
Abstract
Acidic noncaspase proteases-like cathepsins have been introduced as novel mediators of apoptosis. A clear role for these proteases and the acidic endolysosomal compartment in apoptotic signalling is not yet defined. To understand the role and significance of noncaspases in promoting and mediating cell death, it is important to determine whether an intersection of these proteases and the caspase pathway exists. We recently identified the endolysosomal aspartate protease cathepsin D (CTSD) as a target for the proapoptotic lipid ceramide. Here, we show that tumor necrosis factor (TNF)-induced CTSD activation depends on functional acid sphingomyelinase (A-SMase) expression. Ectopic expression of CTSD in CTSD-deficient fibroblasts results in an enhanced TNF-mediated apoptotic response. Intracellular colocalization of CTSD with the proapoptotic bcl-2 protein family member Bid in HeLa cells, and the ability of CTSD to cleave directly Bid in vitro as well as the lack of Bid activation in cathepsin-deficient fibroblasts indicate that Bid represents a direct downstream target of CTSD. Costaining of CTSD and Bid with Rab5 suggests that the endosomal compartments are the common ‘meeting point’. Caspase-9 and -3 activation also was in part dependent on A-SMase and CTSD expression as revealed in the respective deficiency models. Our results link as novel endosomal intermediates the A-SMase and the acid aspartate protease CTSD to the mitochondrial apoptotic TNF pathway.
Similar content being viewed by others


Introduction
Ceramide is a potent proapoptotic second messenger lipid, but the pathway used by ceramide to transmit death-inducing signals is still unknown. Events downstream of ceramide include the induction of caspase-9 and -3 activity,1,2 but ceramide does not interact directly with caspase-2, -3, -8 or -9.3 Thus, this pathway probably requires additional intermediate proteins linking ceramide to caspases.
Ceramide is produced by de novo synthesis or by sphingomyelinases (see, for review, Perry and Hannun4). Acid sphingomyelinase (A-SMase) is part of an apoptosis-inducing signalling pathway, activated by tumor necrosis factor (TNF), and mediated by the p55 TNF receptor (TR55) death domain.5 The inhibition of TNF receptor (TR55) internalization abrogates the TNF-induced activation of A-SMase and apoptosis,6 suggesting an intracellular compartmentation of TNF signalling and ceramide action. Previously, we demonstrated direct targeting of the endolysosomal protease cathepsin D (CTSD) by ceramide derived from A-SMase.3 This links CTSD to the TNF-induced apoptotic pathway.3,7
Specific binding of ceramide results in an autocatalytic activation of CTSD.3 Ceramide induces the maturation of the inactive 52 kDa prepro- to the membrane-bound active 48 kDa pro- and the active soluble 32 kDa CTSD isoform. Furthermore, the substrate directed activity of the 48 and 32 kDa isoforms of CTSD is increased by interaction with ceramide.3
Cathepsins have been implicated in the activation of caspases and apoptosis.8,9,10 CTSD may be part of a proapoptotic protease cascade similar to the caspase cascade, since CTSD has been found to activate cathepsin B (CTSB), an acidic cystein protease.11 CTSB in turn mediates apoptosis induced by various stimuli,10,12 can cleave caspase-11 and -113,14 and is involved in TNF-induced apoptosis.15,16 CTSB knockout mice were found to be resistant to TNF-mediated hepatocyte apoptosis and liver injury, suggesting potential implications for therapeutic applications.17 Thus, the cathepsins, like caspases, may be activated in a cascade-like manner and may work as initiator proteases in apoptosis signalling.
Evidence for the apoptotic potential of CTSD came from overexpression studies and knockout mice. Overexpression of CTSD induces or sensitizes HeLa and PC12 cells to apoptosis upon serum starvation18 and oxidative stress.19 Fibroblasts derived from CTSD-deficient mice were resistant to adriamycin- and etoposide-induced apoptosis.20 Furthermore, microinjection of CTSD-induced caspase-dependent apoptosis in human fibroblasts.21 In addition to the caspase family of proteases, initiating and executing apoptosis, lysosomal noncaspase proteases like CTSD have been recognized recently as a new group of players in the ‘death scene’.22,23
A possible link of cathepsins to the mitochondrial pathway was suggested by observations that Bcl-2 protein family members, like Bid for example, are cleaved by lysosomal extracts at a specific cleavage site differing from the one found for caspase-8.24 Since caspases, Bid, and all other potential targets for cleavage by cathepsin and relevant for apoptosis, are localized in the cytoplasm, the release of cathepsins from the endolysosomes to the cytoplasm is an important prerequisite for the participation of cathepsins in apoptotic signalling. Oxidative stress induced by reactive oxygen species-generating agents as well as oxidized LDL (oxLDL) and photo-oxidative disruption of lysosomes induce the relocation of cathepsin from endolysosomes to the cytoplasm. The induction of cathepsin release results in apoptosis.13,25,26,27 Notably, the oxLDL-induced apoptosis has been shown to depend on A-SMase expression and ceramide production.28
The functional link between TNF, A-SMase-generated ceramide and CTSD activation in endolysosomal compartments, and the connection of CTSD with further downstream apoptotic signalling remains an unsolved issue and is subject of the present study. In particular, we were interested in investigating a possible link to the ‘classical’ caspase cascade and in identifying cytosolic CTSD downstream substrates.
Results
TNF-induced apoptosis is linked to CTSD expression
Involvement of CTSD in apoptotic cell death was suggested by various recent reports.10,11,12,13,14,15,16 To confirm a role of CTSD in TNF-mediated cell death, we compared the effects of TNF on retrovirally tranduced fibroblasts derived from CTSD-deficient mice (CD−/−) carrying empty vectors with TNF effects on CTSD-deficient fibroblasts after ectopic expression of the CTSD gene (CD−/−CatD). As shown in Figure 1a, in the presence of CHX TNF treatment of CD−/− cells for 4 h resulted in 4.1% apoptosis, whereas the same treatment of CD−/−CatD fibroblasts lead to 14% apoptotic cells as estimated by Annexin V-positive staining (Figure 1b). After 12 h treatment, 12.7% of CD−/− cells (Figure 1c) and 45% of CD−/−Cat D cells (Figure 1d) were killed by TNF/CHX as indicated by positive propidium iodide staining. This demonstrates that the expression of CTSD sensitizes TNF-induced apoptotic cell death. Both CD−/− cells and CD−/−Cat D cells did not differ significantly in endogenous ceramide levels (data not shown), ruling out the possibility of an unspecific, sensitizing effect from ceramide overload.

TNF-induced cell death depends on CTSD expression. CTSD-deficient fibroblasts carrying empty vector (a, c) or CTSD−/− fibroblasts reconstituted with CTSD cDNA (b, d) were treated with 3.56 μM CHX alone or with CHX and 100 ng/ml TNF for 4 h (a, b) or 12 h (c, d) and analyzed for apoptosis using the Annexin V–propidium iodide assay, measured by FACS. A representative result of three independent experiments is shown
TNF-induced activation of CTSD depends on the expression of a functional A-SMase
For its role as a lipid second messenger in apoptosis signalling, ceramide needs to interact directly with specific enzymes that in turn transmit its apoptotic signals to further downstream targets. The endolysosomal aspartic protease CTSD was recognized as a ceramide-binding protein.3 Ceramide induces maturation and activation of CTSD both in vitro and in whole cells, and CTSD maturation depends on A-SMase function.
