
Abstract
The nematode Caenorhabditis elegans contains a single ancestral p53 family member, cep-1, which is required to activate apoptosis of germ cells in response to DNA damage. To understand how the cep-1/p53 pathway is regulated in response to genotoxic stress, we performed an RNA interference screen and identified the neddylation pathway and components of an SCF (Skp1/cullin/F-box) E3 ubiquitin ligase as negative regulators of cep-1-dependent germ cell apoptosis. Here, we show that the cullin gene cul-1, the Skp1-related gene skr-1, and the ring box genes rbx-1 and rpm-1 all negatively regulate cep-1-dependent germ cell apoptosis in response to the DNA-alkylating agent N-ethyl-N-nitrosourea (ENU). We also identified the F-box protein FSN-1, previously shown to form an SCF ligase that regulates synapse development, as a negative regulator of cep-1-dependent germline apoptosis. The hypersensitivity of fsn-1 mutants to ENU-induced germline apoptosis was completely suppressed by a cep-1 loss-of-function allele. We further provide evidence that the transcriptional activity, phosphorylation status, and levels of endogenous CEP-1 are higher in fsn-1 mutants compared with wild-type animals after ENU treatment. Our results uncover a novel role for the SCFFSN-1 E3 ubiquitin ligase in the regulation of cep-1-dependent germ cell apoptosis.
Similar content being viewed by others


Main
Tumor suppressor p53 is a transcription factor that coordinates cellular responses to a variety of stresses, such as DNA damage, oncogene activation, nutrient depletion, and hypoxia.1 Depending on the level of damage, p53 can either arrest proliferation and activate repair mechanisms, or trigger a cellular suicide program (apoptosis) that removes potentially hazardous cells from the organism.1 Activation of apoptosis by p53 involves transcription-dependent and transcription-independent mechanisms that are crucial for preventing tumor formation.1, 2 The activity of p53 is regulated by numerous post-translational modifications, including phosphorylation, ubiquitination, acetylation, and neddylation.1, 3 Several ubiquitin ligases have been identified that control the stability of p53, including Mdm2, ARF-BP1, Pirh2, COP-1, and CHIP.3 The best-characterized p53-specific E3 ubiquitin ligase is Mdm2, which maintains low levels of p53 protein under non-stressed conditions through a negative feedback loop.4, 5 Cellular stresses such as DNA damage stimulate the phosphorylation of p53 and Mdm2, which disrupts their association and stabilizes p53.1, 3 As the cellular concentration of p53 rises, it can activate the transcription of genes such as PUMA and NOXA to trigger apoptosis.6, 7
The p53 family is comprised of three members: p53, p63, and p73. While it is currently unclear whether p63 and p73 are involved in tumorigenesis, they share many of the same properties as p53 in the control of cell survival.8 The stability of both p63 and p73 is regulated by the E3 ubiquitin ligase Itch, and recent work has shown that Mdm2-dependent neddylation of p73 antagonizes its transcriptional activity.9, 10, 11 The nematode Caenorhabditis elegans contains a single ancestral p53 family member, cep-1, which is required to activate apoptosis of germ cells in response to DNA-damaging agents.12, 13 We recently found that CEP-1 regulates the expression of many of the same genes as human p53 and p63, suggesting that it may represent a composite of the vertebrate paralogues.14 Structurally, the DNA binding domain of CEP-1 is similar to human p53 but its C terminus contains a SAM domain that resembles the C termini of vertebrate p63 and p73.15, 16 Like its relatives, the apoptotic activity of CEP-1 is controlled by many of the same proteins, such as Akt/PKB, Brca1, and the p53-binding protein iASPP.17, 18, 19 Because C. elegans contains a single p53-like gene, genetic analysis is simplified in this organism and may reveal novel mechanisms by which the vertebrate paralogues are regulated. Here, we show that the neddylation pathway and the Skp1/cullin/F-box (SCF) E3 ubiquitin ligase SCFFSN-1 control the apoptotic activity of CEP-1 in response to genotoxic stress. We propose that neddylation and SCFFSN-1 negatively regulate the transcriptional activity, phosphorylation, and endogenous levels of CEP-1 protein in response to DNA damage through an intermediate protein.
Results
The neddylation pathway regulates cep-1-dependent germline apoptosis
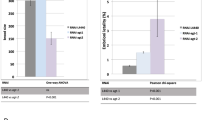
From an RNA interference (RNAi) modifier screen, we found that cep-1(gk138) deletion mutants survived better on bacteria that express double-stranded RNA to the ned-8 gene (Nedd8 in humans) compared with wild-type (N2) controls (data not shown). Recent studies in mammalian cells have implicated a key role for the neddylation pathway in suppressing p53- and p73-dependent apoptosis.9, 20 To investigate how NED-8 modulates CEP-1 activity in vivo, we inhibited components of the neddylation pathway by RNAi and quantified DNA damage-induced apoptosis in the germ line, where cep-1 has a well-established role.12, 13, 17, 21 Ablation of ned-8 caused significantly higher levels of cep-1-dependent germ cell apoptosis in response to the DNA-alkylating agent N-ethyl-N-nitrosourea (ENU) compared with controls (Figure 1a–c). Inhibiting expression of the NED-8 E1-activating genes ula-1 and rfl-1, as well as the NED-8 E2-conjugating gene ubc-12, also caused hypersensitivity to ENU-induced germ cell apoptosis (Figure 1d). In rfl-1(or198ts) mutants, there were elevated levels of germline apoptosis in the absence and presence of ENU at the restrictive temperature (Supplementary Figure 1). Because physiological germline apoptosis was unaffected when the neddylation pathway was inhibited by RNAi, the rfl-1(or198ts) allele might be more potent than RNAi or activates the damage checkpoint in the absence of genotoxic stress.

