
Abstract
Proteolysis of cellular substrates by caspases (cysteine-dependent aspartate-specific proteases) is one of the hallmarks of apoptotic cell death. Although the activation of apoptotic caspases is considered a ‘late-stage’ event in apoptosis signaling, past the commitment stage, one caspase family member, caspase-2, splits the cell death community into half – those searching for evidence of an apical initiator function of this molecule and those considering it as an amplifier of the apoptotic caspase cascade, at best, if relevant for apoptosis at all. This review screens past and present biochemical as well as genetic evidence for caspase-2 function in cell death signaling and beyond.
Similar content being viewed by others

Main
On the basis of structural and functional characteristics, caspases involved in apoptosis are grouped into ‘initiator’ caspases, containing a long prodomain, such as the death effector domain found in mammalian caspases-8 and -10 or the caspase activation and recruitment domain (CARD) found in caspases-2 and -9, as well as short prodomain containing effector caspases-3, -6 and -7. Initiator caspases are activated by autocatalytic processing, which is initiated upon prodomain-dependent dimerization of zymogens at specific activation platforms such as the ‘apoptosome’ or the ‘death-inducing signaling complex’ (DISC).1 The number of substrates cleaved by initiator caspases is thought to be limited to themselves and downstream effector caspases. The BH3-only Bcl-2 family member Bid appears to be the only exception, as it can also be cleaved by initiator caspases, to connect the extrinsic apoptotic pathway with the mitochondrial apoptosis signaling pathway.2, 3
A molecule at odds with current dogma is caspase-2 (earlier synonyms Ich-1: ICE/CED-3 homolog 1 or Nedd-2, for neural precursor cells-expressed, developmentally downregulated), the most conserved caspase across species, sharing 55% similarity with Caenorhabditis elegans caspase Ced-3.4, 5, 6 On the one hand, the predicted cleavage specificity of caspase-2 appears to place it more closely to effector caspases-3 and -7.7 On the other, caspase-2 contains a long CARD prodomain, through which it can interact with adaptor proteins, which is typical for initiator caspases. Overall, biochemical analysis supports a role of caspase-2 as an initiator caspase, but many contradictory findings render the exact function of this enzyme unresolved. In this review, we aim to give an overview on the steadily growing body of literature about this enigmatic enzyme, aiming to provide a synopsis on the present knowledge and to point out important aspects that still need to be tackled in future research.
Activation and Processing of Caspase-2
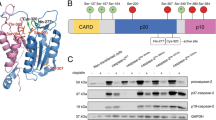
During apoptosis induction, caspase-2 is thought to be activated by CARD-mediated, dimerization-induced intrasubunit cleavage (@D333) and subsequent removal of the prodomain (@D169) as well as of the linker region (@D347), connecting its large p19 subunit with the small p12 fragment. In addition to these cleavage site residues, it was found that cystein C320 in the catalytic site is critical for autoprocessing of the protein.8, 9 Consistent with the ‘initiator’ status of caspase-2, a D333G mutant that remains a monomer, according to the examination by gel filtration, has no enzymatic activity in fluorescence substrate assays.9 In contrast, other mutations that prohibit processing, but not dimerization, retain catalytic and apoptosis-inducing activity, including a C436G mutant,9 which prevents the formation of the unique disulfide bond between two caspase-2 monomers.10
Although caspase-2 can form a stable dimer in solution, this may not be sufficient for full activation that may depend on specific activation platforms, which is analogous to the activation of initiator caspases-9 or -8.11, 12 The fact that caspase-2 is highly active and appears to autoprocess, even when expressed without prodomain, is believed to be due to self-association at high concentrations,9 and this has also been documented for other caspases when bacterially expressed.13 When present at endogenous levels, however, it is believed that the prodomain is essential for autoprocessing, which is supported by the observation that caspase-3 may be converted into an autoprocessing caspase with hugely enhanced killing activity when fused to the prodomain of caspase-2.14 In addition to activation by autoprocessing, several studies showed that caspase-2 is readily cleaved by caspases-3, and -8, and with less efficiency also by others.15, 16, 17 Taken together, these experiments show that for activating caspase-2, and initiator caspases in general, dimerization is the crucial activation step and that, different from effector caspases, the appearance of cleavage products in western blotting analysis is not synonymous with their activation.
Interaction Partners of Caspase-2
The number of molecules reported to interact with caspase-2 through its prodomain has steadily increased in recent years, suggesting the involvement of caspase-2 in different signaling pathways (Figure 1). The first among these proteins was the death adaptor molecule RAIDD/CRADD (receptor-interacting protein (RIP)-associated ICH-1/CED-3 homologous protein with a death domain; CASP2 and RIPK1 domain containing adaptor with death domain),20, 21 which has an N-terminal CARD and a C-terminal death domain (DD). Through its CARD, RAIDD/CRADD can bind to the CARD of caspase-2 by homophilic interaction (as well as to that of the C. elegans caspase Ced-3) that connects caspase-2 to different protein complexes. One such DD-containing protein was found to be RIP-1 kinase, which was in turn associated with TRADD (tumor necrosis factor receptor-1 (TNF-R1)-associated DD protein) and implicated a role for caspase-2 in TNF-R signaling.20 However, although the association of this complex could be documented upon overexpression, its physiological relevance remains doubtful considering that neither catalytically inactive caspase-2 nor dominant-negative RAIDD constructs were able to block TNF-R1-induced cell death,20 nor are cells from the relevant knock-out mice resistant to killing by TNF.22 Nonetheless, some studies have in fact indicated a role for caspase-2 in TNF-induced cell death,23, 24 but that may not require a physical association of the protease with the receptor complex (see below). In addition, a role for caspase-2 in non-apoptotic signaling emanating from the TNF-R remains to be thoroughly investigated.

A summary of experimentally validated and suggested molecules involved in the control of expression and processing of caspase-2, and in the modulation of its enzymatic activity. PKCK2, protein kinase CK2; CAMK2, calcium/calmodulin-dependent protein kinase 2; GMEB1, glucocorticoid modulatory element-binding protein 1; cIAP1, cellular inhibitor of apoptosis protein 1; PIDD-CC, C-terminal fragment of p53-induced protein with a death domain; RAIDD, RIP-associated Ich-1/Ced-3-homolog protein with a death domain; Ubc9, SUMO-conjugating enzyme 9; PACAP, pro-apoptotic caspase adaptor protein; DEFCAP-L, long isoform of death effector filament-forming Ced-4 like apoptosis protein; ARC, apoptosis repressor with a CARD. ? signifies that the mechanism of interaction is unknown
Caspase-2 can also be recruited to the DISC upon Fas ligation in human T- and B-cell lines, and this was reported to associate with its activation.25 The DISC recruitment even persisted in caspase-8-deficient cells but then caspase-2 was no longer processed, suggesting its activation is a downstream event of apoptosis induction.25 In contrast, processing of caspase-8 was independent from caspase-2, and thus the significance of caspase-2 recruitment in this context remains obscure. Furthermore, it should be noted that two other studies detected no association of caspase-2 with the DISC26, 27 and overexpression of dominant-negative versions of neither RIP-1 nor RAIDD, which may mediate the recruitment of caspase-2 to death receptors (DRs), prevents Fas-induced apoptosis in lymphocytes.28
The most prominent, although indirect, interaction partner of caspase-2 is PIDD (p53-induced protein with a DD), which together with RAIDD and caspase-2 forms the so-called PIDDosome.29 This activating platform was found to self-assemble upon co-incubating all three components in vitro (at 37°C but not at 4°C) as well as in various cell lines upon overexpression, but seems to form even spontaneously upon cell rupture. When the induction of enzymatic activity of caspase-2 was examined, estimated from binding of b-VAD-fmk (a biotinylated synthetic caspase inhibitor),30 it was found that for activation by PIDD there was a strict requirement for RAIDD, as RNA interference against RAIDD blocked caspase-2 processing. Consistently, resolution of the crystal structure of the PIDD DD and RAIDD DD complex indicated that five C-terminal PIDD fragments (termed PIDD-CC), seven RAIDD and seven caspase-2 molecules can form a high-molecular-weight complex of ∼700 kDa.31 Thus, the PIDDosome appeared to be the long-sought physiological activation platform for caspase-2. However, the recruitment of caspase-2 into a ∼700-kDa high-molecular-weight complex was already reported earlier, but co-migration of RAIDD was not detected in this study.32 Read and colleagues33 also reported that they were unable to detect the caspase-2 complex formation in etoposide-treated HeLa cells, raising the possibility that temperature shift-induced formation of caspase-2-containing high-molecular-weight complexes may differ in their composition from those that assemble during apoptosis induction. It can be noted that caspase-2 knockout mouse embryonic fibroblasts (MEFs) were only partially resistant to PIDD overexpression, whereas RAIDD knockout cells were fully protected, suggesting that PIDD can also induce cell death independent of caspase-2.