To evaluate the connection of CTSD activation with TNF-induced A-SMase activity and ceramide production, we investigated CTSD activity in A-SMase-deficient lymphoblasts derived from patients with Niemann Pick disease (MS1418 cells) and in MS1418 cells stable transfected with the gene for A-SMase (MS1418/ASM cells). As shown in Figure 2, A-SMase activation (A) as well as ceramide production (B) were not detected in MS1418 parental cells following TNF stimulation (open symbols), but in MS1418/ASM cells retransfected with A-SMase (closed symbols ) (Figure 2b).

TNF-induced activation of CTSD depends on A-SMase expression. A-SMase activation and ceramide production was determined in A-SMase-deficient MS1418 NPD lymphoblasts carrying empty vector (○) or MS1418 cells reconstituted with A-SMase cDNA (MS1418/ASM) (•). Cells were incubated with TNF for indicated times and the activity of A-SMase was evaluated in an in vitro mixed micellar assay using cholin-methyl-1-[14C]sphingomyelin as substrate (a) or the amount of ceramide was measured by HPTLC and charring densitometry (b). Results of three independent experiments performed in triplicates (+S.E.M.) are shown. TNF-induced CTSD activity was measured after stimulation of A-SMase-deficient MS1418 NPD-lymphoblasts (c) and MS1418 cells reconstituted with A-SMase cDNA (d) were stimulated for 1 h with 3.56 μM CHX/100 ng/ml TNF. CTSD activity of the lysates was determined using PTH as CTSD-specific substrate. The degradation of PTH was analyzed from 0–120 min by Western blotting using a monoclonal anti-PTH antibody and quantitated by 2D laser-scanning densitometry (Molecular Dynamics Personal Densitometer). Results of three to five independent experiments (+S.E.M.) are shown. The insets show the protein levels of the CTSD isoforms in both cell lines by Western blotting using anti-CTSD antibodies (Calbiochem, Germany)
In the same cell system, CTSD activity was evaluated using an in vitro assay based on hydrolysis of the CTSD-specific substrate parathyroid hormone (PTH) and its detection by Western blotting. This assay was described previously3 and is highly specific for CTSD since total lysates from cells derived from CTSD knockout mice were unable to cleave PTH.3 As shown in Figure 2c, CTSD is not activated in A-SMase-deficient lymphoblasts after 1 h of TNF/CHX treatment. In contrast, A-SMase retransfection in MS1418/ASM cells resulted in a significant activity of CTSD after TNF treatment (Figure 2d). Notably, the expression of A-SMase resulted in enhanced protein levels of the enzymatically active 48 kDa CTSD isoform (Figure 2d). The effect of A-SMase overexpression on CTSD as a target protein of ceramide is also reflected by an enhanced basal CTSD activity in unstimulated MS1418/ASM cells (40%, Figure 2d) compared to A-SMase-deficient MS1418 parental cells (30%, Figure 2c) This confirms our previous observations of the effect of ceramide on CTSD processing and activation.3
Together, these data provide evidence for the involvement of A-SMase-generated ceramide in the activation of CTSD and a sequencial role of both enzyme systems in TNF signal transduction.
TNF- and ceramide-induced caspase-9 and -3 activities are regulated by CTSD
To evaluate the functional role of ceramide-mediated CTSD activity in TNF-induced apoptotic signalling, we investigated the potential of TNF and ceramide to activate caspase-9. A possible involvement of CTSD in TNF and ceramide stimulation of caspases-9 was assessed in these experiments using the aspartate–protease inhibitor pepstatin A. Cells were treated with TNF/CHX for 4 h or ceramide for 8 h and simultaneously inhibited with pepstatin A. TNF induced cleavage of procaspase-9 generating the 35 kDa caspase-9 fragment, as shown in Figure 3a. A significant inhibition of TNF-dependent procaspase-9 cleavage was observed using pepstatin A, indicating the involvement of CTSD in caspase-9 activation. To confirm that the processing of procaspase-9 is related to caspase-9 activation, we measured the enzymatic activity of caspase-9 using a caspase-9-specific fluorescent substrate (Ac-LEHD-AFC) and a fluoretic microplate ELISA-Reader. As shown in Figure 3b, TNF/CHX treatment of HeLa cells resulted in an enhanced capase-9 activity of approximately 222% (mean value) and pepstatin A pretreatment reduced TNF-induced activity to approximately 162% (mean value). This 27% inhibition by pepstatin A was reproduced in three independent experiments with a standard deviation of 1.9%, demonstrating the significance of the effect. Notably, TNF effects on caspase-9 activity were reproduced by treating HeLa cells with ceramide, the lipid activator of CTSD in TNF signalling. The inhibition of CTSD by pepstatin A again resulted in partial, but significant inhibition of ceramide-induced caspase-9 activation (Figure 3c).

Effects of pepstatin A on TNF and ceramide stimulation on caspase-9 activities. HeLa cells were left untreated or were treated with CHX (3.56 μM) and TNF (100 ng/ml) for 4 h or ceramide (100 μM) for 8 h in the absence or presence of the CTSD inhibitor pepstatin A (PepA) (1.5 μM). Equal amounts of cellular protein of the homogenized cells were analyzed for caspase-9 protein and actin as loading control by Western blotting (a) using anticaspase-9 antibodies (MBL Ltd, Nagoya, Japan) and anti-actin antibodies (Santa Cruz). The data shown are representative of three independent experiments. Lysates were incubated in parallel with fluorogenic caspase-9 (Ac-LEHD-AMC) substrate to measure caspase-9 enzymatic activity (b). The increasing fluorescence was measured by an ELISA-Reader for 4 h and changes in relative AMC fluorescence were calculated as % of CHX (for TNF) or solvent (for ceramide) controls. Data of three to four independent experiments (+S.E.M.) are shown
Together, these results indicate the at least partial involvement of CTSD in the regulation of TNF- and ceramide-induced caspase-9 activation.
TNF-induction of caspase-9 and -3 activity depends on functional CTSD and A-SMase expression
To further validate our data obtained by pharmacological inhibitor studies in an independent system, we investigated the dependency of TNF-induced caspase-9 activity on CTSD expression employing fibroblasts derived from wild-type mice (CD+/+), CTSD-deficient mice (CD−/−) and CD−/− fibroblasts stably transfected with the pro-CTSD cDNA (CD−/−CatD) or carrying empty vectors. TNF/CHX-induced caspase-9 activity was significantly reduced in CD−/− fibroblasts as compared to CD+/+ fibroblasts (Figure 4, upper panel). Caspase-9 activation by TNF/CHX was restored after transfection of CTSD cDNA into CTSD-deficient cells. These findings confirmed our results obtained with HeLa cells and pharmacological inhibition of CTSD using pepstatin A (see Figure 3).

