
Abstract
Thanks to the Nobel Foundation for permission to publish this Lecture (Copyright© The Nobel Foundation 2006). We report here the Nobel Lecture delivered by Professor RD Kornberg describing his research in the understanding of transcription in eucaryotes. The amazing work by Professor Kornberg goes from the discovery of the nucleosome to the structural and functional studies of pol II transcription complexes. His research sheds light on fundamental molecular biology problems such as transcription initiation, fidelity of transcription, RNA release at the end of transcription, and many more. This is a beautiful report on how structural and functional studies can be combined to really understand in an accurate and detailed way how proteins combine in huge molecular complexes to regulate one of the most important cellular processes: gene transcription.
Similar content being viewed by others
Main
I am deeply grateful for the honor bestowed on me by the Nobel Committee for Chemistry and the Royal Swedish Academy of Sciences. It is an honor I share with my collaborators. It is also recognition of the many who have contributed over the past quarter century to the study of transcription.
The Nucleosome
My own involvement in studies of transcription began with the discovery of the nucleosome, the basic unit of DNA coiling in eucaryote chromosomes.1 X-ray studies and protein chemistry led me to propose the wrapping of DNA around a set of eight histone molecules in the nucleosome (Figure 1). Some years later, Yahli Lorch2 and I found that this wrapping of DNA prevents the initiation of transcription in vitro. Michael Grunstein and colleagues3 showed that nucleosomes interfere with transcription in vivo. The nucleosome serves as a general gene repressor. It assures the inactivity of all the many thousands of genes in eucaryotic cells except those whose transcription is brought about by specific positive regulatory mechanisms. What are these positive regulatory mechanisms? How is repression by the nucleosome overcome for transcription? Our recent work has shown that promoter chromatin is transformed from a static to a dynamic state upon gene activation.4 Nucleosomes are rapidly removed and reassembled in the activated state. Promoter DNA is made transiently available for interaction with the transcription machinery.

The nucleosome, fundamental particle of the eucaryote chromosome. Schematic shows the coiling of DNA around a set of eight histones in the nucleosome, the further coiling in condensed (transcriptionally inactive) chromatin, and uncoiling for interaction with the RNA pol II transcription machinery
RNA Polymerase II Transcription
Our studies have focused on the RNA polymerase II (pol II) transcription machinery. Pol II is responsible for all messenger RNA synthesis in eucaryotes. As the first step in gene expression, pol II transcription is an end point of a great many signal transduction pathways. The intricate regulation of pol II transcription underlies cell differentiation and development.
Because nucleosomes are removed from promoter DNA for transcription in vivo, we and others have been able to fractionate the components of the transcription machinery guided by transcription assays performed with naked DNA in vitro. Robert Roeder and colleagues initiated the isolation of pol II transcription proteins from human HeLa cell extracts.5 This effort was brought to fruition by Ronald and Joan Conaway, who took advantage of the greater abundance of starting material available from a rat liver extract.6 We at Stanford isolated the pol II machinery from yeast, in the work begun by Neal Lue7 in 1987, who solved the long-standing problem of preparing an extract active in pol II transcription from yeast. In retrospect, our pursuit of the problem in yeast was a fortunate choice. It proved crucial for unraveling both the structure and regulation of the pol II machinery. There were serious doubts when we began studying whether findings in yeast would prove relevant to human cells. But upon fractionation of yeast and mammalian systems, the results were the same.
Both systems comprise six proteins: pol II and five general transcription factors known as TFIIB, -D, -E, -F, and -H.8 Pol II is capable of unwinding DNA, synthesizing RNA, and rewinding DNA. But pol II alone is incapable of recognizing a promoter and initiating transcription. For these essential functions, the participation of the general transcription factors is required.
Mediator of Transcriptional Regulation
It was at first thought that the set of six proteins constituted a complete transcription system and that it would support not only accurately initiated but also appropriately regulated transcription. Communication from a regulatory protein to the transcription machinery at a promoter was believed to be direct. We found, however, that an additional crude fraction was required for regulation in the yeast system (Figure 2). We referred to this activity as Mediator,9, 10 and in 1994, Stefan Björklund and Young-Joon Kim11 isolated the active protein as an assembly of more than 20 subunits, with a total mass in excess of a million daltons. Thirteen of the subunits were products of genes previously identified in screens for molecules involved in transcriptional regulation. These were disparate screens, done on different promoters in different labs at different times. With the isolation of Mediator, the products of all the screens were united in a common biochemical entity. Still the Mediator idea did not gain wide acceptance, as regulation in higher organisms was believed to be direct, through interaction of gene activator proteins with the so-called TAF subunits of the general transcription factor TFIID. Finally in 1998, we and others isolated mammalian counterparts of the yeast Mediator (reviewed in Kornberg12 and Malik and Roeder13), and TAFs were shown to be important for promoter recognition, not regulation.14, 15, 16 As others have shown, 22 of 25 yeast Mediator subunits have demonstrable homologs in higher cells.17, 18