The neddylation pathway negatively regulates cep-1-dependent apoptosis via transcriptional activation of egl-1 and ced-13. (a and b) Representative images of germline apoptotic corpses after 5 mM ENU treatment in the pachytene region of wild-type animals fed bacteria expressing the empty RNAi vector (L4440) (a) or RNAi to the ned-8 gene (b). (a) Scale bar=20 μm. Germ cell corpses in panels a and b in one focal plane of the pachytene region are indicated by white arrowheads. (c) Effect of increasing concentrations of ENU on germ cell apoptosis in young adults depleted of ned-8 24 h after treatment. (d) Effect of 5 mM ENU on germ cell apoptosis in wild-type animals depleted of rfl-1 and ula-1, the ned-8 E1-activating genes, and ubc-12, which encodes the ned-8 E2-conjugating gene. Between 20 and 35 germ lines were scored for each concentration of ENU in triplicate. Error bars represent the standard error of the mean (S.E.M.). (e and f) Transcript abundance of egl-1 and ced-13 was determined by qPCR 24 h after worms were treated with ENU. egl-1 and ced-13 transcript levels were normalized to γ-tubulin (tbg-1) mRNA. Fold induction was calculated relative to normalized transcript levels in untreated wild-type animals. Error bars=S.E.M.
ned-8 regulates germline apoptosis through the core apoptosis pathway
DNA damage-induced germ cell apoptosis in C. elegans is executed through the core programmed cell death (PCD) signaling pathway, which is composed of egl-1, ced-9, ced-4, and ced-3.22, 23, 24 To determine whether neddylation modulates apoptosis through the core PCD pathway, we inhibited ned-8 in strains carrying the loss-of-function mutations ced-3(n717), ced-4(n1162), and egl-1(n1084n3082), as well as a strain carrying the ced-9(n1950) gain-of-function mutation. In all of these mutants, ENU-induced germ cell apoptosis was abolished (Supplementary Figure 2).
CEP-1 activates the core PCD pathway in the germ line by transactivating the BH3-only proteins EGL-1 and CED-13, which antagonize the antiapoptotic CED-9 protein.23, 24, 25 To assess whether NED-8 regulates the transcriptional activity of CEP-1 in vivo, we quantified egl-1 and ced-13 transcript levels by quantitative real-time PCR (qPCR). At 5 mM ENU, there was a two- to three-fold increase in both egl-1 and ced-13 transcript in wild-type animals depleted of ned-8 compared with controls, but in cep-1(gk138) mutants the expression of these genes was not activated (Figure 1e and f). Together, these results indicate that NED-8 regulates germline apoptosis through the core PCD pathway by affecting the transcriptional activity of CEP-1.
Germline apoptosis is regulated by an SCF ubiquitin ligase
Because we could not detect neddylated CEP-1 from worm extracts (Figure 4, and data not shown), we reasoned that neddylation may regulate CEP-1 through an indirect mechanism. The best-characterized substrates for neddylation are proteins of the cullin family, which act as scaffolds for the assembly of multi-subunit E3 ubiquitin ligases.26 Of the six cullin genes in C. elegans, only cul-1 regulated ENU-induced germ cell apoptosis (Figure 2a). Although cul-1(e1756)/unc-69(e587) heterozygous animals were partially hypersensitive to ENU-induced apoptosis (Supplementary Figure 3), this effect was not statistically significant. It was not possible to examine germline apoptosis in cul-1(e1756) homozygotes because they arrest during larval development.27 Since CUL-1 is a component of the SCF complex, these results suggested that DNA damage-induced germ cell apoptosis is negatively regulated by an SCF E3 ubiquitin ligase. SCF complexes also contain a Skp1 protein, a RING domain protein, and an F-box protein.26 Therefore, we next evaluated the two RING domain proteins, RBX-1 and RPM-1, and found that both of these proteins negatively regulate DNA damage-induced germ cell apoptosis (Figure 2b). Inhibition of rbx-1 in rpm-1(ju44) mutants did not have an additive effect on ENU-induced apoptosis, suggesting that these RING proteins act redundantly in this process (Figure 2b).

Endogenous CEP-1 is phosphorylated in response to ENU and its abundance is higher in fsn-1 mutants. (a) The appearance of a slower migrating form of CEP-1 is detectable in wild-type animals at 6 h after ENU treatment. These bands were not present in cep-1(gk138) deletion mutants. Detection of α-tubulin on the same blot was used as a loading control. (b) The slower migrating form of CEP-1 in response to ENU is due to phosphorylation. Wild-type worm lysates were incubated in the presence or absence of shrimp alkaline phosphatase, then separated by SDS-PAGE and analyzed as above. (c) Wild-type, fsn-1(hp1), fsn-1(gk429), and cep-1(gk138) mutants were treated with ENU, 200 μg of worm lysates was separated by SDS-PAGE and CEP-1 was detected as above. α-Tubulin was used as a loading control. (d) In response to ENU, there is more CEP-1 (unmodified+phosphorylated) in fsn-1(hp1) and fsn-1(gk429) mutants compared with wild-type controls. Relative densitometric units of total CEP-1 to α-tubulin are shown. Data represent three independent experiments. Error bars=S.E.M.

