells in 0.5 ml of PBS were combined with 1.5 ml of 1% low melt agarose (Fisher). After brief mixing, the solution was rapidly transferred to a glass microscope slide and allowed to solidify for 1 min. The slide was then placed into freshly prepared lysing solution (0.03 M NaOH/1 M NaCl) for 1 h at RT. After that, the slide was gently rinsed in distilled water and placed in a horizontal gel electrophoresis apparatus (BioRad) containing 40 mM Tris, 5 mM EDTA, 20 mM acetic acid, pH8.3. The electrophoresis apparatus was set at 150 V for 12 min. Slides were then rinsed with distilled water and incubated for 10 min in the dark with 2.5 μg ml−1 propidium iodide. Slides were rinsed in distilled water and images were taken immediately after staining using a Nikon Eclipse 600 fluorescence microscope using MetaView.
Quantifying condensed nuclei
The number of condensed nuclei was analyzed as described before.33
Analyses of cell type-specific, activated caspase-3 positive (caspase-3+) cells
Cells grown on glass coverslips were fixed and stained as described above. Ten random fields of images were obtained using a × 20 objective on a Nikon Eclipse 600 fluorescence microscope. Cells that were positive for a cell-type specific marker (NeuN+ or nestin+), for activated caspase-3 (caspase-3+), or positively stained for both a marker and the activated caspase-3, were recorded. Total cells per field were obtained by DAPI-stained nuclei. The percentage of NeuN+ or nestin+, and the percentage of caspase-3+ cells per total number of NeuN+ or nestin+ cells, were analyzed by two-tailed t-test and presented in the bar graphs.
Data collection and analyses
All the experiments described in this article were based on the initial observations by western blotting; such a method relies on less human intervention, although it might not be very sensitive. In experiments when cell counts were involved, a naive person collected the images from coded samples to minimize potential observer bias. In such experiments, 10 random fields were examined with about 150 cells per image. Cells in each field were then counted in a blinded manner. The total number of cells was based on DAPI staining. The percentage of a cell type per image field forms the data set (n=10) in each independent experiment. This data set was used for t-test analyses assuming unequal variances using the Excel data analysis tool (Microsoft). Such an experiment was repeated at least three times and a representative experiment was presented showing the mean and standard error in the graph. A statistical significance is declared when a P-value is ⩽0.05.
Western blots from multiple independent experiments were analyzed by t-test assuming unequal variances.33 Briefly, the specific protein band was normalized to the corresponding γ-tubulin after subtracting the background before comparison. The normalized mean and standard error are shown in the graphs.
Abbreviations
- Aβ:
-
beta-amyloid
- Aβ40:
-
Aβ1−40
- Aβ42:
-
Aβ1−42
- AD:
-
Alzheimer's disease
- NPC:
-
neural progenitor cell
- APP:
-
amyloid precursor protein
- PS:
-
presenilin
- MW:
-
molecular weight
References
Fryer JD, Holtzman DM . The bad seed in Alzheimer's disease. Neuron 2005; 47: 167–168.
Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci 2007; 27: 627–633.
Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E et al. Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat 2006; 27: 686–695.
Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J Neurochem 2006; 96: 732–742.
Shen J, Kelleher III RJ . The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA 2007; 104: 403–409.
Deng Y, Tarassishin L, Kallhoff V, Peethumnongsin E, Wu L, Li YM et al. Deletion of presenilin 1 hydrophilic loop sequence leads to impaired gamma-secretase activity and exacerbated amyloid pathology. J Neurosci 2006; 26: 3845–3854.
Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA et al. Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Brain Res Brain Res Rev 2003; 43: 1–16.
Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE et al. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci USA 2004; 101: 13363–13367.
Dong H, Goico B, Martin M, Csernansky CA, Bertchume A, Csernansky JG . Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience 2004; 127: 601–609.
Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ . Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease. J Comp Neurol 2006; 495: 70–83.
Zhang C, McNeil E, Dressler L, Siman R . Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. Exp Neurol 2007; 204: 77–87.
Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG et al. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol 2004; 188: 224–237.
Wang R, Dineley KT, Sweatt JD, Zheng H . Presenilin 1 familial Alzheimer's disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience 2004; 126: 305–312.
Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH . Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol 2005; 167: 151–159.
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC et al. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci USA 2004; 101: 343–347.
Ziabreva I, Perry E, Perry R, Minger SL, Ekonomou A, Przyborski S et al. Altered neurogenesis in Alzheimer's disease. J Psychosom Res 2006; 61: 311–316.
Boekhoorn K, Joels M, Lucassen PJ . Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis 2006; 24: 1–14.
Sugaya K . Possible use of autologous stem cell therapies for Alzheimer's disease. Curr Alzheimer Res 2005; 2: 367–376.
Peterson DA . Stem cells in brain plasticity and repair. Curr Opin Pharmacol 2002; 2: 34–42.
Filippov V, Kronenberg G, Pivneva T, Reuter K, Steiner B, Wang LP et al. Subpopulation of nestin-expressing progenitor cells in the adult murine hippocampus shows electrophysiological and morphological characteristics of astrocytes. Mol Cell Neurosci 2003; 23: 373–382.