Mediator of transcriptional regulation. Schematic shows the transduction of regulatory information from a gene activator protein bound to an enhancer DNA element to the pol II transcription machinery at a promoter
We are just beginning to fathom the complexity of Mediator, but already three points are clear. First, Mediator is not only the basis for regulated transcription, but it is also absolutely required for transcription of almost all pol II promoters.19, 20 Mediator is no less essential for transcription than pol II itself. Second, Mediator interacts directly with both activator proteins and with pol II.13 It forms a tight complex with an activator at an enhancer, and it subsequently contacts pol II and the general transcription factors at the promoter to stimulate the initiation of transcription.12 Third, Mediator is important not only for positive but also for negative regulation of transcription. Although Mediator is commonly referred to as a co-activator, this is a misnomer. Mediator is a co-activator, a co-repressor, and a general transcription factor all in one. Mediator may be viewed as a signal processor.
It transduces regulatory information from enhancers to promoters in the entire range of organisms from yeast to humans.
Structural Studies of the Pol II Transcription Machinery
It has been said that if you wish to understand function, study structure. The challenge in the case of the pol II transcription machinery lies in the great size of the structure. A giant complex of nearly 60 proteins, with a total mass in excess of three million daltons, assembles at every pol II promoter prior to the initiation of transcription (Figure 3). We began with structure determination of pol II because it forms the core of the transcription complex. In retrospect, this was another fortunate choice. It was preferable to pursuing the smaller simpler transcription factors because pol II is the platform upon which all factors are assembled. It emerged from our work that some of the general factor proteins adopt their fully folded structures only upon interaction with pol II. Knowledge of the pol II structure has proved key to understanding eucaryotic gene transcription.

The RNA pol II transcription machinery. Masses are round figures for proteins from the yeast Saccharomyces cerevisiae
Two-Dimensional Protein Crystallography
The story of the pol II structure began with my graduate work in physical chemistry, with nuclear resonance experiments revealing the rapid lateral diffusion of lipid molecules in multilayers.21 Some years later, I thought of exploiting lateral diffusion for the formation of single-layer-thick or two-dimensional (2-D) protein crystals. The idea was to bind a protein to a lipid layer, through interaction with the lipid head groups. The bound protein would be constrained in 2-D but free to diffuse in the plane and crystallize (Figure 4).22 Seth Darst and Al Edwards23 succeeded in forming 2-D crystals of pol II in this way. The crystals were initially small and poorly ordered. They were of little use for structure determination, but they provided a starting point. We could take advantage of the ease, rapidity, and small amount of material required by the 2-D crystallographic approach and use it as a structural assay to guide the preparation of pol II that would form better crystals. We soon found that heterogeneity due to a substoichiometric amount of two small polymerase subunits was the problem – although they accounted for only 8% of the mass of the enzyme, their presence in a variable amount was the impediment to crystallization. We turned to a deletion mutant of yeast from which we could isolate pol II lacking both small subunits. The resulting homogeneous pol II preparation formed very large, exceedingly well-ordered 2-D crystals.24 Even at the low protein concentration used for 2-D crystal growth, about 50 μg/ml, the crystals could often be seen to add additional layers in register with the first. This propensity for epitaxial growth could be exploited and the 2-D crystals used to seed the formation of 3-D crystals for X-ray analysis.

