
Abstract
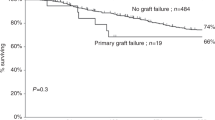
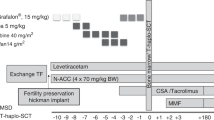
We retrospectively analyzed our results of 30 patients with three distinctive primary immunodeficiency diseases (PIDs) – severe combined immunodeficiency (SCID, n=11), Wiskott–Aldrich syndrome (WAS, n=11) and X-linked hyper-immunoglobulin M (IgM) syndrome (XHIM, n=8) – who underwent hematopoietic SCT (HSCT) during the past 20 years. Until 1995, all donors were HLA-haploidentical relatives with T-cell depletion (TCD) (n=8). Since 1996, the donors have been HLA-matched related donors (MRD) (n=8), unrelated BM (UR-BM) (n=7) and unrelated cord blood (UR-CB) (n=7). Twenty-seven of 30 patients had various pre-existing infections with or without organ damages before HSCT. Conditioning regimen and GVHD prophylaxis were determined according to disease, donor and pretransplant status. Although one of eight patients transplanted with TCD is alive with full engraftment, the other seven died. On the other hand, 18 of 22 patients transplanted without TCD are alive and well, including six of eight transplanted from MRD, seven of seven from UR-BM and five of seven from UR-CB. All 19 survivors did not require Ig supplementation after HSCT. These results indicate that UR-CBT as well as UR-BMT provides good results for PID comparable to MRD-SCT, and that early diagnosis, HSCT at early stage, careful supportive therapy and monitoring for various pathogens are important for the successful HSCT.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Buckley RH, Schiff SE, Schiff RI, Markert L, Williams LW, Roberts JL et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med 1999; 340: 508–516.
Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–99. Lancet 2003; 361: 553–560.
Fischer A, Landais P, Friedrich W, Morgan G, Gerritsen B, Fasth A et al. European experience of bone-marrow transplantation for severe combined immunodeficiency. Lancet 1990; 336: 850–854.
Stephan JL, Vlekova V, Le Deist F, Blanche S, Donadieu J, De Saint-Basile G et al. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr 1993; 123: 564–572.
Hale G, Waldmann H . Risks of developing Epstein–Barr virus-related lymphoproliferative disorders after T-cell-depleted marrow transplants. CAMPATH Users. Blood 1998; 91: 3079–3083.
Kernan NA, Bartsch G, Ash RC, Beatty PG, Champlin R, Filipovich A et al. Analysis of 462 transplantations from unrelated donors facilitated by the National Marrow Donor Program. N Engl J Med 1993; 328: 593–602.
Rubinstein P, Carrier C, Scaradavou A, Kurtzberg J, Adamson J, Migliaccio AR et al. Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med 1998; 339: 1565–1577.
Aversa F, Tabilio A, Velardi A, Cunningham I, Terenzi A, Falzetti F et al. Treatment of high-risk acute leukemia with T-cell-depleted stem cells from related donors with one fully mismatched HLA haplotype. N Engl J Med 1998; 339: 1186–1193.
Takagi S, Minakuchi J, Okawa H, Yata J . Phenotypical and functional heterogeneity of the large granular lymphocytes increased after various treatments in a patient with combined immunodeficiency. J Clin Immunol 1989; 9: 39–47.
Nagasawa M, Morio T, Takagi S, Yata J . Generation and function of gamma delta T cells after allogeneic bone marrow transplantation in humans: comparison in absence or presence of HLA-matched or mismatched thymus. Acta Paediatr Jpn 1991; 33: 146–158.
Nagasawa M, Morio T, Takagi S, Yata J . Differences of LAK-activity and IL-2 responsiveness between alpha/beta and gamma/delta T cells which developed after thymus transplantation. Acta Paediatr Jpn 1994; 36: 396–403.
Tomizawa D, Aoki Y, Nagasawa M, Morio T, Kajiwara M, Sekine T et al. Novel adopted immunotherapy for mixed chimerism after unrelated cord blood transplantation in Omenn syndrome. Eur J Hematol 2005; 75: 441–444.
Tomizawa D, Imai K, Ito S, Kajiwara M, Minegishi Y, Nagasawa M et al. Allogeneic hematopoietic stem cell transplantation for seven children with X-linked hyper-IgM syndrome: a single center experience. Am J Hematol 2004; 76: 33–39.
Nagasawa M, Imai M, Imai K, Itoh S, Kajiwara M, Morio T et al. In vivo class switch of B cells after cord blood stem cell transplantation in severe combined immune deficient (SCID) patient. Am J Hematol 2000; 65: 176–177.
Minegishi Y, Ishii N, Maeda H, Takagi S, Tsuchida M, Okawa H et al. Three novel mutations in the interleukin-2 receptor gamma chain gene in four Japanese patients with X-linked severe combined immunodeficiency. Hum Genet 1995; 96: 681–683.