A member of the mammalian Ced-4 protein family named DEFCAP (death effector filament-forming Ced-4-like apoptosis protein), also known as NALP1 (NACHT, LRR and PYD domains-containing protein 1) or CARD7, identified in a CARD-domain homology database search, is able to interact with capsase-2.34 Similar to Apaf-1, DEFCAP has a nucleotide-binding domain and a CARD, and both isoforms of the protein identified, DEFCAP-L and DEFCAP-S, co-immunoprecipitated with overexpressed caspase-2 or -9, but did not bind to caspases-3, -8 and -10 or the adaptors FADD or RAIDD. Overexpression of the long, but not the short, isoform in MCF-7 cells induced moderate cell killing, which depended on the presence of its CARD and could be inhibited by a pan-caspase inhibitor.34 Its relation to caspase-2 activation has subsequently not been followed up, whereas other studies have identified it as a component of a protein complex required for pro-interleukin-1β processing, the so-called inflammasome, which contains caspase-1, -5, Pycard/Asc and DEFCAP/NALP1.35 Interestingly, delayed caspase-1 processing has been reported in caspase-2−/− macrophages infected with salmonella.36 This leaves us with the possibility that caspase-2 may even be a modulator of inflammasome activity, similar to caspase-11 or -12.37 Further support for this idea can be found in the observation that caspase-2 is recruited through its CARD to a complex with TNF-R-associated factor 2 and RIP-1 kinase. This occurs in a CARD-dependent manner, but does not require its proteolytic activity and contributes to the activation of nuclear factor-κB (NF-κB) and p38 mitogen-activated protein kinase.38 Size exclusion chromatography indicated that this complex again has an apparent molecular mass of approximately 670 kDa, similar to that of the PIDDosome (697 kDa31). Subjecting caspase-2−/− mice to bacterial or viral infection will help in determining the physiological relevance of these observations.
A number of additional caspase-2-binding molecules have been identified on the basis of protein–protein interaction screens (Table 1). As their interaction with caspase-2 was subsequently only observed upon overexpression, we will not further speculate on their putative role, because some may be experimental artefacts.
In summary, all these data suggest that caspase-2 becomes fully activated in a platform similar to the apoptosome, but, so far, no evidence has been presented that the activation of caspase-2 strictly depends on its recruitment into such a complex. It is equally well possible that simple dimerization of caspase-2, either due to an increase in local protein concentration and/or due to post-translational modification, can prompt its full activation.
Fine-Tuning of Caspase-2 Activity
A unique feature of the PIDDosome is that its formation appears to depend on the nature and the severity of the proapoptotic stimulus. It was proposed that the PIDD-CC fragment, generated by autoproteolysis (@S588) and capable of interacting with RAIDD and caspase-2, is formed only when cells are destined to undergo apoptosis, whereas the longer PIDD-C fragment, also generated by autoprocessing (@S446), is preferentially produced when cell repair mechanisms are needed to be switched on in response to low-dose DNA damage.41, 42 How or if autoprocessing is fine-tuned remains unclear at present. The cellular response may simply depend on the relative expression levels of PIDD isoforms that either possess or lack the critical cleavage site @S588 required for the formation of PIDD-CC.43
A more conventional way of keeping caspase-2 activity in check is by inactivation due to phosphorylation at critical residues (Figure 1). As shown in human cancer cell lines, protein kinase CK2 (PKCK2) can phosphorylate caspase-2 at serine-157, and this phosphorylation prevented dimerization, processing and enzymatic activation of caspase-2.44 In addition, after the induction of apoptosis by TRAIL (tumor necrosis factor-related apoptosis-inducing ligand), but also in response to DNA-damaging agents, PKCK2 activity was inhibited, while in parallel caspase-2 activity was enhanced.44 Interestingly, the sequence around serine-157 in caspase-2 is highly conserved in mammals, but not in chicken or Xenopus, indicating that this specific mechanism is a rather new evolutionary twist. In contrast, phosphorylation of caspase-2 at serine-135 by calcium/calmodulin-dependent protein kinase II (CaMKII) is more conserved, as it was also detected in extracts from Xenopus oocytes using either human or Xenopus caspase-2.45 Whether this phosphorylation, similar to that by PKCK2, prevented dimerization of caspase-2 was not tested, but it clearly prohibited the CARD-dependent interaction of caspase-2 with RAIDD, its processing and enzyme activity and, importantly, caspase-2-dependent apoptosis. Upon exhaustion of oocyte nutrients, CaMKII activity was reduced, which resulted in a de-repression of caspase-2 activity and developmentally triggered apoptosis. Consistently, caspase-2−/− mice were reported to show increased oocyte numbers. In this context, it is worth mentioning that caspase-2 expression in various cell lines was induced by lipid-lowering drugs through binding of sterol regulatory element-binding protein at functional response elements within the 5′-flanking region of the caspase-2 gene,46 which led to the suggestion that caspase-2 may act as a more general sensor of cellular nutritional status.
Recently, it was reported that, both in damaged and in non-stimulated cells, a p53/p21-dependent pathway for the repression of caspase-2 expression may exist.47 Although this may seem counterintuitive for apoptotic cells, it supposedly serves to inhibit caspase-2 activity under resting conditions, and this block may be overcome once DNA damage-induced p53 triggers transcription of PIDD.
A further proposed way of controlling caspase-2 activity is differential expression of caspase-2L, referred to as caspase-2, and caspase-2S.5 Caspase-2S is generated by the inclusion of a 61-bp exon in the 3′-region of caspase-2 pre-mRNA, which causes a premature stop of translation and production of a hypothetical 33 kDa isoform with anti-apoptotic properties. Overexpression of caspase-2S was shown to protect Rat-1 and NIH-3T3 cells against cell death induced by serum withdrawal,5, 48 and to delay occurrence of certain apoptotic features, including processing of caspase-2L, in etoposide or Fas-stimulated U937 human leukemic cells. At first sight this may be explained by formation of inactive caspase-2L/S heterodimers at the expense of pro-apoptotic, active caspase-2L homodimers.49 However, mutation of the active site cysteine prevented this protective effect, suggesting that caspase-2S may even selectively process and thereby activate a cytoprotective substrate. It was further shown that relative expression levels of long and short isoform mRNAs were differentially regulated by topoisomerase inhibitors, causing an increase in caspase-2S mRNA, whereas agents causing DNA damage by other means were without effect.50, 51, 52 The control of transcription by topoisomerase inhibitors was in turn regulated in a cell cycle-dependent manner by PKC-ζ,53 which is known to act cytoprotective in various cell lines.54, 55 However, until now no unequivocal western blot analysis has shown the expression of caspase-2S because its predicted mass is very close to that of the intermediate cleavage product of caspase-2L, but 2D-electrophoresis or mass spectrometry analysis may help to resolve that issue. Furthermore, caspase-2S mRNA was shown to be very short-lived and may thus normally not be expressed or only at very low levels.56 However, mRNA encoding both isoforms has been detected readily in the mouse brain.22 Interestingly, the developmental death of facial motor neurons appeared temporarily enhanced in caspase-2−/− mice, whereas factor deprivation-induced apoptosis of sympathetic neurons from newborn mice was normal. This suggests a protective role for caspase-2S in neurodevelopmental apoptosis, whereas it may be redundant once the central nervous system has been established. Taken together, these data indicate that an inhibitory isoform of caspase-2 can be expressed occasionally under certain stress conditions, but may also be limited to specific cell types such as neurons. Its mode of action remains to be determined.
Proteasomal degradation of caspase-2 was reported to be induced by the PKC inhibitor Rottlerin in HeLa and ovarian cancer cells. This effect was apparently independent of kinase inhibition but was considered to contribute to the anti-apoptotic function of this drug.57 Finally, the small ubiquitin-like modifier (SUMO) and the SUMO-conjugating enzyme 9 (Ubc-9), constituents of the PML-NBs(promyelocytic leukemia nuclear bodies), were found to interact with caspase-2.40 Co-immunoprecipitation of overexpressed proteins and microscopic co-localization of caspase-2 and SUMO at PML-NBs corroborated this interaction. In fact, it was shown that SUMO can be conjugated to a lysine residue within the CARD of the protease (@K60), if caspase-2, SUMO and Ubc-9 are co-transfected into 293T cells, and that endogenous sumoylated caspase-2 is detectable in human neuroblastoma cells.40 As caspase-858 and caspase-759 may as well be sumoylated and in this form can locate to the nucleus, the conjugation of caspase-2 may be important for its subcellular localization (see below).
Subcellular Localization of Caspase-2: A Functional Relationship?