Two-dimensional protein crystallization on lipid layers. Schematic shows the binding of a protein of interest (oval objects) to the head groups (red triangles) of lipid molecules in a monolayer at the air–water interface. Rapid lateral diffusion of the lipids leads to protein crystallization
X-ray Crystal Structure of Pol II
We were excited when the first 3-D crystals were obtained. I also recall a chill of anxiety. The largest X-ray structure of an asymmetric particle at the time, 17 years ago, was a fifth the size of pol II, and for good reason: X-ray beam intensities, detectors, and computational capabilities were all limiting. Actually, the limitations of diffraction technology need not have concerned us because the pol II crystals failed to diffract! The project would have ended there, were it not for our noticing that the crystals were faintly tinged with yellow. The problem was oxidation, and the solution was to grow and maintain the crystals in a glove box under argon.
Then it emerged that the crystals were profoundly polymorphic, varying by more than 10 Å along one unit cell direction. This variation, and the sheer size of pol II, posed challenges for phasing the diffraction by multiple isomorphous replacement with heavy atoms. Jianhua Fu,25 who discovered the best diffracting form of pol II crystals, addressed the phase problem by data collection from a large number of crystals and with the use of heavy atom clusters developed by others. He found matched pairs of native and derivative crystals from which phases to 5 Å resolution could be derived. The resulting electron density map corresponded closely to the structure of pol II at 16 Å resolution determined from 2-D crystals by electron microscopy and 3-D reconstruction. This marked a turning point in the solution of the pol II problem. With reliable 5 Å phases it was possible, in principle, to locate individual heavy atoms and solve the structure to near atomic resolution.
Patrick Cramer and Dave Bushnell sought additional heavy atom derivatives. When they changed the mother liquor of the crystals for the purpose, the crystals shrank by 11 Å along the previously variable unit cell direction, eliminating the problem of polymorphism and extending diffraction to 2.8 Å resolution. None of the 50 heavy atom compounds commonly used for phase determination gave useful derivatives, but an iridium compound identified by Fu and rhenium compounds were eventually effective. The resulting structure comprised some 3500 amino acids, with 28 000 non-hydrogen atoms (Figure 5).26,27

Structure of RNA pol II at 2.8 Å resolution. The protein is shown in a ribbon representation, with a color code to the various subunits and interaction diagram at the upper right. A Mg ion at the active center is depicted as a pink sphere
Where do DNA and RNA bind to pol II? The answer came from the structure determination of the polymerase in the form of a transcribing complex. We tried to crystallize such a complex all the while we pursued the structure of the polymerase alone. The problem was that even very highly purified pol II contains many inactive molecules and these would contaminate any preparation of transcribing complexes. Finally, Avi Gnatt discovered a way of removing the inactive molecules and succeeded in growing transcribing complex crystals.28 Transcription had been paused by withholding one of the four nucleoside triphosphates (NTPs), and upon soaking the crystals in the missing NTP, transcription resumed without loss of crystal morphology. The crystals were very thin and gave diffraction complete to only about 6 Å resolution. After years of trying, Avi collected a data set complete to 3.3 Å resolution, which was solved by molecular replacement with the 2.8 Å polymerase structure.29
DNA can be seen entering the transcribing complex in duplex form and unwinding three bases before the active site (Figure 6). Then the template strand makes a sharp bend, and as a result, the next base is flipped, pointing down toward the active site. This base is paired with that of the ribonucleotide just added to the RNA strand. The structure reveals eight more DNA–RNA hybrid base pairs and one additional base on the template DNA strand. The remainder of the template strand, the RNA, and the nontemplate DNA strand are not seen due to motion or disorder.

