
Abstract
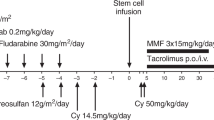
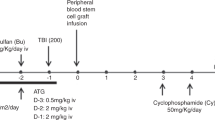
This study was designed to determine the safety of a nonmyeloablative regimen in patients with primary immunodeficiency disorders (PID) who had infections, organ dysfunction or other risk factors that precluded conventional hematopoietic cell (HC) transplant. Fourteen patients received HLA-matched related (n=6) or unrelated (n=8) HC grafts from marrow (n=8), peripheral blood mononuclear cells (n=5) or umbilical cord blood (n=1), either without conditioning (n=1), or after 200 cGy total body irradiation alone (n=3) or with 90 mg/m2 fludarabine (n=10). All patients were given postgrafting immunosuppression with mycophenolate mofetil and cyclosporine. Mixed (n=5) or full (n=8) donor chimerism was established in 13 patients, and one patient rejected the graft. Eight patients developed acute grade III (n=1) and/or extensive chronic GVHD (n=8). With a median follow-up of 4.9 (range, 0.7–8.1) years, the 3-year overall survival, event-free survival and transplant-related mortality were 62, 62 and 23%, respectively. Correction of immune dysfunction was documented in 8 of 10 patients with stable donor engraftment. These preliminary results indicated that this approach was associated with stable donor engraftment and a low incidence of early mortality and, thus, can be considered for certain high-risk patients with PID. However, there was a risk of GVHD, which is an undesirable outcome for this group of patients.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Sanders JE, Guthrie KA, Hoffmeister PA, Woolfrey AE, Carpenter PA, Appelbaum FR . Final adult heights of patients who received hematopoietic cell transplantation in childhood. Blood 2005; 105: 1348–1354.
Sanders JE . Endocrine complications of high-dose therapy with stem cell transplantation. Pediatr Transplant 2004; 8 (Suppl 5): 39–50.
Sanders JE, the Seattle Marrow Transplant Group. Effects of bone marrow transplantation on reproductive function. In: Green DM, D'Angio GJ (eds). Late Effects of Treatment for Childhood Cancer. Wiley-Liss: New York, 1992, pp 95–101.
Sanders JE . Growth and development after hematopoietic cell transplantation. In: Blume KG, Forman SJ, Appelbaum FR (eds). Thomas' Hematopoietic Cell Transplantation. Blackwell Publishing Ltd: Oxford UK, 2004, pp 929–943.
Storb R, Yu C, Wagner JL, Deeg HJ, Nash RA, Kiem H-P et al. Stable mixed hematopoietic chimerism in DLA-identical littermate dogs given sublethal total body irradiation before and pharmacological immunosuppression after marrow transplantation. Blood 1997; 89: 3048–3054.
Niederwieser D, Maris M, Shizuru JA, Petersdorf E, Hegenbart U, Sandmaier BM et al. Low-dose total body irradiation (TBI) and fludarabine followed by hematopoietic cell transplantation (HCT) from HLA-matched or mismatched unrelated donors and postgrafting immunosuppression with cyclosporine and mycophenolate mofetil (MMF) can induce durable complete chimerism and sustained remissions in patients with hematological diseases. Blood 2003; 101: 1620–1629.
Maris MB, Niederwieser D, Sandmaier BM, Storer B, Stuart M, Maloney D et al. HLA-matched unrelated donor hematopoietic cell transplantation after nonmyeloablative conditioning for patients with hematologic malignancies. Blood 2003; 102: 2021–2030.
McSweeney PA, Niederwieser D, Shizuru JA, Sandmaier BM, Molina AJ, Maloney DG et al. Hematopoietic cell transplantation in older patients with hematologic malignancies: replacing high-dose cytotoxic therapy with graft-versus-tumor effects. Blood 2001; 97: 3390–3400.
Fischer A, Landais P, Friedrich W, Morgan G, Gerritsen B, Fasth A et al. European experience of bone-marrow transplantation for severe combined immunodeficiency. Lancet 1990; 336: 850–854.