One remarkable features of caspase-2 is that, different from all other caspases, it can also be found constitutively in the nucleus.60, 61, 62 This unusual subcellular distribution was originally believed to depend on a bipartite nuclear localization signal (NLS) contained in the CARD of the prodomain,63, 64 but subsequent analysis showed that the NLS is contained in the C-terminal linker of the prodomain.65 Formal evidence for this was provided by showing that upon mutation of a single conserved lysine residue in this region, caspase-2–GFP (green fluorescent protein) fusion proteins no longer accumulated in the nucleus, but aggregated in the cytoplasm. Moreover, it was shown that caspase-2 is transported into the nucleus through interaction of this NLS with the α/β-importin heterodimer, which are constituents of the classical nuclear import machinery. Somewhat surprisingly, the subcellular distribution of caspase-2 was shown to be independent of its state of processing, as a protease-dead mutant did not show any localization difference compared with the wild-type form.64 Interestingly, the capacity to enter the nucleus does not correlate with its potency to kill cells when overexpressed.65
It was noted that within the nucleus, caspase-2 often formed dot-like aggregates or filaments,63, 64 and generally the latter are considered overexpression artefacts. However, at least the former structures were supposed to colocalize with PML-NBs (reviewed in more detail in Trends Biochem Sci 2007 of Ref. 68).66 These structures are involved in numerous cell functions, including DNA repair and apoptosis, are dynamic in their composition and at least transiently they seem to contain proteins that could interact with caspase-2.67 One such protein, SP100, a protein involved in transcriptional regulation, contains a putative CARD, by which it might bind to caspase-2, although this has not yet been verified.68 In line with such an interaction, no specific aggregates, but a rather uniform distribution of caspase-2 across the nucleus was observed upon transfection with a CARD-deficient caspase-2 construct.65 Interestingly, in 293 HEK and Jurkat T cells, all caspases expressed as GFP fusions, with the exception of caspase-2, showed exclusive or mainly cytoplasmic localization.64 However, although localization of caspase-2 still prevailed upon expression in other cell types, caspases-1 and -3 appeared to distribute between the cytoplasm and the nucleus, suggesting either cell type specific differences regarding this behavior or experimental artefacts, due to overexpression.
Some early studies also suggested the existence of a mitochondrial pool of caspase-2 (and also caspase-9).60, 69 However, subsequent studies on three cell types and highly purified mitochondrial preparations could not confirm these findings neither under control nor under apoptotic conditions.70 Nonetheless, given that caspase-2 is the only caspase identified with a putative mitochondrial localization signal,70 it appears too early to exclude this possibility.
A unique subcellular localization of caspase-2, not documented for any other caspase, is that at the Golgi complex. In two studies, using a range of independently generated as well as commercial antibodies, confocal scanning microscopy indicated that, besides nuclear and cytoplasmic staining, caspase-2 can specifically localized to Golgi-like structures and in fact colocalizes with the Golgi membrane markers giantin, golgin-16061 and β-coatomer protein.62 Although this was only shown for HeLa cells in both studies, Golgi localization was reportedly also seen in human keratinocytes and salivary gland cells.61 Immunoblot analysis confirmed that a pool of caspase-2 can be detected in membrane fractions enriched in Golgi marker proteins. Closely related to this, one report suggested the presence of caspase-2 at the endoplasmic reticulum (ER), as detected by microscopy and cell fractionation.71 Considering the continuous trafficking between the ER and the Golgi, both compartments may be similarly affected by proapoptotic stimuli generally referred to as ER stress. As several studies suggest caspase-2 to act as an initiator caspase in this case (see below),72, 73 its localization at the ER and the Golgi would thus seem of physiological relevance. It will be interesting to find out if possible activators such as PIDD or RAIDD can also be found at such sites, or may translocate to such sites in response to ER stress. So far, RAIDD has been detected both in the cytosol and nucleus, where it co-localized in nuclear dots with overexpressed caspase-2 in a CARD-dependent manner.74 Somewhat contradictory, both, full length PIDD as well as the fully processed PIDD-CC fragment, which is supposedly rate-limiting for caspase-2 activation, remained mostly in the cytosolic compartment even upon induction of DNA damage.41, 42 It can be noted that in several cell types, caspase-2 remained mainly nuclear even after inducing cell death or its translocation to the cytoplasm occurred only late during apoptosis.75, 76, 77 Thus, neither activators nor inhibitors restricted to the cytosol would be functional. However, in some cell lines, early and stimulus-specific nuclear–cytoplasmic translocation was observed,77 but, overall, it appears that a systematic approach is needed to clarify the question where caspase-2 activation is first initiated and whether cytosolic versus nuclear activation of caspase-2 has distinct requirements.
Can we Learn from Identifying Caspase-2 Substrates?
A common feature of caspases is that they cleave their substrates next to an aspartate residue at a specific recognition sequence of four amino acids, an exception being again caspase-2, which requires a motif of five amino acid residues for optimal cleavage.78, 79 In two seminal papers, it was shown that the preferred substrate for caspase-2 should contain the sequence VDVAD,78, 79 but unfortunately artificial substrates based on this pentapeptide are equally well processed by caspases-3 and -7.80 In line with this, immuno-depleting cell extracts of caspase-2 did not significantly reduce its VDVADase activity, indicating that the main share of this protease activity can in fact be attributed to other caspases. Along that line, we observed that VDVADase activity in caspase-2−/− cell extracts is comparable with that observed in wild-type extracts (GK and AV, unpublished). Similarly, the VDVAD-based inhibitors available are not at all selective for caspase-2,80, 81 and caspase-2 was also found to be insensitive to inhibitor of apoptosis proteins-mediated caspase inhibition,3, 82 although one study suggested sensitivity to cIAP-1 (cellular inhibitor of apoptosis protein 1).83 Thus, it is has proven notoriously difficult to detect proteins that are bona fide caspase-2 substrates. In summary, experiments using VDVAD-based compounds as substrates or inhibitors to determine caspase-2 activity in cells or extracts that contain other caspases do not allow any conclusion about the involvement and/or hierarchical position of this protease in a given signaling pathway.
Nevertheless, from presumed context-specific activation of the protease and from in vitro cleavage of proteins by recombinant caspase-2, a few substrates have been identified (Table 2), the probably most important one being the proapoptotic BH3-only protein Bid. This well-established caspase-8 substrate3, 4 couples the extrinsic and intrinsic apoptotic pathways and upon cleavage to its truncated form tBid, translocates to the mitochondria and participates in the mitochondrial outer membrane permeabilization (MOMP).92 The formation of tBid occurs by cleavage at aspartate-59, and this can be catalyzed by caspases-1,-2, -3 and -8 and by granzyme B in vitro.3, 4 The suggestion that caspase-2 may process Bid in vivo came from the observation that it was apparently activated upstream of cytochrome c release in some cell models and that in these models apoptosis was inhibited upon silencing of caspase-2.26 However, other investigators suggested that MOMP by caspase-2 occurred independent of Bid cleavage, by direct action of processed caspase-2 on either an unidentified membrane protein or lipid.83, 93 Supporting this hypothesis, it was reported that caspase-2 can cause the release of cytochrome c from artificial liposomes and from mitochondria incubated with caspase-2 in the absence of cytosolic components.16, 94 However, this was again challenged because neither Bid-knockout MEFs nor HeLa cells stably transfected with RNA interference against Bid underwent apoptosis or released cytochrome c in response to the overexpression of caspase-2, suggesting that the requirement for Bid may at least be cell type and stimulus-dependent,95, 96 as it appears to be in the context of Fas-induced apoptosis.97
The capsase-2-mediated cleavage of other caspases has been repeatedly investigated, as this would be indicative for its role as an initiator caspase. However, besides processing its own zymogen, most studies found no indication that it could cleave any other caspase.15, 16, 21 The only exception described so far was the cleavage of caspase-7 upon co-expression of both caspases in yeast.98 In addition to caspase-7 processing, this also resulted in enhanced cleavage of the presumed caspase-3/-7-specific substrate DEVD-AFC without a concomitant enhanced VDVADase activity, although results based on this type of readout must be interpreted with caution.80 Furthermore, this caused a dramatic increase in yeast cell death, which was absent when only caspase-7 was expressed or when caspase-2 was co-expressed with other caspases. However, this phenomenon has so far not been confirmed in mammalian cells.
The only caspase-2-specific substrate identified so far is the peripheral Golgi membrane protein golgin-160.61 Although it may be cleaved by other caspases as well at different motives, golgin-160 was shown to possess a caspase-2-specific cleavage site, aspartate-59, not susceptible to other caspases and processing at this site during apoptosis appears to precede that at other residues. Caspase-2-mediated cleavage was confirmed in vitro and, upon overexpression of golgin-160 also in vivo. Expression of a D59E mutant of golgin-160 in HeLa cells delayed disintegration of the Golgi complex,61 and HeLa cells stably expressing a golgin-160 variant mutated at three caspase cleavage sites showed significantly delayed cell death, when exposed to agents inducing ER stress.99 However, assuming that overexpression of cleavage-resistant mutants mimics the effect of synthetic or natural caspase inhibitors by classic substrate inhibition, it is not surprising that expression of cleavage fragments of golgin-160 alone had no impact on cell viability, leaving the mechanistic role of golgin-160 proteolysis unresolved.99
A more relevant substrate of caspase-2 may be DFF-45/ICAD (DNA fragmentation factor 45/inhibitor of caspase-dependent DNase).84, 100 Originally, DFF-45/ICAD was described as a caspase-3 substrate, but early on, it was noticed that other caspases might as well process this protein.101, 102 Recently, it was shown that in HeLa cells exposed to a membrane-permeable constitutively active caspase-2 construct, ICAD was cleaved and subsequent DNA fragmentation was induced.85 As the ICAD/CAD heterodimer is supposedly activated in the nucleus,103 an in vivo role for nuclear caspase-2 in this process appears plausible and it may be interesting to assess DNA fragmentation in cells double-deficient for caspases-3 and -7.