Structure of RNA pol II in the act of gene transcription at near atomic resolution. The polypeptide chain is shown in white, orange (mobile ‘clamp’), and green (bridge helix connecting the two largest subunits). Backbone models of the nucleic acids are shown in blue (template DNA strand), green (nontemplate DNA strand), and red (RNA)
Fidelity of Transcription
How does pol II select the right nucleotide for addition to the RNA chain?
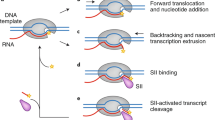
This is the essence of transcription, the accurate readout of the genetic code. Our most recent work has shown how accurate readout is achieved. In the original transcribing complex structure, the nucleotide just added to the RNA was still in the active center. In subsequent structures, we were able to trap the complex following translocation of DNA and RNA across the enzyme surface, creating an empty site at the active center, available for binding the next NTP (Figure 7). Soaking crystals of this ‘post-translocation’ complex with NTPs resulted in additional electron density at two sites.30 All four NTPs were seen to bind an entry or ‘E’ site, whereas only the NTP correctly matched for base pairing with the coding base in the DNA was seen to bind in the active center, at the nucleotide addition or ‘A’ site. The orientation of NTP in the E site was inverted with respect to that in the A site, leading to the suggestion that NTPs in the E site rotate to sample base pairing in the A site.

A cycle of nucleotide addition by RNA pol II. At the upper left, the structure from Figure 6 is shown, omitting all but the DNA and RNA near the active center and the bridge helix. The ribonucleotide in the active center, just added to the RNA chain, is yellow. At the lower left is the structure after translocation of DNA and RNA across the pol II surface. At the lower right is the structure with an unmatched NTP in the entry (E) site. At the upper right is the structure with NTP, matched for pairing to the coding base in the template strand, in the addition (A) site
These structures failed to reveal the basis for the fidelity of transcription. The energy of base pairing, through two or three hydrogen bonds to the template DNA, is far less than required to account for the selectivity of the polymerase reaction. The mystery remained until this year, when screening many hundreds of crystals by Dong Wang and Dave Bushnell led to improved resolution and data quality. The structure of a transcribing complex with correctly matched NTP in the A site now included a feature termed the trigger loop beneath the A site (Figure 8).31 The trigger loop was seen before in many pol II structures, but only in the two solved this year, with correctly matched NTP in the A site, did it appear in proximity to the A site. In all previous structures, it was located 30 Å or more distant from the A site. The trigger loop is evidently a mobile element, swinging like a trap door beneath correctly matched NTP in the A site.

The trigger loop. Transcribing complex structures with purine nucleotide (orange) or pyrimidine nucleotide (dark blue) in the addition site (cf. that in the upper right panel of Figure 7) are shown superimposed. The corresponding trigger loops are purple and yellow, and the bridge helices are green and light blue
The trigger loop contacts all moieties of the NTP – the base, the phosphates and, through other pol II residues, the sugar as well (Figure 9). The resulting network of interactions even includes the 2′-OH group of the nucleotide just added to the end of the RNA. The importance of these interactions is shown by mutations affecting transcription (Figure 9). For example, mutation of asparagine 479, hydrogen bonded to the 3′-OH group of the NTP, results in a loss of discrimination between the normal nucleotide and one lacking a 3′-OH group. The discrimination is not great, only about 10-fold the magnitude expected for the energy of a single hydrogen bond. In contrast, discrimination between a normal ribo NTP and a 2′-deoxy NTP is very great, at least 1000-fold, and is unaffected by mutation of asparagine 479. How is such extraordinary specificity for a single OH group achieved? The answer lies in the alignment of the trigger loop with the NTP and the precise positioning of a histidine side chain, 3.5 Å from the β-phosphate (Figure 10). The histidine promotes the flow of electrons during the nucleophilic attack of the 3′-OH at the chain terminus and phosphoanhydride bond breakage. It serves as a proton donor for the pyrophosphate-leaving group. It literally triggers phosphodiester bond formation. It couples nucleotide selection to catalysis.