Fischer A, Landais P, Friedrich W, Gerritsen B, Fasth A, Porta F et al. Bone marrow transplantation (BMT) in Europe for primary immunodeficiencies other than severe combine immunodeficiency: a report from the European Group for BMT and the European Group for Immunodeficiency. Blood 1994; 83: 1149–1154.
Filipovich AH . Stem cell transplantation from unrelated donors for correction of primary immunodeficiencies. Immunol Allergy Clin North Am 1996; 16: 377–392.
Dupont B, Yang SY . Histocompatibility. In: Forman SJ, Blume KG, Thomas ED (eds). Bone Marrow Transplant. Blackwell Scientific Publications: Boston, 1994, pp 22–40.
Hansen JA, Mickelson EM, Choo SY, Petersdorf EW, Anasetti C, Martin PJ et al. Clinical bone marrow transplantation: donor selection and recipient monitoring. In: Rose NR, De Macario EC, Fahey JL, Friedman H, Penn GM (eds). Manual of Clinical Laboratory Immunology. American Society for Microbiology: Washington, 1992, pp 850–866.
Petersdorf EW, Smith AG, Mickelson EM, Martin PJ, Hansen JA . Ten HLA-DR4 alleles defined by sequence polymorphisms within the DRB1 first domain. Immunogenetics 1991; 33: 267–275.
Petersdorf EW, Gooley TA, Anasetti C, Martin PJ, Smith AG, Mickelson EM et al. Optimizing outcome after unrelated marrow transplantation by comprehensive matching of HLA class I and II alleles in the donor and recipient. Blood 1998; 92: 3515–3520.
Thomas ED, Storb R, Clift RA, Fefer A, Johnson FL, Neiman PE et al. Bone-marrow transplantation. N Engl J Med 1975; 292: 832–843, 895–902.
Bensinger WI, Martin PJ, Storer B, Clift R, Forman SJ, Negrin R et al. Transplantation of bone marrow as compared with peripheral-blood cells from HLA-identical relatives in patients with hematologic cancers. N Engl J Med 2001; 344: 175–181.
Burroughs L, Mielcarek M, Leisenring W, Sandmaier BM, Maloney DG, Baron F et al. Extending postgrafting cyclosporine decreases the risk of severe graft-versus-host disease after nonmyeloablative hematopoietic cell transplantation. Transplantation 2006; 81: 818–825.
Giaccone L, McCune JS, Maris MB, Gooley TA, Sandmaier BM, Slattery JT et al. Pharmacodynamics of mycophenolate mofetil after nonmyeloablative conditioning and unrelated donor hematopoietic cell transplantation. Blood 2005; 106: 4381–4388.
Maris MB, Sandmaier BM, Storer BE, Maloney DG, Shizuru JA, Agura E et al. Unrelated donor granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cell transplantation after nonmyeloablative conditioning: the effect of postgrafting mycophenolate mofetil dosing. Biol Blood Marrow Transplant 2006; 12: 454–465.
Przepiorka D, Weisdorf D, Martin P, Klingemann H-G, Beatty P, Hows J et al. 1994 Consensus conference on acute GVHD grading. Bone Marrow Transplant 1995; 15: 825–828.
Sullivan KM, Agura E, Anasetti C, Appelbaum FR, Badger C, Bearman S et al. Chronic graft-versus-host disease and other late complications of bone marrow transplantation. Semin Hematol 1991; 28: 250–259.
Koc S, Leisenring W, Flowers MED, Anasetti C, Deeg HJ, Nash RA et al. Therapy for chronic graft-versus-host disease: a randomized trial comparing cyclosporine plus prednisone versus prednisone alone. Blood 2002; 100: 48–51.
Mielcarek M, Martin PJ, Leisenring W, Flowers MED, Maloney DG, Sandmaier BM et al. Graft-versus-host disease after nonmyeloablative versus conventional hematopoietic stem cell transplantation. Blood 2003; 102: 756–762.
Goodman JL, Winston DJ, Greenfield RA, Chandrasekar PH, Fox B, Kaizer H et al. A controlled trial of fluconazole to prevent fungal infections in patients undergoing bone marrow transplantation. N Engl J Med 1992; 326: 845–851.