An SCF ubiquitin ligase complex antagonizes CEP-1/p53-mediated germ cell apoptosis in response to DNA damage. (a) Effect of 5 mM ENU on germ cell apoptosis in wild-type animals depleted of the six cullin genes. Only cul-1(RNAi) enhanced ENU-induced germ cell apoptosis. (b) Wild-type animals depleted of rbx-1 by RNAi or rpm-1(ju44) mutants treated with 5 mM ENU exhibit hypersensitivity to germ cell apoptosis. No additive effects were observed when rbx-1 was inhibited in rpm-1(ju44) mutants. (c) Inhibiting the Skp1-related genes skr-1 and skr-7 by RNAi sensitized worms to ENU-induced germ cell apoptosis. (d) The neddylation pathway and components of the SCF ligase regulates germ cell apoptosis in response to ENU through cep-1
The complexity of SCF ligases in C. elegans is highlighted by the presence of 21 Skp1-related (skr) and 520 predicted F-box genes.28, 29, 30 Inhibiting skr-1 or skr-7 by RNAi caused a significant increase in ENU-induced germ cell apoptosis similar to inactivating cul-1 or ned-8 (Figure 2c and Supplementary Table 1). The increased apoptosis caused by inhibiting cul-1 or skr-1 was abolished in cep-1(gk138) mutants (Figure 2d), suggesting that these genes also act upstream of cep-1. Because SKR-1 and SKR-7 bind to CUL-1,29, 30 these results are consistent with the hypothesis that an SCF ligase regulates the apoptotic activity of CEP-1.
fsn-1 regulates cep-1-dependent signaling in response to DNA damage
The specificity of SCF ligases is controlled by F-box proteins, which bind to distinct substrate proteins that are targeted for ubiquitin-mediated proteolysis.26 We systematically inhibited 328 of the 520 predicted F-box genes28 by RNAi and identified FSN-1, an F-box protein conserved from worm to human that physically associates with RPM-1, SKR-1, and CUL-1 to regulate neuromuscular synapse formation.31 Using two different null alleles, fsn-1(hp1) and fsn-1(gk429), we confirmed that FSN-1 is a potent negative regulator of ENU-induced germ cell apoptosis (Figure 3a). The increased apoptosis observed in fsn-1 mutants was completely abolished by the cep-1(gk138) allele (Figure 3a), suggesting that fsn-1 is upstream of cep-1. fsn-1 mutants were also hypersensitive to cep-1-dependent apoptosis induced by ionizing radiation (Supplementary Figure 4), indicating that it may have a general role in genotoxic stress response.

fsn-1 regulates germline apoptosis through cep-1. (a) Hypersensitivity of fsn-1(hp1) and fsn-1(gk429) mutants to ENU-induced germ cell apoptosis is dependent on cep-1. Between 25 and 35 germ lines were scored at each dose of ENU in triplicate. (b) Effect of fsn-1(hp1) and fsn-1(gk429) mutants on cep-1-dependent transcription of egl-1. Fold induction of egl-1 was calculated relative to levels in untreated wild-type hermaphrodites. Error bars=S.E.M.
Since fsn-1 functions in neurons to control synapse development,31 we asked whether expression of fsn-1(+) in neurons or muscle would rescue the sensitivity of fsn-1(hp1) mutants to ENU-induced germline apoptosis. fsn-1(hp1) mutants that express fsn-1(+) in neurons or muscle were treated with 0.5 mM ENU, which has minimal effects on germline apoptosis in wild-type animals but triggers widespread apoptosis in fsn-1 mutants (Figure 3a). Expression of fsn-1(+) in these somatic tissues failed to rescue the sensitivity of fsn-1(hp1) mutants to germline apoptosis (Table 1). These results suggest that FSN-1 functions in a different tissue, such as the germ line, to regulate CEP-1-dependent apoptosis. To address the epistatic relationship between ned-8 and fsn-1, we inhibited ned-8 by RNAi in fsn-1 mutants and quantified germline apoptosis in response to ENU. There was no difference in the levels of ENU-induced germ cell apoptosis when both genes were ablated (Supplementary Table 2), suggesting that ned-8 and fsn-1 act in the same pathway.
In addition to defects in activating cep-1-dependent apoptosis, C. elegans DNA repair mutants also produce a high fraction of dead progeny after treatment with DNA-damaging agents.18, 32, 33 To ascertain whether fsn-1 mutants have defects in DNA repair, the survival of their progeny was quantified after ENU treatment. We found that fsn-1(gk429) mutants were as slightly sensitive to 2.5 mM ENU and cep-1; fsn-1 double mutants were moderately sensitive to 1.0 mM ENU compared with cep-1 single mutants. Therefore, we cannot rule out the possibility that fsn-1 may have a minor role in DNA repair. Interestingly, cep-1(gk138) mutants were sensitive to 1.0 and 2.5 mM ENU (Table 2), suggesting that cep-1 may have a role in the repair of damage caused by alkylating agents.
To determine whether the increased levels of apoptosis observed in fsn-1 mutants was due to a defect in the engulfment of apoptotic corpses, we quantified the time germ cell corpses remained in the gonads of animals treated with ENU before they were engulfed.17, 22 Germ cell corpses persisted for 27±3 min (n=10) in wild type and 26±1 min in fsn-1(hp1) mutants (n=10), indicating that fsn-1 mutants do not have more germ cell corpses in response to ENU because of delayed engulfment.
FSN-1 downregulates cep-1-dependent transcription
Because apoptosis in the fsn-1-null mutants was dependent on cep-1, we next evaluated whether CEP-1 activity was increased in fsn-1 mutants. To measure CEP-1 activity in vivo, we quantified its transcriptional target gene egl-1 by qPCR in wild type and fsn-1(hp1) and fsn-1(gk429) mutants treated with ENU (Figure 3b). In both fsn-1 mutants, there was an ∼2-fold increase in egl-1 transcript over wild-type controls treated with 5 mM ENU (Figure 3b). The increased CEP-1 transcriptional activity in fsn-1 mutants paralleled the higher levels of germ cell apoptosis observed in these mutants treated with ENU. Because apoptosis and CEP-1 transcriptional activity in the fsn-1 mutants were suppressed by the cep-1(gk138) allele (Figure 3), we conclude that FSN-1 normally represses CEP-1 activity in the germ line in response to DNA damage.