The trigger loop network. Trigger loop is magenta, GTP is orange, and the 3′ end of the RNA is red. Other residues of Rpb1 and Rpb2 are indicated in black and cyan. Stars identify residues whose mutation impairs transcript elongation in vivo (blue from the literature, red from an unpublished screen by Craig Kaplan)

The trigger loop couples NTP recognition to phosphodiester bond formation. Color scheme as in Figure 8, with side chains of Rpb2 Arg1020 and Rpb2 Arg766 in yellow. Contacts responsible for alignment and for the contact of histidine 1085 with the NTP that promotes catalysis are indicated by dashed yellow lines. Nucleophilic attack and phosphoanhydride bond breakage are indicated by black arrows
The electronic transactions involved in trigger loop function require precise alignment of the interacting moieties. This is achieved for a correct NTP by formation of the trigger loop network. In the case of an incorrect NTP, for example a 2′-deoxy NTP, misalignment is profound. A double helix formed with a 2′-deoxy nucleotide is 2 Å narrower than that formed by a ribonucleotide. The resulting misalignment with the catalytic histidine residue is as great as in the case of a pyrimidine–pyrimidine base mismatch, leading to a 1000-fold reduction in the rate of phosphodiester bond formation.
Nucleic Acid Translocation
The significance of the trigger loop network extends beyond nucleotide selection and catalysis. The network includes many contacts with the bridge helix, which, in turn, contacts the coding base in the template DNA strand. The structure of bacterial polymerase, determined by Seth Darst, also includes a bridge helix, but in contrast with that in pol II, the bridge helix in the bacterial enzyme is bent (Figure 11).32 The bend produces a movement of about 3 Å in the direction of the template strand, corresponding to one base-pair step along the strand. This led us to suggest that transitions of the bridge helix between straight and bent states underlie the translocation step in transcription. The bridge helix may serve as a kind of molecular ratchet, allowing the polymerase to let go of the DNA and RNA for translocation, while retaining a grip on the growing end of the DNA–RNA hybrid helix to preserve the register of transcription. There is now a good deal of biochemical and genetic evidence to support this idea.

Straight and bent states of the bridge helix in RNA pol II and bacterial RNA polymerase structures, proposed to underlie nucleic acid translocation during transcription. Color code as in Figure 6 except with bridge helix in purple
RNA Release
In the final step of transcription, the RNA is released. The question arises as to how the RNA is peeled off the template DNA. How is the very stable RNA–DNA hybrid helix disrupted and the RNA discharged into solution? Our original transcribing complex structure gave no indication of the mechanism of this important process. A subsequent structure, determined by Ken Westover, revealed RNA release taking place.33 Base pair 7 of the DNA–RNA hybrid in this structure appears normal – the bases are coplanar, with a distance appropriate for hydrogen bonding between them (Figure 12). Base pairs 8, 9, and 10, however, show increasing deviations and consequent splaying apart of the DNA and RNA strands. The strand separation is due to the intervention of three protein loops, termed fork loop 1, rudder, and lid (Figure 12). These loops were disordered in all previous pol II structures. Rudder and lid lie between DNA and RNA strands, with rudder contacting the DNA and lid the RNA. A phenylalanine side chain of the lid serves as an actual wedge to maintain separation of the strands. Fork loop 1 contacts the sugar-phosphate backbone of the hybrid helix at base pairs 6 and 7, stabilizing the helix, limiting strand separation to position 8 and beyond, preventing the DNA–RNA hybrid from unraveling further and inhibiting transcription.