TNF induction of caspase-9 and -3 activity depends on functional CTSD. Vector-transfected fibroblasts from wild-type mice, from CTSD-deficient mice or CTSD−/− fibroblasts reconstituted with CTSD cDNA were left untreated or were treated with CHX (3.56 μM) and TNF (100 ng/ml) for 4 h. Equal amounts of cellular protein of the homogenized cells were incubated with the fluorogenic caspase-9 (Ac-LEHD-AMC) and caspase-3 (Ac-DEVD-AMC) substrate to measure caspase-9 (a) and caspase-3 (b) enzymatic activity. The increasing fluorescence was measured by an ELISA-Reader for 4 h and changes in relative AMC fluorescence were calculated as % of CHX controls. Data of three to four independent experiments (+S.E.M.) are shown
Since caspase-3 represents the classical downstream execution enzyme of caspase-9, we investigated the possible modulation of caspase-3 activation by CTSD. Using the same CTSD-deficient and reconstituted cell system described above, we found a strongly reduced caspase-3 response after TNF/CHX treatment in CD−/− fibroblasts (Figure 4, lower panel), which was restored in CTSD-reconstituted CD−/−CatD fibroblasts. Together, these data suggest partial involvement of CTSD in TNF-induced activation of both caspase-9 and -3.
Subsequently, we investigated the functional role of A-SMase with respect to caspase-9 and -3 activation in TNF signalling. As shown in Figure 5, TNF/CHX-induced caspase-9 as well as caspase-3 activation were significantly lower in A-SMase-deficient MS1418 lymphoblasts compared to MS1418 cells reconstituted with A-SMase cDNA (MS1418/ASM). The activation of caspase-8 by TNF/CHX was slightly, but not significantly, reduced in MS1418 cells. The inhibition of caspase-8 by IETD-fmk resulted in a complete block of A-SMase activation by TNF (supplementary data 1, not shown), thus caspase-8 seems to be located upstream of the A-SMase/CTSD signalling cascade. The slightly reduced caspase-8 activation observed in MS1418 cells may thus reflect a negative feedback effect of reduced caspase-9 and -3 activity.

TNF induction of caspase-9 activity depends on functional A-SMase. A-SMase-deficient MS1418 NPD lymphoblasts or MS1418 cells reconstituted with A-SMase cDNA (MS1418/ASM) were incubated with TNF/CHX for 4 h. Equal amounts of cellular protein from each cell line was incubated for 3 h with caspase-9 (Ac-LEHD-AMC) substrate, caspase-3 (Ac-DEVD-AMC) substrate and caspase-8 (Ac-IETD-AMC) substrate. The data of three independent experiments (+S.E.M.) are shown
Since the metabolic defect in MS1418 cells might result in defective TNF receptor clustering, leading to defective TNF signalling, we determined the distribution of TNF receptors in both MS1418 and MS1418/ASM cells. As shown in Figure 6, no significant differences in TNF receptor clustering were observed. This suggests that a defective A-SMase does not alter the initial events in TNF receptor signal transduction.

Ectopic expression of A-SMase in MS1418 cells does not influence TNF receptor clustering. MS1418 cells (a, b) or MS1418 cells reconstituted with A-SMase cDNA (MS1418/ASM), (c, d) were incubated with biotin–TNF and streptavidin–FITC at 4oC for 1 h and TNF receptor distribution analyzed by confocal laser-scanning microscopy before (a, c) or after a temperature shift to 37°C for 5 min (b, d)
In concert, these observations indicate the involvement of A-SMase (as endogenous ceramide-generating enzyme) as well as CTSD (as ceramide-interacting protein) in the TNF signal transduction pathway regulating caspase-9 and -3 activation downstream of caspase-8.
CTSD is involved in ceramide-induced cytochrome c release
To further assess the involvement of mitochondria in A-SMase/CTSD-dependent downstream activation of caspase-9 and -3, we investigated the ability of ceramide to induce CTSD-dependent cytochrome c release. As shown by Western blotting in Figure 7, ceramide induced a significant increase of cytosolic cytochrome c in HeLa cells, indicating release from mitochondrial stores. Pretreatment of HeLa cells with pepstatin A reduced the ceramide-induced cytochrome c release, suggesting that CTSD is involved in the activation pathway.

Ceramide-induced cytochrome c release is inhibited by pepstatin A. Western blot analysis was performed from cytosolic proteins prepared by a cytochrome c release assay. HeLa cells were left unstimulated or were stimulated with 100 μM ceramide alone or in combination with the CTSD inhibitor pepstatin A for 1 h. Cytosolic proteins were washed out of permeabilized cells by a digitonin-containing buffer, cytochrome c levels were estimated by immunoblotting using mouse anti-cytochrome c antibodies (Pharmingen, Germany) and anti-actin antibodies as loading control. The figure shows one representative Western blot out of four experiment giving similar results
Bid is a target for CTSD
Release of cytochrome c induced by ceramide indicates the involvement of CTSD in the mitochondrial apoptotic pathway. Since the proapoptotic protein Bid of the Bcl-2 protein family plays a significant role in this pathway, we evaluated it as a possible CTSD target. Bid protein was prepared by immunoprecipitation from pancreatic adenocarcinoma cells PancTuI, expressing high levels of Bid protein. Incubation of purified Bid with exogenous mature CTSD from the human liver at pH 4.2 resulted in rapid cleavage of the 23 kDa Bid protein paralleled by the generation of a truncated Bid (tBid) fragment with an apparent molecular mass of 15 kDa (Figure 8a). Bid cleavage and tBid generation were blocked by pepstatin A and also occurred at higher pH values up to 6.2 but did not take place at pH 7.0. These in vitro findings suggest that Bid may be a potential substrate for CTSD even at moderately acidic conditions.

TNF-induced Bid cleavage depends on CTSD activity. (a) Bid was purified from lysates of PancTuI cells by immunoprecipitation using goat anti-Bid antibody C-20 (Santa Cruz) and treated with purified human liver CTSD at various pH as indicated in vitro. Bid levels were analyzed by SDS-PAGE separation and immunoblotting using rabbit anti-Bid antibodies (R&D Systems). The results are representative of three independent experiments giving similar results. (b) Fibroblasts from wild-type mice (CD+/+), CTSD-deficient fibroblasts (CD−/−) or CD−/− fibroblasts reconstituted with CTSD cDNA (CD−/−CatD) were left untreated or were incubated with TNF (100 ng/ml) for 1 h. Cell lysates were analyzed by immunoblotting using polyclonal anti-Bid antibody (R&D Systems) to detect murine 23 kDa Bid and polyclonal goat anti-Bid antibody (Santa Cruz, USA) to detect murine 15 kDa tBid. Equal protein loading was confirmed by anti-actin antibody staining
TNF-induced activation of Bid depends on CTSD expression
We subsequently investigated if Bid is activated by TNF in a CTSD-dependent manner in whole cells. When compared to unstimulated cells, TNF-stimulated fibroblasts derived from wild-type mice (CD+/+) showed a decrease of the 23 kDa Bid protein and a concomitant increase in the amount of 15 kDa tBid, the active cleavage product of 23 kDa Bid (Figure 8b). TNF-induced activation of Bid was not observed in fibroblasts derived from CTSD-deficient mice (CD−/−), but in CD−/− cells reconstituted with CTSD cDNA (CD−/−CatD), a strong activation of Bid was again observed. These data clearly indicate the requirement of functional CTSD for TNF-mediated activation of Bid. Defective Bid cleavage in response to TNF was also observed in A-SMase-deficient MS1418 cells (supplementary data 2, not shown).
Together with our previous data, these observations link TNF- and ceramide-induced activation of CTSD to caspase-9 and -3 activation via the mitochondrial apoptotic pathway and involve Bid as a CTSD target and signalling intermediate.