Endogenous CEP-1 protein phosphorylation levels are regulated by FSN-1
We recently reported that endogenous CEP-1 is phosphorylated in response to ionizing radiation.17 To determine if CEP-1 protein is also affected by ENU treatment, we performed western analysis of worm extracts using an antibody against CEP-1.17, 21 In wild-type extracts, we detected a band of ∼75 kDa (the predicted size of full-length CEP-1) as well as a slower migrating band that was only detectable after ENU treatment (Figure 4a). The levels of modified CEP-1 reached a peak at 12 h and declined 24 h after ENU treatment. To assess whether this slower migrating band represents a phosphorylated form of CEP-1, we treated wild-type worm extracts with shrimp alkaline phosphatase and performed western analysis with anti-CEP-1. Phosphatase treatment caused the abundance of the upper band to dramatically decrease, but in the presence of phosphatase inhibitors this modified form of CEP-1 did not disappear (Figure 4b). Because the modified CEP-1 bands disappear after phosphatase treatment and were not present in extracts isolated from cep-1(gk138) deletion mutants (Figure 4c), we conclude that endogenous CEP-1 is phosphorylated in response to ENU. Since ENU caused the transcriptional activity of CEP-1 to increase in fsn-1 mutants, we next asked whether CEP-1 protein was also augmented. In response to ENU, phosphorylated CEP-1 levels (upper band) increased by two- and three-fold in fsn-1(hp1) and fsn-1(gk429) mutants, respectively, over wild-type controls (Figure 4c and d). In non-stressed fsn-1(gk429) mutants, there were higher levels of unmodified CEP-1 (Figure 4d), suggesting that FSN-1 may also negatively regulate CEP-1 levels under normal conditions.
To address whether the increased levels of CEP-1 detected in fsn-1 mutant extracts were due to ectopic expression outside the germ line, wild-type and fsn-1 mutant worms were stained at various stages of development with CEP-1 antibodies. We observed CEP-1 staining in the same pharyngeal cells and in the germ line as previously reported,13, 21 but did not detect ectopic CEP-1 staining in other somatic tissues in wild-type or fsn-1 mutant worms treated with ENU (Supplementary Figure 5). In the germ line, there was a similar pattern of CEP-1 staining as described by Schumacher et al.,21 where levels were highest in the mitotic and meiotic regions and lower in the transition zone (Supplementary Figure 5D). CEP-1 staining was more intense in the meiotic germ lines of fsn-1 mutants treated with ENU compared to wild-type animals (Figure 5 and Supplementary Figure 6). In the absence of ENU, there was no detectable difference in CEP-1 staining in the germ lines of fsn-1 mutants and wild-type animals, suggesting that CEP-1 levels in the germ line are regulated by FSN-1 in response to genotoxic stress. Interestingly, we also detected CEP-1 staining in the germ line at all stages of larval development and in the male germ line (Supplementary Figure 5) that was not previously reported.13, 21 Therefore, we conclude that CEP-1 is expressed throughout germline development in both hermaphrodites and males, and that FSN-1 negatively regulates endogenous CEP-1 levels in the hermaphrodite germ line in response to DNA damage.

CEP-1 protein levels are higher in fsn-1 mutants treated with ENU. To detect qualitative differences in CEP-1 germline staining intensities, photographs were taken at non-saturating intensities using identical exposure times for all samples. (a) Wild-type hermaphrodites stained with anti-CEP-1 antibodies in the absence of ENU. (b) Wild-type hermaphrodites treated with 5 mM ENU stained with anti-CEP-1 antibodies after 20 h. (c) fsn-1(hp1) mutant hermaphrodites stained with anti-CEP-1 antibodies in the absence of ENU. (d) fsn-1(hp1) mutant hermaphrodites+5 mM ENU stained with anti-CEP-1 antibodies 20 h after ENU treatment. (e) cep-1(lg12501) mutant hermaphrodites do not stain with anti-CEP-1
FSN-1 may regulate CEP-1 through an indirect mechanism
To determine if FSN-1 binds directly to CEP-1, we transiently transfected HEK293 cells with FLAG-tagged CEP-1 and H7-tagged FSN-1. Cell extracts were immunoprecipitated with anti-FLAG or anti-T7 and blots probed with anti-T7 or anti-FLAG, respectively. We were unable to detect FSN-1 binding to CEP-1 in cotransfected cells (Supplementary Figure 7), suggesting that FSN-1 may regulate CEP-1 by an indirect mechanism. Previously, FSN-1 was shown to regulate synaptogenesis by directly binding SCD-2, the worm orthologue of anaplastic lymphoma kinase.31 To determine if SCD-2 functions downstream of FSN-1 in the germ line, we treated scd-2(ok565) deletion mutants with ENU but observed similar levels of germ cell apoptosis as wild-type controls (Supplementary Table 3). The MAP kinase dlk-1 regulates synapse formation downstream of rpm-1 and fsn-1 (unpublished data), but also does not affect ENU-induced germline apoptosis (Supplementary Table 3). To test if scd-2 and dlk-1 function redundantly downstream of fsn-1 to regulate germ cell apoptosis, scd-2 was inhibited by RNAi in dlk-1(ju476) mutants. As with the single mutants, there was no detectable effect on germ cell apoptosis when both genes were ablated (Supplementary Table 3), indicating that the SCFFSN-1 E3 ligase regulates CEP-1 activity in the germ line through a novel target.