Boeckh M, Gooley TA, Myerson D, Cunningham T, Schoch G, Bowden RA . Cytomegalovirus pp65 antigenemia-guided early treatment with ganciclovir versus ganciclovir at engraftment after allogeneic marrow transplantation: a randomized double-blind study. Blood 1996; 88: 4063–4071.
Limaye AP, Huang M-L, Leisenring W, Stensland L, Corey L, Boeckh M . Cytomegalovirus (CMV) DNA load in plasma for the diagnosis of CMV disease before engraftment in hematopoietic stem-cell transplant recipients. J Infect Dis 2001; 183: 377–382.
Limaye AP, Huang M-L, Atienza EE, Ferrenberg JM, Corey L . Detection of Epstein-Barr virus DNA in sera from transplant recipients with lymphoproliferative disorders. J Clin Microbiol 1999; 37: 1113–1116.
Stelzer GT, Shults KE, Loken MR . CD45 gating for routine flow cytometric analysis of human bone marrow specimens. Ann NY Acad Sci 1993; 677: 265–280.
Lin MT, Tseng LH, Frangoul H, Gooley T, Pei J, Barsoukov A et al. Increased apoptosis of peripheral blood T cells following allogeneic hematopoietic cell transplantation. Blood 2000; 95: 3832–3839.
Scharf SJ, Smith AG, Hansen JA, McFarland C, Erlich HA . Quantitative determination of bone marrow transplant engraftment using fluorescent polymerase chain reaction primers for human identity markers. Blood 1995; 85: 1954–1963.
Kasai K, Nakamura Y, White R . Amplification of a variable number of tandem repeats (VNTR) locus (pMCT118) by the polymerase chain reaction (PCR) and its application to forensic science. J Forensic Sci 1990; 35: 1196–1200.
Boerwinkle E, Xiong WJ, Fourest E, Chan L . Rapid typing of tandemly repeated hypervariable loci by the polymerase chain reaction: application to the apolipoprotein B 3′ hypervariable region. Proc Natl Acad Sci USA 1989; 86: 212–216.
Bryant E, Martin PJ . Documentation of engraftment and characterization of chimerism following hematopoietic cell transplantation. In: Thomas ED, Blume KG, Forman SJ (eds). Hematopoietic Cell Transplantation, 2nd edn. Blackwell Science: Boston, 1999, pp 197–206.
Fletcher MA, Urban RG, Asthana D, Walling J, Friedlander A, Page JB . Lymphocyte proliferation. In: Rose NR, De Macario EC, Folds JD, Lane HC, Nakamura RM (eds). Clinical Diagnosis and Management by Laboratory Methods. ASM Press: Washington, DC, 1997, pp 313–319.
Ochs HD, Davis SD, Wedgwood RJ . Immunologic responses to bacteriophage φX 174 in immunodeficiency diseases. J Clin Invest 1971; 50: 2559–2568.
Noel DR, Witherspoon RP, Storb R, Atkinson K, Doney K, Mickelson EM et al. Does graft-versus-host disease influence the tempo of immunologic recovery after allogeneic human marrow transplantation? An observation on 56 long-term survivors. Blood 1978; 51: 1087–1105.
Lee W-I, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD . Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood 2005; 105: 1881–1890.
Richardson MP, Ayliffe MJ, Helbert M, Davies EG . A simple flow cytometry assay using dihydrorhodamine for the measurement of the neutrophil respiratory burst in whole blood: comparison with the quantitative nitrobluetetrazolium test. J Immunol Methods 1998; 219: 187–193.
Kalbfleisch JD, Prentice RL . The Statistical Analysis of Failure Time Data. John Wiley & Sons: New York, 1980.
Kaplan EL, Meier P . Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958; 53: 457–481.
Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood 2007; 109: 383–385.
Filipovich AH . Bone marrow transplantation from unrelated donors for congenital immunodeficiencies. Bone Marrow Transplant 1993; 11 (Suppl 1): 78–80.
Buckley RH, Fischer A . Bone marrow transplantation for primary immunodeficiency diseases. In: Ochs HD, Smith CIE, Puck JM (eds). Primary Immunodeficiency Diseases: A Molecular and Genetic Approach. Oxford University Press Inc.: Oxford, 1999, pp 459–475.