Bid and CTSD colocalize in endosomes
The enzymatically inactive 52 prepro form of CTSD is located in the endoplamic reticulum and trans-Golgi compartment. The further processed the 48 kDa active CTSD enzymes reside in endosomal compartment and is converted to the 32 kDa form in lysosomal vesicles.29,30 The regulation of Bid by CTSD D requires a location within the cell where interaction can occur. By using indirect confocal immunofluorescence analysis, we searched for such ‘meeting points’ of CTSD and Bid. Unexpectedly, we found that Bid is localized at vesicular structures (Figure 9a). Costaining for CTSD D revealed that Bid colocalizes with the endolysosomal protease CTSD (Figure 9b–d). Anti-Bid antibodies as well as anti-CTSD antibodies bound to identical subcellular structures partly. While CTSD is assumed to be located within the vesicles, Bid most likely is located at the outer surface of these structures. Notably, Bid does not colocalize with lysotracker-positive compartments (Figure 10a), suggesting that the organelle containing jointly CTSD and Bid is distinct from lysosomes. As shown in Figure 10c, CTSD is also not located exclusively in lysosomes (showing positive lysotracker staining). Costaining of Bid and CTSD with the endosomal protein Rab5 (Figure 10b and d) suggest that these common compartments are most likely endosomes.

Bid and CTSD colocalize at vesicular structures in HeLa cells. Confocal indirect immunofluorescence images of Bid and CTSD. Methanol-fixed HeLa cells were treated with a rabbit polyclonal anti-CTSD antibodies (Calbiochem) and secondary rhodamine labelled goat-anti-rabbit antibodies in combination with goat polyclonal anti-Bid-antibodies (Santa Cruz, USA) and secondary FITC labelled donkey-anti-goat antibodies. Lysotracker red DND-99 was incubated for 1 h and cells were fixed with paraformaldehyde and stained for Bid. (a) Distribution of vesicular Bid staining. (b–d) Colocalization of Bid with CTSD, marked by arrows. (b) Green CTSD, (c) in red staining of Bid within the same cell, (d) matched images showing colocalization of CTSD and Bid in the same vesicles in yellow

Bid and CTSD colocalize with late endosomal marker Rab 5. HeLa-cells were incubated with Lysotracker red DND-99 for 1 h, fixed with paraformaldehyde and stained for Bid (a) or CTSD (c) using goat polyclonal anti-Bid or goat polyclonal anti-CTSD antibodies (Santa Cruz) and secondary rhodamine labelled donkey-anti-goat antibodies. In (a) Bid and lysotracker do not colocalize. Arrows in (c) indicate CTSD positive (red)/lysotracker negative vesicles. (b, d) HeLa cells were fixed with paraformaldehyde and stained with rabbit polyclonal antibodies for Rab 5 and Bid (b) or Rab 5 and CTSD (d). Arrows indicate colocalization of Bid and CTSD with Rab 5 (yellow)
Our findings demonstrate the existence of a shared endosomal compartment for CTSD and Bid, with conditions for CTSD-mediated cleavage and activation of Bid.
TNF and ceramide induce translocation of CTSD from membrane compartments to the cytosol
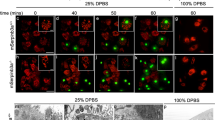
An important prerequisite for the activation of Bid by CTSD is the relocation of CTSD from the lumen of the endosomal compartment to the cytosolic face of the endosomes. We therefore investigated the influence of ceramide and TNF on the intracellular distribution of CTSD. The specificity of the polyclonal CTSD antibody was confirmed by indirect immunofluorescence analysis on methanol-fixed fibroblasts derived from wild-type (CD+/+), CTSD-deficient (CD−/−) mice and CD−/− cells overexpressing CTSD cDNA (CD−/−CatD) (supplementary data 3, not shown). By indirect immunofluorescence imaging of HeLa cells, we then showed the relocation of CTSD in response to ceramide stimulation: in unstimulated cells, CTSD is located in distinct vesicular structures. Stimulation by ceramide results in complete disappearance of the stained vesicular structures accompanied by increased diffuse CTSD staining within the cytosol (Figure 11). These findings were confirmed by analyzing the translocation of CTSD protein after separation of membrane and cytosolic fractions of TNF-treated HeLa cells. In the cytosolic fraction, we observed an increase of the active 32 kDa CTSD isoform (Figure 12a). This effect of TNF was again mimicked by the A-SMase-generated mediator ceramide, since in an identical approach ceramide stimulation of HeLa cells also induced the relocation of 32 kDa CTSD from membranes to cytosol (Figure 12b). Notably, translocation of the endolysosomal protease CTSB did not occur after TNF or ceramide treatment in these cells (Figure 12c), indicating that the effects observed on CTSD were specific and not caused by nonselective disruption of endolysosomal membranes.

Ceramide induces translocation of vesicular CTSD D to the cytosol. Indirect immunofluorescence staining of CTSD. HeLa cell were left untreated (a) or were treated with ceramide (100 μM) for 12 h (b). CTSD was visualized in methanol-fixed HeLa cells by polyclonal anti-CTSD antibodies (Santa Cruz) and FITC-labelled secondary antibodies

TNF and ceramide induce translocation of CTSD from the membrane fraction to the cytosolic fraction. HeLa cells were left unstimulated or were stimulated by TNF for 8 h (a, c) and by ceramide for 1, 2, 8 (b) and 12 h (c). After stimulation cells were permeabilized by digitonin and cytosolic and membrane proteins prepared as described in the Materials and methods section. The cellular fractions were analyzed for CTSD and CTSB levels by Western blotting using polyclonal anti-CTSD and anti-CTSB (Santa Cruz) antibodies, respectively
Discussion
This study identifies CTSD as the missing link between TNF-activated ceramide production by A-SMase and the induction of apoptotic signalling involving the execution caspase-9 and -3. We show that both TNF-induced and ceramide-mediated activation of CTSD results in the induction of mitochondrial apoptosis signalling by cleavage of Bid. TNF-induced activation of CTSD depends on functional A-SMase. The activity of caspase-9, the effector caspase in mitochondrial apoptosis signalling, induced by TNF or exogenous ceramide can be blocked by pepstatin A, an inhibitor specific for aspartate proteases including CTSD. In addition, a strong reduction in TNF stimulation of caspase-9 and -3 activity in CTSD-deficient fibroblasts derived from CTSD knockout mice or in A-SMase-deficient lymphocytes was observed. TNF activation of caspase-9 and -3 could be restored in both cell lines by reconstitution of CTSD and A-SMase, respectively. The bcl-2 protein family member Bid was identified as a direct substrate for CTSD in vitro. TNF-mediated cleavage of Bid in whole cells depends on the expression of CTSD. Since TNF-induced A-SMase activation is sensitive to the inhibition of caspase-8 with IETD, our findings suggest a sequential TNF signalling pathway involving caspase-8-dependent induction of endosomal A-SMase followed by the activation of CTSD in the same intracellular compartment. This pathway is subsequently linked via cleavage of Bid to the mitochondrial apoptosis pathway as summarized in Figure 13.