Release of RNA transcript from DNA–RNA hybrid revealed in the structure of an RNA pol II transcribing complex. The upstream end of the DNA–RNA hybrid helix, 7–10 residues from the active center, is shown on the left, with distances between the DNA and RNA bases indicated. The entire DNA–RNA hybrid helix is shown on the right, along with protein loops involved in helix melting (rudder and lid) and stabilization (fork loop)
RNA pol II–TFIIB Complex
It may be asked how the transcribing complex is formed in the first place. How is straight duplex promoter DNA melted, bent, and inserted in the pol II active center, enabling the initiation of transcription? These DNA transactions are brought about by the general transcription factors TFIIB, -D, -E, -F, and -H. Our solution of the X-ray structures of pol II–TFIIB and pol II–TFIIF complexes has shed light on the initiation mechanism.
The structure of the pol II–TFIIB complex34 revealed distinct functions of the N- and C-terminal domains of TFIIB. A polypeptide chain trace of the N-terminal domain (yellow in Figure 13) begins with a Zn ribbon that binds the pol II surface adjacent to the clamp and wall. Then the polypeptide does an amazing thing: rather than pass back into solution, it continues across the saddle between the clamp and wall and plunges toward the active center, from which it loops back and remerges across the saddle. The loop, which we have termed the B finger, occupies almost the same location as the DNA–RNA hybrid in a transcribing complex. Superimposing the B finger and the DNA–RNA hybrid from the transcribing complex structure reveals no interference with the template DNA strand or with the RNA up to position 5, but a steric clash with the RNA at position 6 and beyond (Figure 14).

Structure of an RNA pol II–TFIIB complex. A surface representation of pol II is shown, with the clamp and wall as in Figure 6, a polypeptide chain trace of the N-terminal region of TFIIB, designated IIBN, in yellow, and the region of the pol II surface interacting with IIBN in green

Biochemical experiments show that the B finger is not only compatible with a hybrid containing five residues of RNA, but is also required for stability of such a complex. When the RNA grows beyond five or six residues, however, it must compete with TFIIB for space on the pol II saddle. If TFIIB wins the competition, initiation is aborted and must be tried again. If the RNA wins, TFIIB is ejected and pol II is released from the promoter to continue and complete transcription. The B finger thus explains two crucial but until now mysterious aspects of pol II transcription, abortive initiation and promoter escape. In these respects, it resembles the sigma factor in bacterial transcription.35,36
Turning to the C-terminal domain of TFIIB, its location in the structure of the pol II–TFIIB complex served as a guide for docking a structure determined long ago37 of a ternary complex of a C-terminal TFIIB fragment, the TATA-binding protein (TBP) subunit of TFIID, and a TATA box DNA fragment (Figure 15). This modeling exercise led to a moment of truth. The TATA box DNA fragment is sharply bent by TBP. What would happen if the ends of the bent fragment were extended with straight B-form DNA? The result was remarkable in two respects. First, the DNA fits snugly against the protein (Figure 15, left panel) – TBP evidently configures promoter DNA to the contours of the pol II surface. Second, the DNA downstream of the TATA box ran past the saddle (Figure 15, right panel). The distance from the TATA box to the saddle is about 1.5 turns of the double helix, or 15 base pairs (bp). We know from the transcribing complex structure that about 12 residues are required to cross the saddle to the active site. The sum of 15 bp from the TATA box and 12 residues to the active site is 27 bp, closely coincident with the spacing of 25–30 bp from the TATA box to the transcription start site of almost all pol II promoters. In this way, pol II–TFIIB interaction may determine the location of the transcription start site.

Model of an RNA pol II–TBP–TFIIB–DNA complex. The structure of the C-terminal region of TFIIB (pink) complexed with TBP (green) and TATA-box containing DNA (red/white/blue atomic model) was docked to the structure of the pol II–TFIIB complex (as shown in Figure 13). The views in the left and right panels are related by a 90° rotation about an axis between them, as indicated by the curved arrow. The direction of view in the right panel is the same as that in Figure 13
RNA pol II–TFIIF Complex
A cocrystal structure of a transcribing complex with the central subunit of TFIIF is also informative about the initiation of transcription. This work in progress of Guillermo Calero was made possible by his mastery of the biochemical behavior of TFIIF. The structure includes a complete transcription bubble – not only the template DNA strand with associated RNA seen in previous structures, but also the nontemplate DNA strand, and the region upstream of the bubble where duplex DNA is reformed following transcription (Figure 16). The nontemplate strand and upstream duplex DNA were revealed because of interaction with TFIIF, constraining their mobility. This interaction of the nontemplate strand with TFIIF may trap a transient bubble in promoter DNA, leading to the initiation of transcription.