A putative substrate of significant clinical relevance is Huntingtin (Htt), the mutant form of which underlies the eponymous disease. Several studies suggest that caspase-mediated cleavage of Htt is critical for inducing neurodegeneration and in vitro Htt was processed by caspase-2, creating a fragment also detected in vivo.87 Htt co-immunoprecipitated with caspase-2 in 293 cells overexpressing both proteins and, more importantly, also in extracts from mouse cortex expressing a human Htt transgene. In addition, expressing dominant-negative caspase-2 protected neurons against Htt expression induced cell death. Both proteins colocalized within cells, and caspase-2 mRNA was found enhanced in brain regions showing early signs of the disease.87 However, interaction, partial protection and co-localization were also detected for caspases-6 and -7, and a more recent study indicates that caspase-6-mediated cleavage of Htt is of critical importance for pathology.104 Nonetheless, it cannot be excluded that caspase-2 is important in the early stages of the disease and may act in synergy with caspase-6 by promoting cleavage of mutant Htt at distinct sites.
Additional substrates that can also be cleaved by caspase-2 include poly-ADP-ribose polymerase,88 the cortical membrane skeleton proteins αII/βII-spectrin89 and plakins, which are proteins involved in cell junction complexes.90 Finally, histone deacetylase-4 is an in vitro substrate of caspase-2, releasing fragments identical to those seen during ultraviolet radiation-induced apoptosis.91 However, although processing of all these substrates would appear appropriate in an apoptotic context, these proteins are also in vitro targets of several other caspases, and therefore they are only of limited use to identify the physiological role of caspase-2.
Initiator versus Effector Function and Possible Physiological Roles of Caspase-2
Caspase-2 in the DNA damage response
It has been repeatedly proposed that caspase-2 plays an important role in cell death induced by genotoxic stress (reviewed in more detail in Zhivotovsky and Orrenius82), although, depending on the specific mode of DNA damage induced or the cell type investigated, caspase-2 appeared to act either upstream or downstream of other caspases (Figure 2). For example, when treated with DNA-damaging agents etoposide or cisplatin, caspase-2 appears to act as an initiator in various human tumor cell lines, as caspase-2 knockdown by RNA interference or antisense oligonucleotides suppressed Bax translocation, release of mitochondrial proteins and cell death to a variable degree.93, 105, 106, 107 Furthermore, siRNA against caspase-2 was at least partially protective against DNA damage-induced apoptosis in A549 human lung epithelial cells,107, 108 osteosarcoma U2-OS cells,109 primary mouse renal tubular epithelial cells,110 colon adenocarcinoma H1299 cells111 and human prostate cancer cell lines.112 Consistently, cell death induced by the overexpression of caspase-2 was abrogated in HeLa cells with silenced Bid and, not surprisingly, MEFs deficient for Bax/Bak.95

A schematic model summarizing the most important apoptotic pathways with a suggested involvement of caspase-2 activity, i.e. cell death in response to DNA damage, ER stress, heat shock and death receptor (DR) ligation. ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3 related; PIDDosome, protein complex consisting of PIDD, RAIDD and caspase-2; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; FADD, Fas-associated death domain
In summary, all these results suggest that caspase-2 can act upstream of mitochondrial events as an initiator that triggers cytochrome c release and subsequent apoptosome formation, either directly or through generation of tBid (Figure 2). At least two things are worth mentioning here. Some of these data have been generated using siRNA against caspase-2, which apparently affects cell death responses independent of its ability to repress caspase-2 expression105 and, second, similar phenomena, including delayed Bax translocation and cytochrome c release, have been observed in MEFs double-deficient for both caspases-3 and -7, indicating that effector caspases can feed back on events considered to be at the level of or upstream of MOMP.113
However, a large number of other studies contradict the idea that caspase-2 may act as a initiator of DNA damage-induced cell death and assign caspase-2 the role of a downstream caspase in genotoxic cell death, or question their relevance for this process at all. Caspase-9-deficient Jurkat cells reportedly resist apoptosis induction by etoposide or staurosporine, but despite of displaying normal Bax translocation and cytochrome c release, neither caspase-2 processing nor its enzymatic activity was detected. Both features were restored upon re-expression of caspase-9.114 Furthermore, silencing of caspase-2 in normal Jurkat T cells afforded no protection against DNA damage, whereas expression of a dominant-negative caspase-9 construct or of non-cleavable Bid inhibited processing of caspase-2 and apoptosis.3 It seems worth mentioning that, other than the contradictive findings on DNA damage induced by genotoxic agents, most studies agree that, in response to ultraviolet radiation-induced damage, caspase-2 acts as a downstream caspase in a number of cell systems, including MCF-7, HCT-116, MEFs and mouse thymocytes.62, 75, 96, 115 This may be related to the fact that ultraviolet radiation-mediated DNA damage preferentially leads to the activation of ATR (ataxia telangiectasia and Rad3 related), whereas double-strand breaks induced by irradiation and many anticancer agents also engage ATM (ataxia telangiectasia mutated) to activate the DNA damage response machinery and may engage the caspase cascade differently.
Recently, the hierarchical position of caspase-2 in DNA damage-induced apoptosis was also addressed using an ‘in situ trapping’ approach, where Jurkat cells were pre-incubated with a cell-permeable, biotinylated pan-caspase inhibitor (b-VAD-fmk), which then supposedly binds to the first caspase activated in response to a particular stimulus. Analysis of the caspase trapped in Jurkat cells exposed to a range of etoposide concentrations revealed that caspases-9 and -8 were trapped, suggesting that both can be activated upstream of caspase-2.30
Equally relevant, however, is the fact that primary cells from caspase-2 knockout mice respond to genotoxic stress similar to wild-type cells22, 62 (own unpublished data). Accordingly, Marsden et al.116 investigated caspase-2/-9 double-deficient primary MEFs and lymphocytes, but also in these cells, no protection against DNA damage was detected, which was also not very pronounced in caspase-9 knockout cells, but compensatory phenomena, as observed in hepatocytes derived from caspase-9−/− or caspase-3−/− mice, may in part account for these observations.117 However, in thymocytes lacking either Apaf-1 or caspase-9, processing of caspase-2 was not observed after γ-irradiation.62 Furthermore, lymphocyte apoptosis was also reported to be normal in raidd−/− mice.33 In addition, although myeloid cells lacking caspases-2 and -9 simultaneously showed somewhat delayed cell death when challenged with etoposide or doxorubicin, a clonogenic survival advantage was not observed.118 This strongly contradicts the idea that caspase-2 may act upstream of the mitochondria because clonal survival is clearly provided by overexpression of Bcl-2 after DNA damage or growth factor deprivation119 as well as by loss of caspase-8, overexpression of the viral caspase-8 inhibitor CrmA or a dominant-negative version of FADD after Fas ligation.120, 121
Taken together, these genetic studies place caspase-2 downstream of apoptosome formation and caspase-9 activation, suggesting a highly redundant role for caspase-2 in cell death initiation after DNA damage, but there seems to be a new twist to the tale. In zebrafish embryos bearing a loss-of-function mutation of p53, radiosensitivity can be restored by knockdown or pharmacological inhibition of the checkpoint kinase 1 (Chk1).122 Interestingly, cell death observed in embryos with impaired Chk1 function exposed to radiation could specifically be blocked by concomitant ablation of caspase-2 expression, but not any other evolutionary conserved cell death mediator. Intriguingly, similar observations were made in human cancer cell lines, although with more varying effects, leading to the conclusion that this Chk1-suppressed caspase-2 activation pathway may be conserved in vertebrates. Somewhat puzzling, this type of caspase-2-mediated cell death appeared to occur in the absence of any detectable release of mitochondrial factors or even caspase-3 activation, raising the question which substrates may be directly cleaved by caspase-2 to promote such cell death. As mentioned before, DFF45/ICAD may be such a target.85 This study actually places caspase-2 neither upstream nor downstream but in parallel to the mitochondrial as well as the DR pathways of apoptosis (Figure 2). If true, conditional expression of caspase-2 overexpression should be sufficient to induce apoptosis in bax−/−bak−/− MEFs, but this has been excluded earlier by others.95 One additional caveat, however, remains. Pharmacological sensitization to radiation by Go 6976 in cells with inactive p53 is unlikely solely because of specific inhibition of Chk1, as it was originally developed as a PKC-selective inhibitor, targeting classical as well as novel PKCs in the nanomolar range.123 Hence, the effects observed using this drug may not solely be due to inhibition of Chk1. However, investigating radiation responses in chk1/p53 or caspase-2/p53 double-mutants will help to clarify this issue.