Role of CTSD in TNF apoptosis signalling. TNF binding to the p55 TNF receptor results in receptor internalization and caspase-8-dependent activation of A-SMase in endolysosomal compartments. Ceramide generated by A-SMase in turn activates CTSD and mediates CTSD translocation from the endolysosomes to the cytosol. CTSD activates tBid by proteolysis of Bid, resulting in cytochrome c release from mitochondria and activation of caspase-9 and -3, leading to apoptotic cell death. Caspase-8 also directly activates caspase-9 and -3 independently of the endosomal compartment (dashed line)
Numerous reports documented that the intracellular second messenger lipid ceramide transduces the effects of various stimuli including cytokines like TNF, interferon-γ, interleukin-1, nerve growth factor or CD28 and CD95 (Fas/APO-1), and other stimuli and agents like ionizing radiation, serum deprivation, glucocorticoids and anticancer drugs (reviewed in Rijnboutt et al.30 and Hannun and Luberto31). Among the broad range of cellular responses including proliferation, differentiation and cell-cycle arrest, the most prominent role of ceramide is the induction of apoptosis in various cell types (reviewed in Kolesnick and Krönke32 and Kolesnick and Hannun33). The identity of the enzyme generating ceramide and thereby responsible for the proapoptotic activity of the lipid is not yet clearly defined and obviously depends on the stimuli and cell type investigated. A role for A-SMase in radiation- and CD95-mediated cell death was observed mainly in endothelial cells, hepatocytes, oocytes and lymphoblasts,34,35,36,37,38,39,40 and an involvement of A-SMase in TNF-induced apoptosis was recently found in mouse embryonic fibroblasts.40 In hepatocytes, A-SMase contributes to TNF-induced apoptosis by inducing mitochondrial targeting of ganglioside GD3.41
Searching for ceramide-binding proteins transmitting ceramide effects further downstream, we excluded direct binding of ceramide to caspases3 as shown in our previous report. However, we demonstrated specifical binding of the lysosomal protease CTSD to exogenous ceramide and the activation of CTSD in whole cells by A-SMase-derived ceramide. Elsewhere, CTSD was implicated in TNF-induced apoptosis.16 Here, we confirm the role of CTSD in TNF-mediated apoptosis using a different cell system and we show that TNF-induced CTSD activity depends on the expression of functional A-SMase.
TNF induces apoptotic signalling by triggering receptor clustering, followed by rapid receptor internalization,15 and recruitment of the TNF receptor-associated proteins TRADD and FADD,42,43 leading to the activation of the initiator caspase-8 as well A-SMase.14 Our data link CTSD to TNF-induced ceramide production by A-SMase and to the type II apoptosis signalling pathway, as shown by TNF- and ceramide-induced activation of caspase-9 and -3 partly depending on CTSD function. The residual CTSD-independent activation of caspase-9 and -3 observed after pepstatin A treatment as well as in CD−/− cells most likely results from direct caspase-8 activity bypassing the endosomal compartment as outlined in Figure 13.
It should be noted, however, that targeting CTSD is obviously not the only apoptotic pathway activated by ceramide because CD−/− cells were not resistant to ceramide-induced apoptosis (data not shown).
To participate in apoptotic signalling – occurring mainly outside of cellular vesicles in the cytoplasm – the active CTSD isoform has to translocate from intravesicular structures to the cytoplasm. Numerous studies outline the possibility of translocation of lysosomal proteases and their participation in apoptotic signalling. Oxidative stress induced by the redox cycling quinone naphtazarin (5,8-dihydroxy-1,4-naphthoquinone) leads to the relocation of CTSD with ensuing apoptosis in rat cardiomyocytes26 and to the relocation of cathepsins D, B and L.18 Furthermore, there is evidence for a lysosomal pathway of apoptosis induced by the synthetic retinoid CD437, triggering lysosomal leakage and CTSD relocation.44 Recently, TNF-mediated translocation of CTSD in vascular endothelial cells was reported.45
Our present report describes for the first time a physiological mechanism of CTSD translocation involving an endogenous enzyme generating ceramide. TNF activates sphingomyelinase and increases the amount of ceramide in endosomal membranes. Indirect immunofluorescence investigations and CTSD release assays demonstrate that CTSD is translocated from endosomal compartments to the cytosol by exogenous ceramide, too, mimicking the effect observed using TNF. The ability of TNF and ceramide to trigger translocation appears to be specific for CTSD, since CTSB analyzed under identical experimental conditions did not leave the endolysosomal compartment. Thus, the observed effects concerning CTSD are not related to a nonspecific physical disruption of the membranes. A recent report by Kargedal et al.46 describes the induction of lysosomal permeabilization by sphingosine, a biologically active metabolite of ceramide. This precedes caspase activation and changes in mitochondrial membrane potential.
The release of CTSD seems to be part of the TNF-induced signalling pathway, leading to Bid cleavage and caspase-9 activation via induction of cytochrome c release. Ceramide-induced cytochrome c release in HeLa cells is regulated by CTSD, as shown by its partial inhibition by pepstatin A. An important prerequisite for the activation of Bid by CTSD is the relocation of CTSD from the lumen of the endosomal compartment to the cytosol. After release of CTSD from organelles, its interaction with cytosolic components of the type II apoptosis signalling pathway (like Bid) requires an intracellular locus shared by the interacting partners. By confocal immunofluorescence analysis, we indeed showed that Bid colocalizes with CTSD-positive vesicles, and both CTSD and Bid are located in (rab 5-positive, lysotracker-negative) endosomes. Our observation implicates that Bid is located at the subcellular site of CTSD activation and release. We identified this subcellular site as endosomes containing mainly the membrane-bound 48 kDa CTSD form. While CTSD resides within endosomal compartments, Bid protein is most likely located at the outer surface of these vesicles.
In an independent approach, we detected costaining of internalized biotin–TNF with CTSD in receptosomes and found that magnetically isolated TNF receptosomes contain both CTSD and Bid protein. Bid is released from these vesicles upon prolonged TNF stimulation (S. Schütze et al., manuscript in preparation).
After translocation of CTSD and mild acidification of the cytosol, Bid can be cleaved by CTSD. Effective Bid cleavage by CTSD occurred already at pH 6.2. Acidification of the cytoplasm is a well-documented phenomenon in the progression of apoptosis, preceding morphological changes47 and required for caspase activation in the mitochondrial pathway of apoptosis.48,49 Thus, early apoptotic changes in the cell provide the required environmental conditions for CTSD activity in the cytosol.
Endolysosomal proteases can be understood, like caspases, as a group of proteases that are activated in a cascade-like manner. These proteases may execute apoptosis either independent of caspases or may individually participate in different apoptotic or cell death signalling cascades by connecting the endosomal compartment to the classical apoptosis signalling pathways. Once released to the cytoplasm, endolysosomal proteases can interact with proteins involved in type I and the type II apoptosis signalling.
In summary, our present report describes a novel apoptotic signalling cascade, involving and connecting two distinct intracellular compartments. This pathway is induced by TNF and mediated by ceramide produced by endosomal A-SMase. Ceramide activates and mediates translocation of the aspartate protease CTSD from the endosomal compartment into the cytosol. Cleavage of Bid by CTSD subsequently links this endosomal signalling pathway to type II apoptosis signalling via the induction of cytochrome c release from mitochondria and activation of effector caspase-9 and -3. The relevance and functional implications of this novel pathway in comparison to the direct caspase-8-dependent (type I and type II) pathway is currently under investigation.