Discussion
The C. elegans hermaphrodite germ line is an excellent model for studying the regulation of DNA damage-induced apoptosis. Like its vertebrate relatives, the C. elegans p53-like gene cep-1 rests at the hub of a complex signaling network to make life or death decisions in response to genotoxic stress.14 CEP-1 is tightly regulated by a number of conserved proteins, including the Akt/PKB serine/threonine kinase AKT-1, the p53-binding protein APE-1/iASPP, and the GLD-1 protein.17, 19, 21 Dysregulation of any of these inputs to CEP-1 leads to abnormal apoptosis in the germ line. Using the powerful genetics and functional genomics tools available in C. elegans, we identified the neddylation pathway and the E3 ubiquitin ligase SCFFSN-1 as important regulators of CEP-1-dependent apoptosis.
The p53-directed E3 ubiquitin ligase Mdm2 can conjugate Nedd8 to mammalian p53 and p73 to attenuate their transcriptional activity.9, 20 Because there is no clear Mdm2 orthologue in C. elegans and we did not detect neddylated CEP-1 from worm extracts, we asked if a cullin-based E3 ubiquitin ligase might regulate CEP-1. Previous work in C. elegans demonstrated that NED-8 regulates the mitosis to meiosis transition in early embryos by conjugating to CUL-3 to increase ubiquitin-mediated proteolysis of the microtubule severing protein MEI-1.34 Thus, the regulation of cullin-based ligases by neddylation is well conserved in C. elegans. In mammals, the p53 family is regulated by several different E3 ubiquitin ligases, but it is not clear whether SCF ligases are involved in the regulation of these proteins. Here, we show that the neddylation pathway and the SCFFSN-1 E3 ligase regulate the apoptotic activity of CEP-1 in response to genotoxic stress. Our genetic epistasis results suggest that neddylation and SCFFSN-1 act in the same pathway (Supplementary Table 2), but we do not know the molecular mechanism by which these proteins regulate CEP-1 (Figure 6). It is possible that small amounts of CEP-1 are neddylated in vivo, which could not be detected with our antibody on western blots. Consistent with this possibility is the recent report demonstrating that p53 can be neddylated by the F-box protein FBX011.35 If CEP-1 was directly regulated by neddylation and ubiquitin-mediated proteolysis through the SCFFSN-1 ligase, we might expect higher CEP-1 activity and increased germline apoptosis when these genes are ablated in the absence of genotoxic stress. For example, GLD-1 directly controls CEP-1 protein levels by binding to the 3-UTR of cep-1 mRNA to repress its translation.21 In gld-1(op236) hypomorphic mutants, CEP-1 protein levels accumulate and trigger massive apoptosis of germ cells, which is further enhanced by DNA-damaging agents.21 Similarly, inhibiting the iASPP orthologue ape-1 by RNAi leads to increased levels of physiological germ cell apoptosis (i.e., in the absence of genotoxic stress) that is dependent on cep-1.19 CEP-1 can also be induced to trigger germline apoptosis indirectly by inhibiting genes that normally prevent DNA damage under normal physiological conditions, such as the Brca1 orthologue brc-1 or the Bloom's helicase him-6.36 In contrast, akt-1 negatively regulates DNA damage signals to cep-1 but does not affect physiological germ cell apoptosis or CEP-1 activity in the absence of DNA-damaging agents.17 This is likely because AKT-1 functions to buffer damage signals to CEP-1 but has no role in the repair of endogenously (or exogenously) generated DNA damage. It is unlikely that SCFFSN-1 directly regulates AKT-1 because the levels of this kinase would be expected to increase in the absence of its negative regulator, and we have shown that activated AKT-1 suppresses damage-induced germ cell apoptosis.17

Model showing the regulation of CEP-1 by SCFFSN-1. In the germ line, the SCFFSN-1 E3 ubiquitin ligase negatively regulates CEP-1. In response to ENU, the negative regulation of CEP-1 is disrupted, perhaps by inhibiting neddylation, and CEP-1 activates expression of the BH3-only protein EGL-1 and CED-13. SCFFSN-1 regulates CEP-1 independent of the kinases SCD-2 and DLK-1. Alternatively, SCFFSN-1 may directly bind and degrade phosphorylated CEP-1
The SCFFSN-1 E3 ligase is conserved from nematodes to humans and was previously shown to regulate neuromuscular synapse development in C. elegans.31 Although the neddylation pathway was not examined in this study, we suspect that NED-8 activates the SCFFSN-1 ligase in the germ line because inhibiting ned-8 in fsn-1(hp1) mutants did not enhance the levels of ENU-induced apoptosis compared with fsn-1(hp1) single mutants (Supplementary Table 2). The role of fsn-1 in the DNA damage response is not just restricted to alkylating agents because fsn-1 mutants are also hypersensitive to ionizing radiation (Supplementary Figure 4). Expression of fsn-1(+) in neurons and muscle did not rescue the germline apoptosis defects of fsn-1 mutants (Table 1), suggesting that it acts in a different tissue (perhaps the germ line) to modulate CEP-1 activity. Because we could not immunoprecipitate CEP-1 from worm extracts to determine if it interacts directly with FSN-1, we coexpressed tagged proteins in HEK293 cells and carried out coimmunoprecipitation experiments. Under these conditions, we did not detect binding by CEP-1 and FSN-1 (Supplementary Figure 7). Taken together, this evidence suggests that CEP-1 is regulated by SCFFSN-1 through an intermediate protein.