Caspase-2 and ER stress
A cell death initiating function for caspase-2 was repeatedly suggested for ER stress.71, 72, 73, 124 Given that caspase-2 seems to localize at the ER and the functionally linked Golgi complex,61, 62, 71 its role as an apical sensor of stress signals in this compartment would make sense (Figure 2). How ER stress translates into caspase-2 activation is still unknown, but once activated, it seems to exert its effects again through cleavage of Bid. Thus, caspase-2-directed siRNA as well as pharmacological inhibition (although less selective) protected SV40-transformed MEFs against ER stressors, significantly reduced Bid cleavage, cytochrome c release and cell death.73 Partial resistance was also seen in Bid-deficient MEFs and thymocytes. However, as resistance of caspase-2 knockdown cells remained incomplete, even in the absence of Bid, it is very likely that other BH3-only proteins such as Bim and Puma can contribute significantly to this form of cell death,109, 125 presumably in a caspase-2-independent manner.73 Further evidence for a role of caspase-2 in ER stress was provided by exposing caspase-8-deficient SH-SY5Y neuroblastoma cells to thapsigargin, which led to processing of caspase-2, but not caspase-9.72 ER stress stimulated caspase-2 activation and cell death in HeLa cells,71 and PC12 pheochromocytoma126 was ameliorated by caspase-2 siRNA or Bcl-x(L) or Bcl-2 overexpression. All these studies yielded contradicting results regarding the question whether caspase-2 activation occurs upstream or downstream of MOMP in ER stress. Using the ‘in situ trapping’ approach (see above), the first caspase activated by ER stressors in thymocytes, however, was caspase-9.30 Overall, caspase-2 seems to respond to ER stress in a cell-type-dependent manner and is clearly not the sole molecule relaying ER stressors to MOMP. In some instances, caspase-2 may merely act as an amplifier of stress stimuli, participating in a pro-apoptotic feedback loop or may be redundant (Figure 2). Along that line, loss of caspase-2 also failed to protect from neuronal death caused by forebrain ischemia or in a model of amyotrophic lateral sclerosis,22 a progressive neurodegenerative disorder triggered by ER stress-mediated loss of motor neurons.127
DR-mediated apoptosis
The exact contribution of caspase-2 to DR-mediated apoptosis remains controversial, because genetic evidence for additive or synergistic action with caspase-8 is still lacking, as is genetic evidence showing that DR-induced apoptosis is impaired in cells from caspase-2 knockout mice. However, in a number of leukemic cells stably transfected with a caspase-2 antisense construct, Fas-mediated apoptosis was significantly delayed.26 Caspase-2 knockdown also inhibited or delayed the activation of caspase-826, 44 and was partly protective against TRAIL-induced apoptosis in Jurkat cells,3 and caspase-2 activation also reportedly preceded that of caspase-8 in TNF-induced apoptosis in breast cancer cells.23 However, the proximal caspase trapped using b-VAD-fmk in Jurkat cells stimulated with death ligands was caspase-8.30 Nevertheless, in some type II cells (cells that require caspase-mediated Bid cleavage for efficient apoptosis induction), such as primary mouse hepatocytes, caspase-2 was supposed to be required for optimal DR-induced cell death.24, 128 Taken together, these studies place caspase-2 again upstream of caspase-8 or suggest its parallel activation (Figure 2). However, caspase-2 processing in the DISC of both type I (do not require Bid cleavage) and type II cells appears to depend on caspase-8,25 suggesting that in either case caspase-2 may mainly act as driver of a positive feedback loop through Bid cleavage. As caspase-8 can readily cleave Bid, the requirement for caspase-2 to amplify DR-mediated apoptosis remains questionable. However, DR-mediated activation of caspase-8 is also required for proliferative responses, for example in T cells, and hence it remains possible that caspase-2, recruited to the DISC along with caspase-8, may be relevant for non-apoptotic signaling events induced by DR ligation.
Caspase-2 and heat shock
Using the Jurkat b-VAD model system mentioned earlier, caspase-2 was reported to be the most apically activated caspase identified in heat-stressed cells (Figure 2) and cell death ensuing from this was partially dependent on RAIDD and Bid processing.30, 96, 129 Surprisingly, it was shown that heat shock also led to Fas ligand expression and activation of caspase-8.30 Neutralizing Fas before heat stress conferred additional protection to already partly resistant caspase-2 knockout MEFs, suggesting synergy between caspase-2 and -8 during heat stress-induced apoptosis. However, this finding may as well have a different molecular basis, as Fas ligation using neutralizing antibodies may impinge on cell death susceptibility through the activation of nuclear factor-κB signaling.130, 131 The idea that caspase-2 is relevant for heat stress-induced apoptosis was recently challenged by Milleron and Bratton,132 who observed that heat shock-induced apoptosis can occur independently from any known initiator caspase activation. In their study, they failed to observe cytoprotection by loss of caspase-2 or -8 in MEFs, as well as after siRNA-mediated knockdown of these caspases in Jurkat cells. Similarly, several attempts to reproduce the above-mentioned experiments using primary thymocytes, activated splenocytes or immortalized MEFs from caspase-2−/− or raidd−/− mice failed to support this hypothesis (our own unpublished results), consistent with the idea that heat shock may trigger oligomerization of Bax and or Bak directly.133
Other stress stimuli
Numerous other stressors were described to activate caspase-2-dependent cell death including cytoskeletal disruption in MEFs,134 trophic factor deprivation of neurons22, 62, 135, 136 or unknown toxic drug action induced by the grape constituent resveratrol137 or mycotoxin T2, a toxin from filamentous fungi.138 Interestingly, otherwise unrelated articles implying caspase-2 in cell death have in common that increased levels of free radicals were produced,139, 140, 141, 142, 143, 144 proposing a role for caspase-2 in reactive oxygen species-induced cell death. It can be noted here that caspase-2 knockout mice were reported to show age-related defects, leading to reduced lifespan and elevated level of liver oxidized proteins.145 In line, caloric restriction concurrently diminished caspase-2 processing and activity in rat.146
Conclusions
Summarizing the studies on caspase-2 activation, it still remains unclear if caspase-2 really requires an activation platform or if it may dimerize, autoprocess and autoactivate in the absence of such a platform, as seen when overexpressed in various cell systems. This may be facilitated by one of the many interacting proteins that could serve to activate this protease in a stimulus and/or cell-type-dependent manner (Figure 1). A particular and so far unresolved difficulty in this regard is that several studies suggest that for initiator caspases, processing is not a prerequisite that they attain enzymatic activity.29, 32 Thus, detecting the processed form of caspase-2 (or any other initiator caspase) by western blot analysis may not at all reflect the time course of its activation. Even worse, estimating the enzymatic activity of individual caspases by using artificial, supposedly caspase-specific peptide substrates may lead to even more erroneous results because commercially available caspase-specific substrates and inhibitors are not specific for a given caspase or, at best, highly promiscuous. Thus, it seems that the activity of caspase-2 can currently only be assessed in an indirect way, either in vitro using highly purified proteins or by carefully analyzing caspase-2 activation in cells lacking other caspases such as caspase-3 and, ideally, also caspase-7 or -8. However, the caveat here is that deficiency of one specific caspase may sometimes be compensated by increased activity of another caspase, as shown for caspases-2, -6 and -7 during acute liver cell apoptosis in hepatocytes from caspase-3 or caspase-9-deficient mice.117 Such phenomena may mask or even invert effects that can be readily seen upon an acute downregulation of caspase-2 by RNA interference or pharmacological inhibition in vitro, suggesting that caspase-2 may be important in a much wider context than delineated from caspase-2 knockout mice. Importantly, specific functions of caspase-2 in newly emerging, not yet fully understood cell death pathways122 indicate that efforts aiming at a deeper insight into the role of this enigmatic protein may provide unexpected and exciting earnings.
Abbreviations
- ARC:
-
apoptosis repressor with a CARD
- ATM:
-
Ataxia telangiectasia mutated
- ATR:
-
Ataxia telangiectasia and Rad3 related
- b-VAD-fmk:
-
N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
- CaMKII:
-
calcium/calmodulin-dependent protein kinase II
- CARD:
-
caspase activation and recruitment domain
- Chk1:
-
check-point kinase 1
- clAP1:
-
cellular inhibitor of apoptosis protein
- CRADD:
-
Caspase-2 and RIPK1 domain containing adaptor with death domain
- DED:
-
death effector domain
- DEFCAP:
-
death effect or filament-forming Ced-4-like apoptosis protein
- DFF-45/ICAD:
-
DNA fragmentation factor 45/inhibitor of caspase-dependent DNase
- DISC:
-
death-inducing signalling complex
- DR:
-
death receptor
- FADD:
-
Fas-associated death domain
- GMEB1:
-
glucocorticoid modulatory element-binding protein 1
- Htt, Huntigtin; Ich-1:
-
ICE/CED-3 homologue 1
- MEF:
-
mouse embryonic fibroblast
- MOMP:
-
mitochondrial outer membrane permeabilization
- Nedd-2, neural precursor cells-expressed:
-
developmentally down-regulated
- NLS:
-
nuclear localization signal
- PACAP:
-
pro-apoptotic caspase adaptor protein
- PIDD:
-
p53-induced protein with a death domain
- PKCK2:
-
protein kinase casein kinase 2
- PML-NBs:
-
promyelocytic leukemia nuclear bodies
- RAIDD:
-
RIP-associated ICH-1/CED-3 homologous protein with a death domain
- RIP:
-
receptor-interacting protein
- SUMO:
-
small ubiquitin-like modifier
- TRADD:
-
TNF-R1-associated death domain protein
- TNF-R1:
-
tumour necrosis factor receptor 1
- TRAIL:
-
tumor necrosis factor related apoptosis inducing ligand
- Ubc9:
-
SUMO-conjugating enzyme 9
References
Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM et al. A unified model for apical caspase activation. Mol Cell 2003; 11: 529–541.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Werner AB, Tait SW, de Vries E, Eldering E, Borst J . Requirement for aspartate-cleaved bid in apoptosis signaling by DNA-damaging anti-cancer regimens. J Biol Chem 2004; 279: 28771–28780.
Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR . The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 1993; 75: 641–652.
Wang L, Miura M, Bergeron L, Zhu H, Yuan J . Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell 1994; 78: 739–750.
Kumar S, Kinoshita M, Noda M, Copeland NG, Jenkins NA . Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev 1994; 8: 1613–1626.
Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 1997; 272: 17907–17911.
Butt AJ, Harvey NL, Parasivam G, Kumar S . Dimerization and autoprocessing of the Nedd2 (caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J Biol Chem 1998; 273: 6763–6768.
Baliga BC, Read SH, Kumar S . The biochemical mechanism of caspase-2 activation. Cell Death Differ 2004; 11: 1234–1241.
Schweizer A, Briand C, Grutter MG . Crystal structure of caspase-2, apical initiator of the intrinsic apoptotic pathway. J Biol Chem 2003; 278: 42441–42447.
Riedl SJ, Salvesen GS . The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 2007; 8: 405–413.
Walczak H, Haas TL . Biochemical analysis of the native TRAIL death-inducing signaling complex. Methods Mol Biol 2008; 414: 221–239.
Stennicke HR, Salvesen GS . Biochemical characteristics of caspases-3, -6, -7, and -8. J Biol Chem 1997; 272: 25719–25723.
Colussi PA, Harvey NL, Shearwin-Whyatt LM, Kumar S . Conversion of procaspase-3 to an autoactivating caspase by fusion to the caspase-2 prodomain. J Biol Chem 1998; 273: 26566–26570.
Van de Craen M, Declercq W, Van den brande I, Fiers W, Vandenabeele P . The proteolytic procaspase activation network: an in vitro analysis. Cell Death Differ 1999; 6: 1117–1124.
Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES . Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem 2002; 277: 13430–13437.
Henderson C, Mizzau M, Paroni G, Maestro R, Schneider C, Brancolini C . Role of caspases, Bid, and p53 in the apoptotic response triggered by histone deacetylase inhibitors trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA). J Biol Chem 2003; 278: 12579–12589.
Tsuruma K, Nakagawa T, Shirakura H, Hayashi N, Uehara T, Nomura Y . Regulation of procaspase-2 by glucocorticoid modulatory element-binding protein 1 through the interaction with caspase recruitment domain. Biochem Biophys Res Commun 2004; 325: 1246–1251.
Bonfoco E, Li E, Kolbinger F, Cooper NR . Characterization of a novel proapoptotic caspase-2- and caspase-9-binding protein. J Biol Chem 2001; 276: 29242–29250.
Duan H, Dixit VM . RAIDD is a new ‘death’ adaptor molecule. Nature 1997; 385: 86–89.
Ahmad M, Srinivasula SM, Wang L, Talanian RV, Litwack G, Fernandes-Alnemri T et al. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP. Cancer Res 1997; 57: 615–619.
Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 1998; 12: 1304–1314.
Chen H, Chung S, Sukumar S . HOXA5-induced apoptosis in breast cancer cells is mediated by caspases 2 and 8. Mol Cell Biol 2004; 24: 924–935.
Guicciardi ME, Bronk SF, Werneburg NW, Yin XM, Gores GJ . Bid is upstream of lysosome-mediated caspase 2 activation in tumor necrosis factor alpha-induced hepatocyte apoptosis. Gastroenterology 2005; 129: 269–284.
Lavrik IN, Golks A, Baumann S, Krammer PH . Caspase-2 is activated at the CD95 death-inducing signaling complex in the course of CD95-induced apoptosis. Blood 2006; 108: 559–565.
Droin N, Bichat F, Rebe C, Wotawa A, Sordet O, Hammann A et al. Involvement of caspase-2 long isoform in Fas-mediated cell death of human leukemic cells. Blood 2001; 97: 1835–1844.
Micheau O, Tschopp J . Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003; 114: 181–190.
Villunger A, Huang DC, Holler N, Tschopp J, Strasser A . Fas ligand-induced c-Jun kinase activation in lymphoid cells requires extensive receptor aggregation but is independent of DAXX, and Fas-mediated cell death does not involve DAXX, RIP, or RAIDD. J Immunol 2000; 165: 1337–1343.
Tinel A, Tschopp J . The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004; 304: 843–846.
Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR . In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol 2006; 8: 72–77.
Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 2007; 128: 533–546.
Read SH, Baliga BC, Ekert PG, Vaux DL, Kumar S . A novel Apaf-1-independent putative caspase-2 activation complex. J Cell Biol 2002; 159: 739–745.
Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R et al. Apoptosis caused by p53-induced protein with death domain (PIDD) depends on the death adapter protein RAIDD. Proc Natl Acad Sci USA 2005; 102: 14314–14320.
Hlaing T, Guo RF, Dilley KA, Loussia JM, Morrish TA, Shi MM et al. Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J Biol Chem 2001; 276: 9230–9238.
Martinon F, Burns K, Tschopp J . The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10: 417–426.
Jesenberger V, Procyk KJ, Yuan J, Reipert S, Baccarini M . Salmonella-induced caspase-2 activation in macrophages: a novel mechanism in pathogen-mediated apoptosis. J Exp Med 2000; 192: 1035–1046.
LeBlanc PM, Yeretssian G, Rutherford N, Doiron K, Nadiri A, Zhu L et al. Caspase-12 modulates NOD signaling and regulates antimicrobial peptide production and mucosal immunity. Cell Host Microbe 2008; 3: 146–157.
Lamkanfi M, D’hondt K, Vande WL, van Gurp M, Denecker G, Demeulemeester J et al. A novel caspase-2 complex containing TRAF2 and RIP1. J Biol Chem 2005; 280: 6923–6932.
Mendelsohn AR, Hamer JD, Wang ZB, Brent R . Cyclin D3 activates caspase 2, connecting cell proliferation with cell death. Proc Natl Acad Sci USA 2002; 99: 6871–6876.
Shirakura H, Hayashi N, Ogino S, Tsuruma K, Uehara T, Nomura Y . Caspase recruitment domain of procaspase-2 could be a target for SUMO-1 modification through Ubc9. Biochem Biophys Res Commun 2005; 331: 1007–1015.
Janssens S, Tinel A, Lippens S, Tschopp J . PIDD mediates NF-kappaB activation in response to DNA damage. Cell 2005; 123: 1079–1092.
Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B et al. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J 2007; 26: 197–208.
Cuenin S, Tinel A, Janssens S, Tschopp J . p53-induced protein with a death domain (PIDD) isoforms differentially activate nuclear factor-kappaB and caspase-2 in response to genotoxic stress. Oncogene 2008; 27: 387–396.
Shin S, Lee Y, Kim W, Ko H, Choi H, Kim K . Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J 2005; 24: 3532–3542.
Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 2005; 123: 89–103.
Logette E, Solary E, Corcos L . Identification of a functional DNA binding site for the SREBP-1c transcription factor in the first intron of the human caspase-2 gene. Biochim Biophys Acta 2005; 1738: 1–5.
Baptiste-Okoh N, Barsotti AM, Prives C . Caspase 2 is both required for p53-mediated apoptosis and downregulated by p53 in a p21-dependent manner. Cell Cycle 2008; 7: 1133–1138.
Kumar S, Kinoshita M, Dorstyn L, Noda M . Origin, expression and possible functions of the two alternatively spliced forms of the mouse Nedd2mRNA. Cell Death Differ 1997; 4: 378–387.
Droin N, Rebe C, Bichat F, Hammann A, Bertrand R, Solary E . Modulation of apoptosis by procaspase-2 short isoform: selective inhibition of chromatin condensation, apoptotic body formation and phosphatidylserine externalization. Oncogene 2001; 20: 260–269.
Wotawa A, Solier S, Logette E, Solary E, Corcos L . Differential influence of etoposide on two caspase-2 mRNA isoforms in leukemic cells. Cancer Lett 2002; 185: 181–189.
Logette E, Wotawa A, Solier S, Desoche L, Solary E, Corcos L . The human caspase-2 gene: alternative promoters, pre-mRNA splicing and AUG usage direct isoform-specific expression. Oncogene 2003; 22: 935–946.
Solier S, Lansiaux A, Logette E, Wu J, Soret J, Tazi J et al. Topoisomerase I and II inhibitors control caspase-2 pre-messenger RNA splicing in human cells. Mol Cancer Res 2004; 2: 53–61.
Solier S, De Cian MC, Bettaieb A, Desoche L, Solary E, Corcos L . PKC zeta controls DNA topoisomerase-dependent human caspase-2 pre-mRNA splicing. FEBS Lett 2008; 582: 372–378.
Leroy I, de Thonel A, Laurent G, Quillet-Mary A . Protein kinase C zeta associates with death inducing signaling complex and regulates Fas ligand-induced apoptosis. Cell Signal 2005; 17: 1149–1157.