Materials and Methods
Cell culture
HeLa cells were obtained from the ATCC and maintained in CLICK's RPMI culture medium (Biochrome) supplemented with 10% fetal calf serum, 10 mM glutamine, 0.1 mM β-mercaptoethanol and 50 μg/ml each of streptomycin and penicillin. EBV-transformed human lymphoblasts from patients with Niemann-Pick disease (clone MS1418) were kindly provided by D Green (La Jolla, USA).34 Fibroblasts derived from CTSD-deficient mice (C57BL/6 CTSD−/−) were kindly provided by Dr Saftig (Kiel, Germany).50 The lymphoblasts and the fibroblast were maintained in DMEM (GIBCO) culture medium supplemented as described for HeLa cells.
Retroviral transfections with A-SMase and CTSD cDNA
cDNAs of human A-SMase and human prepro-CTSD were subcloned into the retroviral vector pLSXN or pBabe puro, respectively. Transient transfection of the retroviral producer line FLYA13 was performed by calcium phosphate precipitation with 10 μg pLXSN-ASMase or pLXSN-CTSD and 5 μg pCMV/VSV-G expressing the G protein of vesicular stomatitis virus to produce pseudotyped amphotropic retroviral particles. A total of 1 × 106 cells were plated on 100 mm dishes in DMEM without HEPES the night before transfection. Medium was changed 12 h post-transfection. Transduction of MS1418 Niemann-Pick lymphoblasts or CTSD−/− fibroblasts was performed by incubating 5 × 104 cells with recombinant viral supernatant harvested 48 h post-transfection and containing 8 μg/ml polybrene at 37°C overnight. Transduced cells were selected using G418 for 10–14 days (MS1418 lymphoblasts) or puromycin for 4–6 days (CD−/− fibroblasts) and pooled cell populations were used for further experiments. The parental cell line MS1418 and CD+/+ fibroblasts were transfected with empty vectors as controls.
Cell treatments
HeLa cells, fibroblasts from C57BL/6 wild-type mice (CD+/+), C57BL/6 CTSD-deficient mice (CD−/−) and CD−/− fibroblasts retransfected with CTSD cDNA (CD−/−CatD), MS1418-lymphoblasts and MS1418-lyphoblasts transfected with A-SMase cDNA (MS1418/ASM) were treated with 3.56 μM cycloheximide (CHX), 100 ng/ml TNF or 100 μM C6-ceramide (Biomol, Germany) for times indicated in the presence of 10% serum. TNF was a kind gift of the Knoll AG, Germany. Cells were preincubated with pepstatin A (1.5 μM) (Boehringer Mannheim, Germany) 12–16 h before TNF or ceramide treatment.
Apoptosis assay
Cell death was monitored by the Annexin V–propidium iodide assay as follows: 0.5 × 106 cells were seeded in six-well plates, incubated over night and treated with 100 ng/ml TNF and/or 3.56 μM CHX for 4 and 12 h, respectively. The cells were then harvested and stained with Annexin V-Fluos (Roche) and 10 ng/ml propidium iodide according to the manufacturer's instructions. Stained cells were analyzed using the FACS-Calibur (BD) and the Cell-Quest 3.3 software.
CTSD activity assay
For estimating the activity of cellular CTSD, cells were lysed in homogenization buffer (40 mM HEPES, 150 mM KCl, 0.05% NP-40, pH 7.4) and 12 μg of cellular protein was incubated for times indicated with 50 ng PTH (SIGMA-ALDRICH, Germany) at 37°C in a volume of 20 μl acidic buffer (100 mM sodium acetate, 100 mM potassium chloride, pH 4.2) as described previously.3 The inhibitor pepstatin A (1.5 μM) was added to control samples. Proteins were separated by SDS-PAGE and PTH was detected by immunoblotting using monoclonal anti-PTH antibodies (DPC, Biermann, Germany). Quantification of PTH digestion was performed by 2D laser-scanning densitometry (Personal Densitometer, Molecular Dynamics) and CTSD activity calculated from the amount of PTH digested/h.
Western blotting
Bid, PTH, caspase-9, cytochrome c and CTSD were detected on 15% SDS-PAGE, using monoclonal anti-PTH (1–34) clone 1F8 (#BM154, DPC Biermann, Germany) and polyclonal anti-human CTSD (#219361, Calbiochem, Germany), anti-CTSB (#SC-6490, Santa Cruz, USA) anti-human and mouse caspase-9 (#M054/3, MBL Ltd, Nagoya, Japan) and anti-Bid (C-20, #SC-6486, Santa Cruz, USA and #AF846, R&D Systems), anti-cytochrome c (#556433, BD Pharmingen, Germany), anti-TRADD (H-278, #SC-7868 Santa Cruz, USA), anti-FADD (12E7, #804-319-C100, Alexis biochemicals, USA) anti-caspase-8 (p20, H-134, #SC-7890 p20, H-134, sc-7890) antibodies and a secondary horseradish-peroxidase conjugate (Santa Cruz, USA). Blots were developed using the ECL detection reagent (Amersham-Pharmacia Corp.) and documented by 2D laser-scanning (Personal Densitometer, Molecular Dynamics).
Fluorescence labelling of TNF receptors
MS1418 and MS1418/ASM cells were incubated with biotinylated TNF (RD Systems GMBH, Wiesbaden, Germany) for 60 min at 4°C. Avidin–FITC reagent was added and cells were incubated for another 30 min at 4°C in the dark. Cells were either kept on ice for detection of surface staining or the temperature was shifted to 37°C for 5 min to allow receptor internalization. For analysis by confocal laser-scanning microscopy (Zeiss LSM 510), cells were fixed with paraformaldehyde (4% in PBS) for 20 min.
Caspase activity assays
To measure caspase activity, fluorogenic substrates specific for caspase-3 (Biomol; Ac-DEVD-AMC), caspase-8 (Biomol; Ac-IETD-AMC) and caspase-9 (Biomol; Ac-LEHD-AMC) were used. In all, 40 μg (MS1418, MS1418-ASM cells) or 50 μg (CD+/+, CD−/−, CD−/−CatD fibroblasts) of cellular protein were used according to the manufacturer's protocol. The cleaved, fluorogenic active AMC was measured at an excitation of 355 nm and an emission of 460 nm by a fluorimetric ELISA-Reader (FLUOROSCAN II, Labsystems). TNF- and ceramide-induced caspase activation was calculated as % change in relative AMC fluorescence compared to CHX- or solvent-treated controls.
Cytochrome c release
Cytochrome c release from mitochondria was analyzed as described.51 Briefly, cells were homogenized by passing through a 26-gauge needle in buffer containing 20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin and 250 mM sucrose. A postnuclear supernatant was centrifuged at 10 000 × g for 15 min at 4°C. The resulting mitochondrial pellet was lysed in 20 mM Tris, pH 7.4, 100 mM NaCl and the protease inhibitors as above. The postmitochondrial supernatant was centrifuged at 100 000 × g for 60 min at 4°C and the supernatant was used for preparation of the cytosol by concentration through a Microcon YM-10 Centrifugal Filter Device (Millipore, Bedford, MA, USA).