Structure of an RNA pol II transcribing complex with the central subunit of TFIIF (work in progress of Guillermo Calero). Pol II and the nucleic acids only are shown on the left, while the structure including the TFIIF subunit (known as Tfg2, in yellow) is shown on the right. Direction of view and color scheme as in Figure 6
RNA pol II Transcription Initiation Complex
The available structural information may be assembled in a preliminary picture of the pol II transcription initiation complex (Figure 17). The structures of pol II, TBP, and TFIIB come from X-ray crystallography, as I have described. The structures of TFIIE, TFIIF, and TFIIH are from electron crystallography and from cryo-electron microscopy and single particle analysis.

The picture of the complete complex provides a solution, in outline, of the transcription initiation problem. Each of the general factors is seen to play a simple, essential role in the initiation process: TBP bends the promoter DNA around the polymerase and the C-terminal domain of TFIIB. The N-terminal domain of TFIIB brings the DNA to a point on the polymerase surface from which it need only follow a straight path and, by virtue of the conserved spacing from TATA box to transcription start site in pol II promoters, the start site is juxtaposed with the active center. TFIIE enters the complex and recruits TFIIH, whose ATPase/helicase subunit introduces negative superhelical tension in the DNA. Thermal unwinding produces a transient bubble, which is captured by TFIIF binding to the nontemplate strand. The DNA can now bend in the single-stranded region and descend into the pol II active center. Initiation and the synthesis of RNA ensue, initially stabilized by the B finger. Synthesis of a transcript greater than about 6 residues in length leads to the displacement of TFIIB, promoter escape, and the completion of transcription.
Mediator and the Regulation of Transcription
It remains to solve the Mediator, and thus to understand transcriptional regulation. The structure of a pol II–Mediator complex has been determined at low resolution by Francisco Asturias, by cryo-electron microscopy and single particle analysis (Figure 18).38 The portion of the structure due to pol II could be identified by docking the atomic model of the polymerase. The remainder of the structure, due to Mediator, was in the form of a crescent, largely enveloping pol II, with many points of contact through which regulatory information may be transmitted. Extension of the structure to atomic resolution will one day reveal the regulatory mechanism.