Perfetti A, Oriente F, Iovino S, Alberobello AT, Barbagallo AP, Esposito I et al. Phorbol esters induce intracellular accumulation of the anti-apoptotic protein PED/PEA-15 by preventing ubiquitinylation and proteasomal degradation. J Biol Chem 2007; 282: 8648–8657.
Solier S, Logette E, Desoche L, Solary E, Corcos L . Nonsense-mediated mRNA decay among human caspases: the caspase-2S putative protein is encoded by an extremely short-lived mRNA. Cell Death Differ 2005; 12: 687–689.
Basu A, Adkins B, Basu C . Down-regulation of caspase-2 by rottlerin via protein kinase C-delta-independent pathway. Cancer Res 2008; 68: 2795–2802.
Besnault-Mascard L, Leprince C, Auffredou MT, Meunier B, Bourgeade MF, Camonis J et al. Caspase-8 sumoylation is associated with nuclear localization. Oncogene 2005; 24: 3268–3273.
Hayashi N, Shirakura H, Uehara T, Nomura Y . Relationship between SUMO-1 modification of caspase-7 and its nuclear localization in human neuronal cells. Neurosci Lett 2006; 397: 5–9.
Zhivotovsky B, Samali A, Gahm A, Orrenius S . Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ 1999; 6: 644–651.
Mancini M, Machamer CE, Roy S, Nicholson DW, Thornberry NA, Casciola-Rosen LA et al. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J Cell Biol 2000; 149: 603–612.
O’Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL et al. Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ 2002; 9: 832–841.
Colussi PA, Harvey NL, Kumar S . Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor. A novel function for a caspase prodomain. J Biol Chem 1998; 273: 24535–24542.
Shikama Y, U M, Miyashita T, Yamada M . Comprehensive studies on subcellular localizations and cell death-inducing activities of eight GFP-tagged apoptosis-related caspases. Exp Cell Res 2001; 264: 315–325.
Baliga BC, Colussi PA, Read SH, Dias MM, Jans DA, Kumar S . Role of prodomain in importin-mediated nuclear localization and activation of caspase-2. J Biol Chem 2003; 278: 4899–4905.
Tang J, Xie W, Yang X . Association of caspase-2 with the promyelocytic leukemia protein nuclear bodies. Cancer Biol Ther 2005; 4: 645–649.
Bernardi R, Pandolfi PP . Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 2007; 8: 1006–1016.
Sanchez-Pulido L, Valencia A, Rojas AM . Are promyelocytic leukaemia protein nuclear bodies a scaffold for caspase-2 programmed cell death? Trends Biochem Sci 2007; 32: 400–406.
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N et al. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med 1999; 189: 381–394.
van Loo G, Saelens X, Matthijssens F, Schotte P, Beyaert R, Declercq W et al. Caspases are not localized in mitochondria during life or death. Cell Death Differ 2002; 9: 1207–1211.
Cheung HH, Lynn KN, Liston P, Korneluk RG . Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: a role for the IAPs. Exp Cell Res 2006; 312: 2347–2357.
Dahmer MK . Caspases-2, -3, and -7 are involved in thapsigargin-induced apoptosis of SH-SY5Y neuroblastoma cells. J Neurosci Res 2005; 80: 576–583.
Upton JP, Austgen K, Nishino M, Coakley KM, Hagen A, Han D et al. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cell Biol 2008; 28: 3943–3951.
Shearwin-Whyatt LM, Harvey NL, Kumar S . Subcellular localization and CARD-dependent oligomerization of the death adaptor RAIDD. Cell Death Differ 2000; 7: 155–165.
Paroni G, Henderson C, Schneider C, Brancolini C . Caspase-2 can trigger cytochrome C release and apoptosis from the nucleus. J Biol Chem 2002; 277: 15147–15161.
Zuzarte-Luis V, Berciano MT, Lafarga M, Hurle JM . Caspase redundancy and release of mitochondrial apoptotic factors characterize interdigital apoptosis. Apoptosis 2006; 11: 701–715.
Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B . DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ 2008; 15: 555–566.
Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D et al. Substrate specificities of caspase family proteases. J Biol Chem 1997; 272: 9677–9682.
Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 1997; 272: 17907–17911.
McStay GP, Salvesen GS, Green DR . Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ 2008; 15: 322–331.
Chauvier D, Ankri S, Charriaut-Marlangue C, Casimir R, Jacotot E . Broad-spectrum caspase inhibitors: from myth to reality? Cell Death Differ 2007; 14: 387–391.
Zhivotovsky B, Orrenius S . Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun 2005; 331: 859–867.
Robertson JD, Gogvadze V, Kropotov A, Vakifahmetoglu H, Zhivotovsky B, Orrenius S . Processed caspase-2 can induce mitochondria-mediated apoptosis independently of its enzymatic activity. EMBO Rep 2004; 5: 643–648.
Sakahira H, Enari M, Nagata S . Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998; 391: 96–99.
Dahal GR, Karki P, Thapa A, Shahnawaz M, Shin SY, Lee JS et al. Caspase-2 cleaves DNA fragmentation factor (DFF45)/inhibitor of caspase-activated DNase (ICAD). Arch Biochem Biophys 2007; 468: 134–139.
Panaretakis T, Laane E, Pokrovskaja K, Bjorklund AC, Moustakas A, Zhivotovsky B et al. Doxorubicin requires the sequential activation of caspase-2, protein kinase Cdelta, and c-Jun NH2-terminal kinase to induce apoptosis. Mol Biol Cell 2005; 16: 3821–3831.
Hermel E, Gafni J, Propp SS, Leavitt BR, Wellington CL, Young JE et al. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ 2004; 11: 424–438.
Gu Y, Sarnecki C, Aldape RA, Livingston DJ, Su MS . Cleavage of poly(ADP-ribose) polymerase by interleukin-1 beta converting enzyme and its homologs TX and Nedd-2. J Biol Chem 1995; 270: 18715–18718.
Rotter B, Kroviarski Y, Nicolas G, Dhermy D, Lecomte MC . AlphaII-spectrin is an in vitro target for caspase-2, and its cleavage is regulated by calmodulin binding. Biochem J 2004; 378: 161–168.
Aho S . Plakin proteins are coordinately cleaved during apoptosis but preferentially through the action of different caspases. Exp Dermatol 2004; 13: 700–707.
Paroni G, Mizzau M, Henderson C, Del Sal G, Schneider C, Brancolini C . Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol Biol Cell 2004; 15: 2804–2818.
Green DR, Kroemer G . The pathophysiology of mitochondrial cell death. Science 2004; 305: 626–629.
Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S . Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem 2002; 277: 29803–29809.
Enoksson M, Robertson JD, Gogvadze V, Bu P, Kropotov A, Zhivotovsky B et al. Caspase-2 permeabilizes the outer mitochondrial membrane and disrupts the binding of cytochrome c to anionic phospholipids. J Biol Chem 2004; 279: 49575–49578.
Gao Z, Shao Y, Jiang X . Essential roles of the Bcl-2 family of proteins in caspase-2-induced apoptosis. J Biol Chem 2005; 280: 38271–38275.
Bonzon C, Bouchier-Hayes L, Pagliari LJ, Green DR, Newmeyer DD . Caspase-2-induced apoptosis requires bid cleavage: a physiological role for bid in heat shock-induced death. Mol Biol Cell 2006; 17: 2150–2157.
Barnhart BC, Alappat EC, Peter ME . The CD95 type I/type II model. Semin Immunol 2003; 15: 185–193.
Ho PK, Jabbour AM, Ekert PG, Hawkins CJ . Caspase-2 is resistant to inhibition by inhibitor of apoptosis proteins (IAPs) and can activate caspase-7. FEBS J 2005; 272: 1401–1414.
Maag RS, Mancini M, Rosen A, Machamer CE . Caspase-resistant Golgin-160 disrupts apoptosis induced by secretory pathway stress and ligation of death receptors. Mol Biol Cell 2005; 16: 3019–3027.
Liu X, Zou H, Slaughter C, Wang X . DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997; 89: 175–184.
Tang D, Kidd VJ . Cleavage of DFF-45/ICAD by multiple caspases is essential for its function during apoptosis. J Biol Chem 1998; 273: 28549–28552.
Janicke RU, Sprengart ML, Wati MR, Porter AG . Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 1998; 273: 9357–9360.
Lechardeur D, Xu M, Lukacs GL . Contrasting nuclear dynamics of the caspase-activated DNase (CAD) in dividing and apoptotic cells. J Cell Biol 2004; 167: 851–862.
Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006; 125: 1179–1191.
Lassus P, Opitz-Araya X, Lazebnik Y . Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 2002; 297: 1352–1354.
Lin CF, Chen CL, Chang WT, Jan MS, Hsu LJ, Wu RH et al. Sequential caspase-2 and caspase-8 activation upstream of mitochondria during ceramide and etoposide-induced apoptosis. J Biol Chem 2004; 279: 40755–40761.
Lin CF, Chen CL, Chang WT, Jan MS, Hsu LJ, Wu RH et al. Bcl-2 rescues ceramide- and etoposide-induced mitochondrial apoptosis through blockage of caspase-2 activation. J Biol Chem 2005; 280: 23758–23765.