CTSD release
The release of CTSD from the endolysosomal compartment was analyzed by a selective digitonin permeabilization method. At indicated time points, cells were subjected to permeabilization buffer (210 mM D-manitol, 70 mM sucrose, 10 mM HEPES, 5 mM succinate, 0.2 mM EGTA, 0.15% BSA, 80 μg/ml digitonin, pH 7.2, 4°C). After 5 min at 4°C, the permeabilization buffer was removed and cells centrifuged for 10 min at 13 000 × g. Protein in the supernatant was precipitated with 5% trichloroacetic acid and analyzed on 15% SDS-PAGE.
Confocal immunofluorescence imaging
Cells were cultured on poly-L-lysine-coated glass coverslips and fixed with cold methanol at –20°C for 5 min and subsequently incubated with primary antibodies (anti-human Bid: Santa Cruz; anti-human CTSD: Calbiochem, Germany; anti-rab 5: Santa Cruz) at room temperature for 1 h. The appropriate secondary antibodies (anti-goat-FITC, anti-rabbit-FITC, anti-rabbit-rhodamine: Santa Cruz, USA) were incubated for 45 min at room temperature. Lysotracker red DND-99 (Molecular Probes, Eugene, USA) was added to cell cultures 1 h prior to fixation with paraformaldehyde at 100 nM. Indirect immunofluorescence was analyzed using a confocal laser-scanning microscope (LSM 510, equipped with an Axiovert 100 M: Zeiss, Germany).
Immunoprecipitation of Bid and in vitro digestion by CTSD
PancTuI cells were lysed in 50 mM Tris-HCl pH 7.5; 1% NP-40; 150 mM NaCl; 5 mM EDTA; 2 mM Na3VO4; 100 nM ocadaic acid; protease inhibitor cocktail complete (Amersham). Immunoprecipitation of Bid from 250 μg of lysate was carried out with 2 μg anti-Bid-antibody (goat anti-Bid-antibody C-20, SC-6538, Santa Cruz) and 50 μl of protein G–sepharose 4 fast flow (Amersham Pharmacia Biotech). After washing 4 × with lysis buffer and 1 × with CTSD reaction buffer (100 mM Na acetate, 100 mM KCl, pH 4.2) beads were resuspended in 40 μl of CTSD buffer containing 10 ng of CTSD in the absence or presence of 1.5 μM of pepstatin A. To determine the pH profile of Bid cleavage by CTSD, reactions were performed in buffers of pH 4.2, 5.0, 6.2 and 7.0 at 37°C for times indicated in the legend to Figure 10a. Samples were separated by Tris-glycine gel electrophoresis (4–20% Tris-glycine Novex precast gel, Invitrogen). Bid and tBid were detected by Western blotting using rabbit-anti-Bid-antibodies (R&D Systems, Wiesbaden, Germany).
Abbreviations
- A-SMase:
-
acid sphingomyelinase
- CTSD:
-
cathepsin D
- TNF:
-
tumor necrosis factor
- TR55:
-
p55 TNF receptor
- PTH:
-
parathyroid hormone
References
Mizushima N, Koike R, Kohsaka H, Kushi Y, Handa S, Yagita H and Miyasaka N (1996) Ceramide induces apoptosis vie CPP32 activation. FEBS Lett. 395: 267–271
Ito A, Uehara T, Tokumitsu A, Okuma Y and Nomura Y (1999) Possible involvement of cytochrome c release and sequential activation of caspases in ceramide-induced apoptosis in SK-N-MC cells. Biochim. Biophys. Acta 1452: 263–274
Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J, Krönke M and Schütze S (1999) Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 18: 5252–5263
Perry DK and Hannun YA (1998) The role of ceramide in cell signaling. Biochim. Biophys. Acta 1436: 233–243
Schwandner R, Wiegmann K, Bernado K, Kreder D and Krönke M (1998) TNF receptor death domain-associated proteins TRADD and FADD signal activation of acid sphingomyelinase. J. Biol. Chem. 273: 5916–5922
Schütze S, Machleidt T, Adam D, Schwandner R, Wiegmann K, Kruse M-L, Heinrich M, Wickel M and Krönke M (1999) Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor death domain signalling. J. Biol. Chem. 274: 10203–10212
Deiss LP, Galinka H, Berissi H, Cohen O and Kimchi A (1996) Cathepsin D protease mediates programmed cell death induced by interferon-γ, Fas/Apo-1 and TNF-α. EMBO J. 15: 3861–3870
Roberg K, Johansson U and Öllinger K (1999) Lysosomal release of cathepsin D precedes relocation of cytochrome c and loss of mitochondrial transmembrane potential during apoptosis by oxidative stress. Free Radic. Biol. Med. 27: 1228–1237
Öllinger K (2000) Inhibition of cathepsin D prevents free-radical-induced apoptosis in rat cardiomyocytes. Arch. Biochem. Biophys. 373: 346–351
Turk B, Turk D and Turk V (2000) Lysosomal cystein proteases: more than scavengers. Biochem. Biophys. Acta 1477: 98–111
Roberts LR, Adjei PN and Gores GJ (1999) Cathepsins as effector proteases in hepatocyte apoptosis. Cell. Biochem. Biophys. 30: 71–88
Roberts LR, Kurusawa H, Bronk SF, Fesmier PJ, Agellon LB, Leung W-Y, Mao F and Gores GJ (1997) Cathepsin B contributes to bile salt induced apoptosis of rat hepatocytes. Gastroenterology 113: 1714–1726
Vancompernolle K, Van Herreweghe F, Pynaert G, Van de Craen M, De Vos K, Totty N, Sterling A, Fiers W, Vandernabeele P and Grooten J (1998) Atractyloside-induced release of cathepsin B, a protease with caspase-processing activity. FEBS Lett. 438: 150–158
Schotte P, Van Criekinge W, Van de Craen M, Van Loo G, Desmedt M, Grooten J, Cornelissen M, De Ridder L, Vandekerckhove J, Fiers W, Vandanabeele P and Beyaert R (1998) Cathepsin B-mediated activation of the proinflammatory caspase-11. Biochem. Biophys. Res. Commun. 251: 379–387
Guicciardi ME, Deussing J, Miyoshi H, Bronk SF, Svingen PA, Peters C, Kaufmann SH and Gores GJ (2000) Cathepsin B contributes to TNF-α-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 106: 1127–1137
Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M, Elling F, Leist M and Jäätelä M (2001) Cathepsin B acts as a dominant executioner protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 153: 999–1009
Guicciardi ME, Miyoshi H, Bronk SF and Gores GJ (2001) Cathepsin B knock out mice are resistant to tumor necrosis factor-α-mediated hepatocyte apoptosis and liver injury. Am. J. Pathol. 159: 2045–2054
Shibata M, Kanamori S, Isahara K, Ohsawa Y, Konishi A, Kametaka S, Watanabe T, Ebisu S, Ishido K, Kominami E and Uchiyama Y (1998) Participation of cathepsin B and D in apoptosis of PC12 cells following serum deprivation. Biochem. Biophys. Res. Commun. 251: 199–203
Kargedal K, Johansson U and Öllinger K (2001) The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. FASEB J. 15: 1592–1594
Wu GS, Saftig P, Peters C and El-Deiry WS (1998) Potential role for cathepsin D in p53-dependent tumor suppression and chemosensitivity. Oncogene 16: 2177–2183
Roberg K, Kagedal K and Öllinger K (2002) Microinjection of cathepsin D induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 161: 89–96
Salvesen GS (2001) A lysosomal protease enters the death scene. J. Clin. Invest. 107: 21–22