At present, we do not know the identity of the germline SCFFSN-1 target protein(s). Because DNA damage-induced germline apoptosis was unaffected in scd-2 or dlk-1 mutants, we propose that FSN-1 targets a unique substrate in the germ line that modulates damage signals to CEP-1 (Figure 6). Furthermore, RPM-1 may regulate distinct targets in different cellular processes.37 Although the viability of progeny from fsn-1 mutants treated with ENU is similar to wild type, we noticed a modest decrease in the survival of progeny from fsn-1 mutants at 2.5 mM ENU (Table 2). Thus, it is possible that partial S-phase progression or chromosome segregation defects of mitotic germ cells could sensitize fsn-1 mutants to ENU-dependent apoptosis. In this regard, SCFFSN-1 may target a protein in the germ line that has a minor role in DNA repair, which feeds into CEP-1. However, F-box proteins typically interact with phosphorylated substrates,26 and since we were unable to detect phosphorylated CEP-1 when it was expressed in HEK293 cells, it is possible that phospho-CEP-1 is the target of FSN-1 in the germ line. It will be interesting to know whether the human FSN-1 orthologue, FBX045, has a conserved role in regulating the apoptotic activity of the p53 family. These studies may reveal conserved mechanisms by which the p53 family is regulated in response to genotoxic stress that could be exploited therapeutically for the treatment of human cancer.
Materials and Methods
Strains and genetics
C. elegans strains were cultured and maintained using standard procedures.37 The following strains were kindly provided by the Caenorhabditis Genetics Center (University of Minnesota, St. Paul, MN, USA): wild-type var. Bristol (N2), ced-3(n717), ced-4(n1162), ced-9(n1950), cep-1(gk138), cep-1(lg12501), cul-1(e1756)/unc-69(e587), egl-1(n1084n3082), fsn-1(gk429). Double mutant strains were generated using standard genetic methods.38
RNAi
Gene expression was inhibited using the feeding method.39 Bacteria expressing double-stranded RNA (dsRNA) to a specific C. elegans gene were grown on the nematode growth media (NGM) plates supplemented with antibiotics (50 μg/ml ampicillin and 12.5 μg/ml tetracycline) and 0.1 mM isopropyl-β-D-thiogalactopyranoside to induce dsRNA production.39 Bacteria expressing an empty RNAi vector (L4440) were used as controls. Larval stage 2 (L2) hermaphrodites were placed on the RNAi-producing plates and allowed to develop until the young adult stage. We did not observe any morphological defects in the germ lines of animals treated with RNAi to the neddylation pathway but observed penetrant and reproducible embryonic lethality in the F1 generation (data not shown), consistent with the essential role of this pathway in the embryo.34 All RNAi clones were sequenced to confirm their identity.
Germline apoptosis and ENU sensitivity
Germline apoptosis was quantified by morphology as described.13 Young adult animals (12 h after the L4 stage) were treated with the DNA-alkylating agent ENU for 4 h at 20°C. After ENU treatment, worms were transferred to fresh plates and allowed to recover for 24 h. Germ cell corpses were quantified in the pachytene region of one gonad arm per animal at 12, 24 and 36 h after ENU treatment. Apoptotic nuclei were scored based on their refractile appearance at × 630 magnification using Nomarski optics. Germ cell corpses were confirmed using the fluorescent dye acridine orange, which is specifically retained in the nuclei of apoptosis germ cells.17
Preparation of RNA and qPCR
Approximately 2000–3000 young adult worms were treated with ENU and total RNA was isolated using GibcoBRL Trizol kit as described previously.17 RNA was dissolved in diethyl pyrocarbonate-treated water and stored at −20°C. For all RNA samples, the ratio of the optical density at 260 nm to the optical density at 280 nm was >1.50. Total RNA (4 μg) was converted to cDNA using SuperScript reverse transcriptase. qPCR was performed with a SYBR Green PCR Master Mix kit (Applied Biosystems), according to the manufacturer's instructions. The final PCR volume was 25 μl. Primer pairs (for egl-1 878 5′-tactcctcgtctcaggactt-3′ and 880 5′-catcgaagtcatcgcacat-3′; for ced-13 706 5′-acggtgtttgagttgcaagc-3′ and 707 5′-gtcgtacaagcgtgatggat-3′; for tbg-1 710 5′-cgtcatcagcctggtagaaca-3′ and 711 5′-tgatgactgtccacgttgga-3′) were designed to generate intron-spanning products of 110–150 bp.24 A standard curve was prepared as a test for the sensitivity of the assay and as a reference for quantification. The standard curve was generated from six points, and each primer pair was tested with a logarithmic dilution of a cDNA mixture to generate a linear standard curve. For normalization of the quantitative results, γ-tubulin (tbg-1) mRNA was used as an external standard.
Western blot analysis
Approximately 2000–3000 adult worms were washed from plates with M9 buffer. After centrifugation at 5000 r.p.m. for 1 min, the supernatant was removed. The pelleted worms were resuspended and washed three times with M9 buffer and centrifuged as above. The worm pellet was resuspended in an equal volume of 2 × Laemmli sample buffer, sonicated for 2 min, and then boiled for 5 min. After a final centrifugation at 13 000 r.p.m. for 1 min, the extracts were stored at −80°C. 200 μg of worm lysate was separated by SDS-PAGE and then transferred to nitrocellulose membranes (Perkin-Elmer Life Science, Boston, MA, USA). The membrane was blocked with 5% skimmed milk in Tris-buffered saline containing 0.1% Tween-20 and subsequently incubated with diluted (1 : 100) preabsorbed goat anti-CEP-1 antibody (a generous gift from Dr. A Gartner)21 or our own rabbit anti-CEP-1 antibody. After washing three times, the membrane was incubated with peroxidase-conjugated rabbit anti-goat immunoglobulin G antibody (Amersham Biosciences Co.), and the specific bands were detected using the Amersham ECL enhanced chemiluminescence reagent. An α-tubulin antibody (Sigma) was used as a loading control.