Cao X, Bennett RL, May WS . c-Myc and caspase-2 are involved in activating Bax during cytotoxic drug-induced apoptosis. J Biol Chem 2008; 283: 14490–14496.
Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007; 129: 1337–1349.
Seth R, Yang C, Kaushal V, Shah SV, Kaushal GP . p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J Biol Chem 2005; 280: 31230–31239.
Baptiste-Okoh N, Barsotti AM, Prives C . A role for caspase 2 and PIDD in the process of p53-mediated apoptosis. Proc Natl Acad Sci USA 2008; 105: 1937–1942.
Taghiyev AF, Guseva NV, Glover RA, Rokhlin OW, Cohen MB . TSA-induced cell death in prostate cancer cell lines is caspase-2 dependent and involves the PIDDosome. Cancer Biol Ther 2006; 5: 1199–1205.
Lakhani SA, Masud A, Kuida K, Porter Jr GA, Booth CJ, Mehal WZ et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 2006; 311: 847–851.
Samraj AK, Sohn D, Schulze-Osthoff K, Schmitz I . Loss of caspase-9 reveals its essential role for caspase-2 activation and mitochondrial membrane depolarization. Mol Biol Cell 2007; 18: 84–93.
He Q, Huang Y, Sheikh MS . Bax deficiency affects caspase-2 activation during ultraviolet radiation-induced apoptosis. Oncogene 2004; 23: 1321–1325.
Marsden VS, Ekert PG, Van Delft M, Vaux DL, Adams JM, Strasser A . Bcl-2-regulated apoptosis and cytochrome c release can occur independently of both caspase-2 and caspase-9. J Cell Biol 2004; 165: 775–780.
Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW et al. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med 2000; 6: 1241–1247.
Ekert PG, Read SH, Silke J, Marsden VS, Kaufmann H, Hawkins CJ et al. Apaf-1 and caspase-9 accelerate apoptosis, but do not determine whether factor-deprived or drug-treated cells die. J Cell Biol 2004; 165: 835–842.
Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams JM . Constitutive Bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci USA 1999; 96: 14943–14948.
Ekert PG, Silke J, Vaux DL . Inhibition of apoptosis and clonogenic survival of cells expressing crmA variants: optimal caspase substrates are not necessarily optimal inhibitors. EMBO J 1999; 18: 330–338.
Pellegrini M, Bath S, Marsden VS, Huang DC, Metcalf D, Harris AW et al. FADD and caspase-8 are required for cytokine-induced proliferation of hematopoietic progenitor cells. Blood 2005; 106: 1581–1589.
Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008; 133: 864–877.
Qatsha KA, Rudolph C, Marme D, Schachtele C, May WS . Go 6976, a selective inhibitor of protein kinase C, is a potent antagonist of human immunodeficiency virus 1 induction from latent/low-level-producing reservoir cells in vitro. Proc Natl Acad Sci USA 1993; 90: 4674–4678.
Gurfinkel DM, Chow S, Hurren R, Gronda M, Henderson C, Berube C et al. Disruption of the endoplasmic reticulum and increases in cytoplasmic calcium are early events in cell death induced by the natural triterpenoid asiatic acid. Apoptosis 2006; 11: 1463–1471.
Kieran D, Woods I, Villunger A, Strasser A, Prehn JH . Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice. Proc Natl Acad Sci USA 2007; 104: 20606–20611.
Murakami Y, Aizu-Yokota E, Sonoda Y, Ohta S, Kasahara T . Suppression of endoplasmic reticulum stress-induced caspase activation and cell death by the overexpression of Bcl-xL or Bcl-2. J Biochem 2007; 141: 401–410.
Turner BJ, Atkin JD . ER stress and UPR in familial amyotrophic lateral sclerosis. Curr Mol Med 2006; 6: 79–86.
Wagner KW, Engels IH, Deveraux QL . Caspase-2 can function upstream of bid cleavage in the TRAIL apoptosis pathway. J Biol Chem 2004; 279: 35047–35052.
Moulin M, Arrigo AP . Caspases activation in hyperthermia-induced stimulation of TRAIL apoptosis. Cell Stress Chaperones 2008; 13: 313–326.
Kreuz S, Siegmund D, Rumpf JJ, Samel D, Leverkus M, Janssen O et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol 2004; 166: 369–380.
Brenner D, Golks A, Kiefer F, Krammer PH, Arnold R . Activation or suppression of NFkappaB by HPK1 determines sensitivity to activation-induced cell death. EMBO J 2005; 24: 4279–4290.
Milleron RS, Bratton SB . Heat shock induces apoptosis independently of any known initiator caspase-activating complex. J Biol Chem 2006; 281: 16991–17000.
Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM et al. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc Natl Acad Sci USA 2005; 102: 17975–17980.
Ho LH, Read SH, Dorstyn L, Lambrusco L, Kumar S . Caspase-2 is required for cell death induced by cytoskeletal disruption. Oncogene 2008; 27: 3393–3404.
Troy CM, Shelanski ML . Caspase-2 redux. Cell Death Differ 2003; 10: 101–107.
Chauvier D, Lecoeur H, Langonne A, Borgne-Sanchez A, Mariani J, Martinou JC et al. Upstream control of apoptosis by caspase-2 in serum-deprived primary neurons. Apoptosis 2005; 10: 1243–1259.
Mohan J, Gandhi AA, Bhavya BC, Rashmi R, Karunagaran D, Indu R et al. Caspase-2 triggers Bax–Bak-dependent and -independent cell death in colon cancer cells treated with resveratrol. J Biol Chem 2006; 281: 17599–17611.
Huang P, Akagawa K, Yokoyama Y, Nohara K, Kano K, Morimoto K . T-2 toxin initially activates caspase-2 and induces apoptosis in U937 cells. Toxicol Lett 2007; 170: 1–10.
Dirsch VM, Kirschke SO, Estermeier M, Steffan B, Vollmar AM . Apoptosis signaling triggered by the marine alkaloid ascididemin is routed via caspase-2 and JNK to mitochondria. Oncogene 2004; 23: 1586–1593.
Fu YC, Chi CS, Yin SC, Hwang B, Chiu YT, Hsu SL . Norepinephrine induces apoptosis in neonatal rat cardiomyocytes through a reactive oxygen species-TNF alpha-caspase signaling pathway. Cardiovasc Res 2004; 62: 558–567.
Yeung BH, Huang DC, Sinicrope FA . PS-341 (bortezomib) induces lysosomal cathepsin B release and a caspase-2-dependent mitochondrial permeabilization and apoptosis in human pancreatic cancer cells. J Biol Chem 2006; 281: 11923–11932.
Prasad V, Chandele A, Jagtap JC, Sudheer KP, Shastry P . ROS-triggered caspase 2 activation and feedback amplification loop in beta-carotene-induced apoptosis. Free Radic Biol Med 2006; 41: 431–442.
Tamm C, Zhivotovsky B, Ceccatelli S . Caspase-2 activation in neural stem cells undergoing oxidative stress-induced apoptosis. Apoptosis 2008; 13: 354–363.
Chen WJ, Huang YT, Wu ML, Huang TC, Ho CT, Pan MH . Induction of apoptosis by Vitamin D2, ergocalciferol, via reactive oxygen species generation, glutathione depletion, and caspase activation in human leukemia cells. J Agric Food Chem 2008; 56: 2996–3005.
Zhang Y, Padalecki SS, Chaudhuri AR, De Waal E, Goins BA, Grubbs B et al. Caspase-2 deficiency enhances aging-related traits in mice. Mech Ageing Dev 2007; 128: 213–221.
Shelke RR, Leeuwenburgh C . Lifelong caloric restriction increases expression of apoptosis repressor with a caspase recruitment domain (ARC) in the brain. FASEB J 2003; 17: 494–496.
Acknowledgements
The work in our laboratory was supported by fellowships and grants to from the Austrian Science Fund (FWF): Y212-B13 START, the Doctoral College MCBO, the SFB021, and by the Association for International Cancer Research (AICR), EU-FP7 (ApopTrain) and the Tyrolean Science Fund (TWF). We apologize to the many scientists in this field whose excellent research was not cited but was only referred to indirectly through reviews.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by G Melino
Conflict of interest
The authors have no competing financial interest.
Rights and permissions
About this article
Cite this article
Krumschnabel, G., Sohm, B., Bock, F. et al. The enigma of caspase-2: the laymen's view. Cell Death Differ 16, 195–207 (2009). https://doi.org/10.1038/cdd.2008.170
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.170
Keywords
This article is cited by
-
Modeled microgravity alters apoptotic gene expression and caspase activity in the squid-vibrio symbiosis
BMC Microbiology (2022)
-
Potent and selective caspase-2 inhibitor prevents MDM-2 cleavage in reversine-treated colon cancer cells
Cell Death & Differentiation (2019)
-
Bacterial Pore-Forming Toxins Promote the Activation of Caspases in Parallel to Necroptosis to Enhance Alarmin Release and Inflammation During Pneumonia
Scientific Reports (2018)
-
Apoptosis regulation by subcellular relocation of caspases
Scientific Reports (2018)
-
Expression of adenoviral E1A throws the PIDD switch
Cell Death & Disease (2017)