Filipovich AH, Stone JV, Tomany SC, Ireland M, Kollman C, Pelz CJ et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood 2001; 97: 1598–1603.
Amrolia P, Gaspar HB, Hassan A, Webb D, Jones A, Sturt N et al. Nonmyeloablative stem cell transplantation for congenital immunodeficiencies. Blood 2000; 96: 1239–1246.
Horwitz ME, Barrett AJ, Brown MR, Carter CS, Childs R, Gallin JI et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med 2001; 344: 881–888.
Rao K, Amrolia PJ, Jones A, Cale CM, Naik P, King D et al. Improved survival after unrelated donor bone marrow transplantation in children with primary immunodeficiency using a reduced-intensity conditioning regimen. Blood 2005; 105: 879–885.
Kahl C, Leisenring W, Deeg HJ, Chauncey TR, Flowers MED, Martin PJ et al. Cyclophosphamide and antithymocyte globulin as a conditioning regimen for allogeneic marrow transplantation in patients with aplastic anaemia: a long-term follow-up. Br J Haematol 2005; 130: 747–751.
Blaise D, Kuentz M, Fortanier C, Bourhis JH, Milpied N, Sutton L et al. Randomized trial of bone marrow versus lenograstim-primed blood cell allogeneic transplantation in patients with early-stage leukemia: a report from the Société Française de Greffe de Moelle. J Clin Oncol 2000; 18: 537–571.
Schmitz N, Beksac M, Hasenclever D, Bacigalupo A, Ruutu T, Nagler A et al. Transplantation of mobilized peripheral blood cells to HLA-identical siblings with standard-risk leukemia. Blood 2002; 100: 761–767.
Mielcarek M, Burroughs L, Leisenring W, Diaconescu R, Martin PJ, Sandmaier BM et al. Prognostic relevance of ‘early-onset’ graft-versus-host disease following nonmyeloablative hematopoietic cell transplantation. Br J Haematol 2005; 129: 381–391.
Le Deist F, Hivroz C, Partiseti M, Thomas C, Buc HA, Oleastro M et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood 1995; 85: 1053–1062.
Acknowledgements
We thank Paul Hoffmeister, Chris Davis, Gary Schoch, Deborah Bassuk and Heather Hildebrant for data management; the research nurses Mary Hinds and John Sedgwick; the nursing and clinical staff for their dedicated care of patients; and Bonnie Larson, Helen Crawford, Debbie Sessions, Karen Carbonneau and Sue Carbonneau for help with manuscript preparation.
Supported in part by Grants CA78902, CA15704, HL36444, HD17427, HD043376, CA076930 and HL085288 from the NIH, Bethesda, MD, USA and grants from the Immunodeficiency Foundation and Jeffrey Modell Foundation. LB is the recipient of an award from the Fighting Children's Cancer Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Burroughs, L., Storb, R., Leisenring, W. et al. Intensive postgrafting immune suppression combined with nonmyeloablative conditioning for transplantation of HLA-identical hematopoietic cell grafts: results of a pilot study for treatment of primary immunodeficiency disorders. Bone Marrow Transplant 40, 633–642 (2007). https://doi.org/10.1038/sj.bmt.1705778
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1705778
Keywords
This article is cited by
-
Prophylaxis and treatment with mycophenolate mofetil in children with graft-versus-host disease undergoing allogeneic hematopoietic stem cell transplantation: a nationwide survey in Japan
International Journal of Hematology (2019)
-
Paediatric reduced intensity conditioning: analysis of centre strategies on regimens and definitions by the EBMT Paediatric Diseases and Complications and Quality of Life WP
Bone Marrow Transplantation (2015)
-
B-cell function after unrelated umbilical cord blood transplantation using a minimal-intensity conditioning regimen in patients with X-SCID
International Journal of Hematology (2013)
-
Treatment of Fanconi anemia patients using fludarabine and low-dose TBI, followed by unrelated donor hematopoietic cell transplantation
Bone Marrow Transplantation (2011)
-
Reduced intensity conditioning and allogeneic stem cell transplantation in childhood malignant and nonmalignant diseases
Bone Marrow Transplantation (2008)