Phosphatase treatment
Approximately 2000–3000 adult worms were washed with M9 buffer as above. The worm pellet was resuspended in an equal volume of sample buffer, sonicated for 2 min, and then dialyzed overnight in shrimp alkaline phosphatase reaction buffer (0.01 M Tris-HCl, pH 7.5, 0.01 M MgCl2, and 0.1 mg/ml BSA) in the presence or absence of inhibitors (0.8 mM Na3VO4+25 mM NaF) at 4°C. Next, 200 μg of dialyzed worm lysate was incubated with or without 15 U of shrimp alkaline phosphatase for 1 h at 37°C, and then separated by SDS-PAGE and analyzed as above.
Worm fixation
The fixation method was based on the protocol of Finney et al.40 Worms were synchronized at the first larval (L1) stage on 100 mm NGM plates, grown to young adults at 20°C. Worms were washed off plates with M9 buffer into a 15 ml conical tube and pelleted at 3000 r.p.m. The supernatant was aspirated and pellets were washed with 10 ml water twice. Supernatant was removed and the worm suspension was quickly transferred to an Eppendorf tube. The worm suspension was washed two more times and pellets were resuspended in 300 μl water. Next, 500 μl of cold 2 × ‘Witches Brew’ buffer (160 mM KCl, 40 mM NaCl, 20 mM EGTA, 10 mM spermidine-HCl, 30 mM Na-Pipes, pH 7.4, 50% methanol) and 200 μl cold 20% paraformaldehyde were added to the worm suspension. Worms were then mixed by inversion and freeze–thawed in liquid nitrogen. Immediately after thawing, worm mixtures were put on ice for 2 h and inverted every 15 min. Worm mixtures were washed with Tris–Triton buffer twice (5 min for each wash) and then pelleted at 4000 r.p.m. for 1 min. Worms were then incubated in 1 ml B03 buffer (25 mM H3BO3, 12.5 mM NaOH, pH 9.3)+10 μl β-mercaptoethanol for 2 h at 37°C on rotator (∼280 r.p.m.). Next, worm suspensions were pelleted at 4000 r.p.m. for 1 min and washed in 1 ml of B03 buffer. Worm suspensions were pelleted after washing and incubated in 1 ml B03 buffer+10 μl of 1 M DTT for 15 min at room temperature on rotator. Next, worm suspensions were pelleted as above, washed with 1 ml B03 buffer for 5 min, and then incubated in 1 ml B03 and 10 μl H2O2 for 15 min at room temperature (repeated twice). Worm suspensions were pelleted and incubated for 15 min in buffer B (1 × PBS, 0.1% BSA (Pentax Fraction V), 0.5% Triton X-100, 5 mM sodium azide, 1 mM EDTA) on rotator. Worms were pelleted, supernatant was removed, and worms were incubated in buffer A (same as buffer B, except 1% BSA) for ∼1.5 h.
Antibody staining and immunofluorescence
To the fixed worms, 100 μl buffer A and 2 μl anti-CEP-1 antibody (1 : 50 dilution) were added and incubated overnight at room temperature without agitation. Worms were pelleted and supernatant was removed by aspiration. Worms were washed with buffer B 4–5 times (10 min each) and pelleted at 3000 r.p.m. between washes. Secondary anti-rabbit Cy3 was added (1 : 400 dilution) and incubated for 3 h in a 37°C water bath with intermittent inversion. Worms were pelleted and washed 4–5 times in buffer B (10 min each). Worms were carefully pipetted onto poly-L-lysine-coated slides and sealed for immunofluorescence microscopy. cep-1(lg12501) mutants were used for negative staining controls because there was less nonspecific staining in this strain using our CEP-1 antibody.
Abbreviations
- PCD:
-
programmed cell death
- ENU:
-
N-ethyl-N-nitrosourea
- SCF:
-
Skp1/cullin/F-box
- RNAi:
-
RNA interference
- NGM:
-
nematode growth media
References
Levine AJ . p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323–331.
Moll UM, Wolff S, Speidel D, Deppert W . Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 2005; 17: 631–636.
Brooks CL, Gu W . p53 ubiquitination: Mdm2 and beyond. Mol Cell 2006; 21: 307–315.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ . The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69: 1237–1245.
Momand J, Wu HH, Dasgupta G . MDM2 – master regulator of the p53 tumor suppressor protein. Gene 2000; 242: 15–29.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000; 288: 1053–1058.
Nakano K, Vousden KH . PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7: 683–694.
Moll UM, Slade N . p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2: 371–386.
Watson IR, Blanch A, Lin DC, Ohh M, Irwin MS . Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J Biol Chem 2006; 281: 34096–34103.
Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci USA 2006; 103: 12753–12758.
Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J 2005; 24: 836–848.
Schumacher B, Hofmann K, Boulton S, Gartner A . The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 2001; 11: 1722–1727.
Derry WB, Putzke AP, Rothman JH . Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 2001; 294: 591–595.
Derry WB, Bierings R, van Iersel M, Satkunendran T, Reinke V, Rothman JH . Regulation of developmental rate and germ cell proliferation in Caenorhabditis elegans by the p53 gene network. Cell Death Differ 2007; 14: 662–670.