Johnson DE (2000) Noncaspase proteases in apoptosis. Leukemia 14: 1695–1730
Stoka V, Turk B, Schendel SL, Kim T-H, Cirman T, Snipas SJ, Ellerby LM, Bredesen D, Freeze H, Abrahamson M, Brömme D, Krajewsky S, Reed JC, Yin X-M, Turk V and Salvesen GS (2001) Lysosomal proteases pathways to apoptosis. Cleavage of Bid, not caspases, is the most likely route. J. Biol. Chem. 276: 3149–3157
Brunk UT, Dalen H, Roberg K and Hellquist HB (1997) Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic. Biol. Med. 23: 616–626
Li W, Yuan XM, Olsson AG and Brunk UT (1998) Uptake of oxidative LDL by macrophages results in partial lysosomal enzyme inactivation and relocation. Arterioscler. Thromb. Vasc. Biol. 18: 177–184
Roberg K and Öllinger K (1998) Oxidative stress causes relocation of the lysosomal enzyme cathepsin D with ensuing apoptosis in neonatal rat cardiomyocytes. Am J. Pathol. 152: 1151–1156
Deigner H-P, Claus R, Bonaterra GA, Gehrke C, Bibak N, Blaess M, Cantz M, Metz J and Kinscherf R (2001) Ceramide induced aSMase expression: implication for oxLDL-induced apoptosis. FASEB J. 15: 807–814
Gieselmann V, Pohlmann R, Hasilik A and Von Figura K (1983) Biosynthesis and transport of cathepsin D in cultured human fibroblasts. J. Cell Biol. 97: 1–5
Rijnboutt S, Stoorvogel W, Geuze HJ and Strous GJ (1992) Identification of subcellular compartments involved in biosynthetic processing of cathepsin D. J. Biol. Chem. 267: 15665–15672
Hannun YA and Luberto C (2000) Ceramide in the eukaryotic stress response. Trends Cell Biol. 10: 73–80
Kolesnick RN and Krönke M (1998) Regulation of ceramide production and apoptosis. Annu. Rev. Physiol. 60: 643–665
Kolesnick R and Hannun Y (1999) Ceramide and apoptosis. Trends Biol. Sci. 24: 224–225
Santana P, Pena LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z and Kolesnick R (1996) Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 86: 189–199
Kolesnick R and Gulbins E (2000) CD95-mediated apoptosis in vivo involves acid sphingomyelinase. J. Biol. Chem. 275: 27316–27323
Lin T, Genestier L, Pinkoski MJ, Castro A, Nicholas S, Mogil R, Paris F, Fuks Z, Schuchmann EH, Kolesnick R and Green DR (2000) Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J. Biol. Chem. 275: 8657–8663
Morita Y, Perez GI, Paris F, Mirando SR, Ehleiter D, Haimovitz-Friedman A, Fuks Z, Xie Z, Reed JC, Schuchman EH, Kolesnick R and Tilly JL (2000) Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingosine-1-phosphate therapy. Nat. Med. 6: 1109–1114
Zhang Y, Mattjus P, Schmid PC, Dong Z, Zhoong S, Ma WY, Brown RE, Bode AM, Schmid HO and Dong Z (2001) Involvement of the acid sphingomyelinase pathway in UVA-induced apoptosis. J. Biol. Chem. 276: 11775–11782
Paris F, Grassme H, Cremesti A, Zager J, Fong Y, Haimovitz-Friedman A, Fuks Z, Gulbins E and Kolesnick R (2001) Natural ceramide reverses Fas resistance of acid sphingomyelinase (−/−) hepatocytes. J. Biol. Chem. 276: 8297–8305
Lozano J, Menendez S, Morales A, Ehleiter D, Liao WC, Wagman R, Haimovitz-Friedman A, Fuks Z and Kolesnick R (2001) Cell autonomous apoptosis defects in acid sphingomyelinase knockout fibroblasts. J. Biol. Chem. 276: 442–448
Garcia-Ruiz C, Colell A, Mari M, Morales A, Calvo M, Enrich C and Fernandez-Checa JC (2003) Defective TNF-alpha-mediated hepatocellular apotosis and liver damage in acidic sphingomyelinase knockout mice. J. Clin. Invest. 111: 197–208
Chinnaiyan AM, O'Rourke K, Tewari M and Dixit VM (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81: 505–512
Hsu H, Xiong J and Goeddel DV (1995) The TNF receptor 1-associated protein TRADD signals cell death and NFkB activation. Cell 81: 495–504
Zang Y, Beard RL, Chandraratna RA and Kang JX (2001) Evidence of a lysosomal pathway for apoptosis induced by the synthetic retinoid CD437 in human leukemia HL-60 cells. Cell Death Differ. 8: 477–485
Madge LA, Li JH, Choi J and Pober JS (2003) Inhibition of PI-3 kinase sensitizes vascular endothelial cells to cytokine-initiated cathepsin-dependent apoptosis. J. Biol. Chem. 278: 21295–21306
Kargedal K, Zhao MSvensson I and Brunk UT (2001) Sphingosine-induced apoptosis is dependent on lysosomal proteases. Biochem. J. 359: 335–343
Gottlieb RA, Nordberg J, Skowronski E and Babior BM (1996) Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification. Proc. Natl. Acad. Sci. USA 93: 654–658
Matsuyama S, Llopis J, Deveraux QL, Tsien RY and Reed JC (2000) Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2: 318–325
Liu D, Martino G, Thangaraju M, Sharma M, Halwani F, Shen SH, Patel YC and Srikant CB (2000) Caspase-8-mediated intracellular acidification precedes mitochondrial dysfunction in somatostatin-induced apoptosis. J. Biol. Chem. 275: 9244–9250
Saftig P, Hetman M, Schmahl W, Weber K, Heine L, Mossmann H, Köster A, Hess B, Evers M, von Figura K and Peters C (1995) Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atropy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 14: 3599–3608
Tafani M, Karpinich NO, Hurster K, Pastorino JG, Schneider T, Russo MA and Farber JL (2002) Cytochrome c release upon Fas receptor activation depends on translocation of full length Bid and the induction of the mitochondrial permeability transition. J. Biol. Chem. 277: 10073–10082
Acknowledgements
We thank P Saftig for providing us with fibroblasts from CTSD knockout mice and D Green for lymphoblasts from Niemann-Pick patients (MS1418). This work was supported by the Deutsche Forschungsgemeinschaft (DFG), SFB 415, Project A11.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by ME Peter
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd).
Supplementary information
Rights and permissions
About this article
Cite this article
Heinrich, M., Neumeyer, J., Jakob, M. et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 11, 550–563 (2004). https://doi.org/10.1038/sj.cdd.4401382
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401382
Keywords
This article is cited by
-
Functional roles of sphingolipids in immunity and their implication in disease
Experimental & Molecular Medicine (2023)
-
How cytosolic compartments play safeguard functions against neuroinflammation and cell death in cerebral ischemia
Metabolic Brain Disease (2021)
-
Palmitoylation is required for TNF-R1 signaling
Cell Communication and Signaling (2019)
-
Mass spectrometry imaging reveals ganglioside and ceramide localization patterns during cerebellar degeneration in the Npc1−/− mouse model
Analytical and Bioanalytical Chemistry (2019)
-
DPubChem: a web tool for QSAR modeling and high-throughput virtual screening
Scientific Reports (2018)