Wada T, Toma T, Okamoto H, Kasahara Y, Koizumi S, Agematsu K et al. Oligoclonal expansion of T lymphocytes with multiple second-site mutations leads to Omenn syndrome in a patient with RAG1-deficient severe combined immunodeficiency. Blood 2005; 106: 2099–2101.
Imai K, Morio T, Zhu Y, Jin Y, Itoh S, Kajiwara M et al. Clinical course of patients with WASP gene mutations. Blood 2004; 103: 456–464.
Bolinger AM, Zangwill AB, Slattery JT, Glidden D, DeSantes K, Heyn L et al. An evaluation of engraftment, toxicity and busulfan concentration in children receiving bone marrow transplantation for leukemia or genetic disease. Bone Marrow Transplant 2000; 25: 925–930.
Reisner Y, Kapoor N, Kirkpatrick D, Pollack MS, Cunningham-Rundles S, Dupont B et al. Transplantation for severe combined immunodeficiency with HLA-A,B,D,DR incompatible parental marrow cells fractionated by soybean agglutinin and sheep red blood cells. Blood 1983; 61: 341–348.
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974; 18: 295–304.
McDonald GB, Hinds MS, Fisher LD, Schoch HG, Wolford JL, Banaji M et al. Veno-occlusive disease of the liver and multiorgan failure after bone marrow transplantation: a cohort study of 355 patients. Ann Intern Med 1993; 118: 255–267.
Nagasawa M, Maeda H, Okawa H, Yata J . Pulmonary miliary tuberculosis and T-cell abnormalities in a severe combined immunodeficient patient reconstituted with haploidentical bone marrow transplantation. Int J Hematol 1994; 59: 303–309.
Bittencourt H RV, Chevret S . Association of CD34 cell dose with hematopoietic recovery, infections, and other outcomes after HLA-identical sibling bone marrow transplantation. Blood 2002; 99: 2726–2733.
Rao K, Amrolia PJ, Jones A, Cale CM, Naik P, King D et al. Improved survival after unrelated donor bone marrow transplantation in children with primary immunodeficiency using a reduced-intensity conditioning regimen. Blood 2005; 105: 879–885.
Gennery AR, Khawaja K, Veys P, Bredius RG, Notarangelo LD, Mazzolari E et al. Treatment of CD40 ligand deficiency by hematopoietic stem cell transplantation: a survey of the European experience, 1993–2002. Blood 2004; 103: 1152–1157.
Lenarsky C, Weinberg K, Kohn DB, Parkman R . Unrelated donor BMT for Wiskott–Aldrich syndrome. Bone Marrow Transplant 1993; 12: 145–147.
Filipovich AH, Stone JV, Tomany SC, Ireland M, Kollman C, Pelz CJ et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott–Aldrich syndrome: collaborative study of the International Bone Marrow Transplant Registry and the National Marrow Donor Program. Blood 2001; 97: 1598–1603.
Knutsen AP, Steffen M, Wassmer K, Wall DA . Umbilical cord blood transplantation in Wiskott–Aldrich syndrome. J Pediatr 2003; 142: 519–523.
Chan K, Puck JM . Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol 2005; 115: 391–398.
Myers LA, Patel DD, Puck JM, Buckley RH . Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood 2002; 99: 872–878.
Acknowledgements
This study is in part supported by a grant from Ministry of Health, Labour and Welfare, Japan Defense Agency, Japan Intractable Diseases Research Foundation, and Kawano Masanori Foundation for Promotion of Pediatrics.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tsuji, Y., Imai, K., Kajiwara, M. et al. Hematopoietic stem cell transplantation for 30 patients with primary immunodeficiency diseases: 20 years experience of a single team. Bone Marrow Transplant 37, 469–477 (2006). https://doi.org/10.1038/sj.bmt.1705273
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1705273
Keywords
This article is cited by
-
Conditioning regimens for inborn errors of immunity: current perspectives and future strategies
International Journal of Hematology (2022)
-
Hematopoietic Cell Transplantation for Severe Combined Immunodeficiency Patients: a Japanese Retrospective Study
Journal of Clinical Immunology (2021)
-
Hematopoietic Cell Transplantation with Reduced Intensity Conditioning Using Fludarabine/Busulfan or Fludarabine/Melphalan for Primary Immunodeficiency Diseases
Journal of Clinical Immunology (2021)
-
Un-manipulated haploidentical transplant in Wiskott-Aldrich syndrome
Indian Pediatrics (2017)
-
A Stable Mixed Chimera After SCT with RIC in an Infant with IκBα Hypermorphic Mutation
Journal of Clinical Immunology (2017)