Fukuda S, Kato F, Tozuka Y, Yamaguchi M, Miyamoto Y, Hisatsune T . Two distinct subpopulations of nestin-positive cells in adult mouse dentate gyrus. J Neurosci 2003; 23: 9357–9366.
Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH . A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell 2004; 116: 779–793.
McConnell SK, Kaznowski CE . Cell cycle dependence of laminar determination in developing neocortex. Science 1991; 254: 282–285.
Desai AR, McConnell SK . Progressive restriction in fate potential by neural progenitors during cerebral cortical development. Development 2000; 127: 2863–2872.
Cameron HA, McKay RD . Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol 2001; 435: 406–417.
Raponi E, Agenes F, Delphin C, Assard N, Baudier J, Legraverend C et al. S100B expression defines a state in which GFAP-expressing cells lose their neural stem cell potential and acquire a more mature developmental stage. Glia 2007; 55: 165–177.
Becker KA, Ghule PN, Therrien JA, Lian JB, Stein JL, van Wijnen AJ et al. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J Cell Physiol 2006; 209: 883–893.
Iwasaki K, Sunderland T, Kusiak JW, Wolozin B . Changes in gene transcription during a beta-mediated cell death. Mol Psychiatry 1996; 1: 65–71.
LaFerla FM, Hall CK, Ngo L, Jay G . Extracellular deposition of beta-amyloid upon p53-dependent neuronal cell death in transgenic mice. J Clin Invest 1996; 98: 1626–1632.
Zhu X, Mei M, Lee HG, Wang Y, Han J, Perry G et al. P38 activation mediates amyloid-beta cytotoxicity. Neurochem Res 2005; 30: 791–796.
Masliah E, Mallory M, Alford M, Tanaka S, Hansen LA . Caspase dependent DNA fragmentation might be associated with excitotoxicity in Alzheimer disease. J Neuropathol Exp Neurol 1998; 57: 1041–1052.
Jin K, Xie L, Mao XO, Greenberg DA . Alzheimer's disease drugs promote neurogenesis. Brain Res 2006; 1085: 183–188.
Chen Y, Liu W, Naumovski L, Neve RL . ASPP2 inhibits APP-BP1-mediated NEDD8 conjugation to cullin-1 and decreases APP-BP1-induced cell proliferation and neuronal apoptosis. J Neurochem 2003; 85: 801–809.
Lopez-Toledano MA, Shelanski ML . Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci 2004; 24: 5439–5444.
Mazur-Kolecka B, Golabek A, Nowicki K, Flory M, Frackowiak J . Amyloid-beta impairs development of neuronal progenitor cells by oxidative mechanisms. Neurobiol Aging 2006; 27: 1181–1192.
Herrup K, Neve R, Ackerman SL, Copani A . Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci 2004; 24: 9232–9239.
Serra HG, Duvick L, Zu T, Carlson K, Stevens S, Jorgensen N et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 2006; 127: 697–708.
Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE . Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res 1989; 24: 548–557.
Dahlgren KN, Manelli AM, Stine Jr WB, Baker LK, Krafft GA, LaDu MJ . Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 2002; 277: 32046–32053.
Olive PL, Banath JP, Durand RE . Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the ‘comet’ assay. Radiat Res 1990; 122: 86–94.
Acknowledgements
We thank Maojie Yang and Nelson Tauro for technical support, Drs. Fred H. Gage and Tomoko Kuwabara for generously supplying the luciferase reporter constructs, Dr. Steven Pfeiffer and Dr. Linda Van Eldik for providing O4 and S100B antibodies, respectively, Drs. Steven Barger, Robert Mrak, and Dr. Sue Griffin for helpful discussions, and Helena Liu for editorial assistance. We also like to express our gratitude to the anonymous reviewers who made constructive suggestions for revising this article. This project was initiated by a pilot fund from the Alzheimer's disease center Grant (AG19606RL) and made possible by the Inglewood foundation. Owing to editorial limitations, we apologize for not being able to cite all relevant publications.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by N Bazan
Conflict of interest
The authors declare no competing interest.
Authors’ contributions
YC conceptualized, designed and carried out most of the experiments, and prepared this article. CD carried out and collected some initial immunofluorescence data.
Rights and permissions
About this article
Cite this article
Chen, Y., Dong, C. Aβ40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ 16, 386–394 (2009). https://doi.org/10.1038/cdd.2008.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.94
Keywords
This article is cited by
-
Amyloid β-based therapy for Alzheimer’s disease: challenges, successes and future
Signal Transduction and Targeted Therapy (2023)
-
Molecular Mechanisms of Metal Toxicity in the Pathogenesis of Alzheimer’s Disease
Molecular Neurobiology (2021)
-
Partial reduction of amyloid β production by β-secretase inhibitors does not decrease synaptic transmission
Alzheimer's Research & Therapy (2020)
-
Aβ40 Oligomers Promote Survival and Early Neuronal Differentiation of Dentate Gyrus-Isolated Precursor Cells Through Activation of the Akt Signaling Pathway
Neurotoxicity Research (2020)
-
Deciphering the neuroprotective and neurogenic potential of soluble amyloid precursor protein alpha (sAPPα)
Cellular and Molecular Life Sciences (2020)