Cryo-EM structure of an RNA pol II–Mediator complex. The pol II structure was docked in the central density, and is shown in a similar direction of view and color scheme as Figure 5
References
Kornberg RD . Chromatin structure: a repeating unit of histones and DNA. Science 1974; 184: 868–871.
Lorch Y et al. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell 1987; 49: 203–210.
Han M, Grunstein M . Nucleosome loss activates yeast downstream promoters in vivo. Cell 1988; 55: 1137–1145.
Boeger H et al. Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell 2003; 11: 1587–1598.
Matsui T et al. Multiple factors required for accurate initiation of transcription by purified RNA polymerase II. J Biol Chem 1980; 255: 11992–11996.
Conaway JW et al. Transcription initiated by RNA polymerase II and purified transcription factors from liver. Cooperative action of transcription factors tau and epsilon in initial complex formation. J Biol Chem 1990; 265: 7552–7558.
Lue NF, Kornberg RD . Accurate initiation at RNA polymerase II promoters in extracts from Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1987; 84: 8839–8843.
Conaway R, Conaway J . General transcription factors for RNA polymerase II. Prog Nucleic Acid Res Mol Biol 1997; 56: 327–346.
Kelleher IRJ et al. A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell 1990; 61: 1209–1215.
Flanagan PM et al. A mediator for activation of RNA polymerase II transcription in vitro. Nature 1991; 350: 436–438.
Kim YJ et al. A multiprotein mediator of transcriptional activation and its interaction with the C-terminal repeat domain of RNA polymerase II. Cell 1994; 77: 599–608.
Kornberg RD . Mediator and the mechanism of transcriptional activation. Trends Biochem Sci 2005; 30: 235–239.
Malik S, Roeder RG . Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem Sci 2005; 30: 256–263.
Moqtaderi Z et al. TBP-associated factors are not generally required for transcriptional activation in yeast. Nature 1996; 383: 188–191.
Walker SS et al. Transcription activation in cells lacking TAFIIs. Nature 1996; 383: 185–188.
Shen W-C, Green MR . Yeast TAFII145 functions as a core promoter selectivity factor, not a general coactivator. Cell 1997; 90: 615–624.
Conaway RC et al. The mammalian Mediator complex and its role in transcriptional regulation. Trends Biochem Sci 2005; 30: 250–255.
Bourbon HM et al. A unified nomenclature for protein subunits of mediator complexes linking transcriptional regulators to RNA polymerase II. Mol Cell 2004; 14: 553–557.
Thompson CM, Young RA . General requirement for RNA polymerase II holoenzymes in vivo. Proc Natl Acad Sci USA 1995; 92: 4587–4590.
Takagi Y, Kornberg RD . Mediator as a general transcription factor. J Biol Chem 2006; 281: 80–89.
Kornberg RD, McConnell HM . Lateral diffusion of phospholipids in a vesicle membrane. Proc Natl Acad Sci USA 1971; 68: 2564–2568.
Uzgiris EE, Kornberg RD . Two-dimensional crystallization technique for imaging macromolecules, with an application to antigen–antibody-complement complexes. Nature 1983; 301: 125–129.
Edwards AM et al. Purification and lipid-layer crystallization of yeast RNA polymerase II. Proc Natl Acad Sci USA 1990; 87: 2122–2126.
Darst SA et al. Two-dimensional and epitaxial crystallization of a mutant form of yeast RNA polymerase II. J Mol Biol 1991; 221: 347–357.
Fu J et al. Yeast RNA polymerase II at 5 Åresolution. Cell 1999; 98: 799–810.
Acknowledgements
The charge for this lecture was to tell the story of the work leading to the Prize. It is also the story of many students and postdoctoral fellows, whose skill, great effort, and willing suspension of disbelief, over a quarter century, transformed a scientific dream into a reality. I could only mention a few by name without interrupting the flow of the story, but the contributions of others were not less important. I pay heartfelt tribute to them, and to all who have shared part of their scientific lives with me (Figure 19), in pursuit not only of the pol II structure, but also of chromatin, transcription biochemistry, and EM methodology. I should add that this is a lecture, not a review, and so is personal rather than referential. The pol II structure is a culmination of research done in many laboratories, and I am deeply indebted to those involved.

Figure 19
Author information
Authors and Affiliations
Corresponding author
Additional information
From Les Prix Nobel. The Nobel Prizes 2006, Editor Karl Grandin, (Nobel Foundation), Stockholm, 2007 Copyright© The Nobel Foundation 2006
Rights and permissions
About this article
Cite this article
Kornberg, R. The molecular basis of eucaryotic transcription. Cell Death Differ 14, 1989–1997 (2007). https://doi.org/10.1038/sj.cdd.4402251
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402251
Keywords
This article is cited by
-
A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin
Cancer & Metabolism (2019)
-
Implication of transcriptional repression in compound C-induced apoptosis in cancer cells
Cell Death & Disease (2013)
-
Identification of valid housekeeping genes for quantitative RT-PCR analysis of cardiosphere-derived cells preconditioned under hypoxia or with prolyl-4-hydroxylase inhibitors
Molecular Biology Reports (2012)
-
MicroRNAs meet cell death
Cell Death & Differentiation (2010)
-
Position of the general transcription factor TFIIF within the RNA polymerase II transcription preinitiation complex
The EMBO Journal (2010)