Ou HD, Löhr F, Vogel V, Mantele W, Dötsch V . Structural evolution of C-terminal domains in the p53 family. EMBOJ 2007; 26: 3463–3473.
Huyen Y, Jeffrey PD, Derry WB, Rothman JH, Pavletich NP, Stavridi ES et al. Structural differences in the DNA binding domains of human p53 and its C. elegans ortholog Cep-1. Structure (Camb) 2004; 12: 1237–1243.
Quevedo C, Kaplan DR, Derry WB . AKT-1 regulates DNA-damage-induced germline apoptosis in C. elegans. Curr Biol 2007; 17: 286–292.
Boulton SJ, Martin JS, Polanowska J, Hill DE, Gartner A, Vidal M . BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr Biol 2004; 14: 33–39.
Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 2003; 33: 162–167.
Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP . Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004; 118: 83–97.
Schumacher B, Hanazawa M, Lee MH, Nayak S, Volkmann K, Hofmann R et al. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 2005; 120: 357–368.
Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO . A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 2000; 5: 435–443.
Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L et al. Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol 2002; 12: 1908–1918.
Schumacher B, Schertel C, Wittenburg N, Tuck S, Mitani S, Gartner A et al. C.elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ 2005; 12: 153–161.
del Peso L, González VM, Núñez G . Caenorhabditis elegans EGL-1 disrupts the interaction of CED-9 with CED-4 and promotes CED-3 activation. J Biol Chem 1998; 273: 33495–33500.
Willems AR, Schwab M, Tyers M . A Hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta 2004; 1695: 133–170.
Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM . cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell 1996; 85: 829–839.
Thomas JH . Adaptive evolution in two large families of ubiquitin-ligase adapters in nematodes and plants. Genome Res 2006; 16: 1017–1030.
Nayak S, Santiago FE, Jin H, Lin D, Schedl T, Kipreos ET . The Caenorhabditis elegans Skp1-related gene family: diverse functions in cell proliferation, morphogenesis, and meiosis. Curr Biol 2002; 12: 277–287.
Yamanaka A, Yada M, Imaki H, Koga M, Ohshima Y, Nakayama K . Multiple Skp1-related proteins in Caenorhabditis elegans: diverse patterns of interaction with cullins and F-box proteins. Curr Biol 2002; 12: 267–275.
Liao EH, Hung W, Abrams B, Zhen M . An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 2004; 430: 345–350.
Garcia-Muse T, Boulton SJ . Distinct modes of ATR activation after replication stress and DNA double-strand breaks in Caenorhabditis elegans. EMBO J 2005; 24: 4345–4355.
Stergiou L, Doukoumetzidis K, Sendoel A, Hengartner MO . The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differ 2007; 14: 1129–1138.
Pintard L, Kurz T, Glaser S, Willis JH, Peter M, Bowerman B . Neddylation and deneddylation of CUL-3 is required to target MEI-1/Katanin for degradation at the meiosis-to-mitosis transition in C. elegans. Curr Biol 2003; 13: 911–921.
Abida WM, Nikolaev A, Zhao W, Zhang W, Gu W . FBXO11 promotes the neddylation of p53 and inhibits its transcriptional activity. J Biol Chem 2006; 282: 1797–1804.
Kim YM, Yang I, Lee J, Koo HS . Deficiency of Bloom's syndrome protein causes hypersensitivity of C. elegans to ionizing radiation but not to UV radiation, and induces p53-dependent physiological apoptosis. Mol Cells 2005; 20: 228–234.
Fulga TA, Van Vactor D . Synapses and growth cones on two sides of a highwire. Neuron 2008; 57: 339–344.
Brenner S . The genetics of Caenorhabditis elegans. Genetics 1974; 77: 71–94.
Timmons L, Court DL, Fire A . Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 2001; 263: 103–112.
Finney M, Ruvkun G, Horvitz HR . The C. elegans cell lineage and differentiation gene unc-86 encodes a protein with a homeodomain and extended similarity to transcription factors. Cell 1988; 55: 757–769.
Acknowledgements
We thank the Caenorhabditis Genetics Center and the C. elegans Gene Knockout Consortium for worm strains and Dr. Anton Gartner (University of Dundee) for CEP-1 antibody. We also thank Dr. Peter Roy, Dr. Frank Sicheri and Dr. Lionel Pintard for helpful discussions on this manuscript. This work was supported by a Restracomp postdoctoral fellowship from The Hospital for Sick Children Research Training Center (to MXG), NSERC Canada Graduate Scholarship (to EHL), grants from CIHR (to WBD and MZ), Canada Foundation for Innovation and the Ontario Innovation Trust (to WBD), and start-up funds from the Hospital for Sick Children Research Institute (to WBD).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by RA Knight
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Gao, M., Liao, E., Yu, B. et al. The SCFFSN-1 ubiquitin ligase controls germline apoptosis through CEP-1/p53 in C. elegans. Cell Death Differ 15, 1054–1062 (2008). https://doi.org/10.1038/cdd.2008.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2008.30
Keywords
This article is cited by
-
Precoce and opposite response of proteasome activity after acute or chronic exposure of C. elegans to γ-radiation
Scientific Reports (2018)
-
VHL deficiency augments anthracycline sensitivity of clear cell renal cell carcinomas by down-regulating ALDH2
Nature Communications (2017)
-
Identification of factors that function in Drosophila salivary gland cell death during development using proteomics
Cell Death & Differentiation (2013)
-
The cullin protein family
Genome Biology (2011)
-
The EEL-1 ubiquitin ligase promotes DNA damage-induced germ cell apoptosis in C. elegans
Cell Death & Differentiation (2011)
