
Summary:
Our objective was to study the outcome of allogeneic hematopoietic stem cell transplantation (HSCT) in patients with severe congenital neutropenia (SCN). Among 101 cases of SCN included in the French Severe Chronic Neutropenia Registry, nine patients received HSCT between 1993 and 2003, in seven institutions. The indications were nonresponse to G-CSF therapy in four cases, bone marrow failure in one case, and myelodysplastic syndrome or leukemia in four cases. The conditioning regimen consisted of total body irradiation in two cases and chemotherapy alone in the other seven cases. Seven patients received stem cells from unrelated donors and two from identical siblings. Engraftment occurred in all but one of the patients. Three patients died. The respective causes of death were graft-versus-host disease, infection, and EBV post-transplant lymphoproliferative disease. Six patients are alive and in complete remission, with a median follow-up of 3.1 years. These results indicate that HSCT is feasible for patients with SCN who do not respond to G-CSF, who have malignant transformation, or who are at a high risk of malignant transformation, even if an HLA-identical sibling donor is not available.
Similar content being viewed by others
Main
Severe congenital neutropenia (SCN) is a rare and heterogeneous inherited hematologic disorder characterized by profound neutropenia diagnosed early in life and complicated by recurrent severe and life-threatening bacterial infections.1,2 It is often caused by mutations of the ELA2 gene3,4,5 or, less frequently, of the GFI1 gene.6 Other major complications are leukemia and the myelodysplastic syndrome (MDS), the latter affecting about 10% of patients.7 The use of G-CSF therapy has radically changed these patients' outlook.8 However, a minority of patients is refractory to G-CSF, and some undergo malignant transformation. Allogeneic hematopoietic stem cell transplantation (HSCT) appears to be the only curative treatment for such patients. In all, 39 HSCT procedures have been reported in this setting, of which 15 cases were described in detail.9,10,11,12 We examined data on nine patients with SCN who underwent HSCT and were included in the French Severe Chronic Neutropenia Registry during the last 10 years.
Patients and methods
Registry organization and disease definition
The French Severe Chronic Neutropenia Registry was created in 1994, with approval from the French computer watchdog commission (CNIL certificate no. 97.075). The patients' files were monitored by clinical research associates who visited each center at least once a year. Patients were eligible for enrollment in the registry if they met the following criteria: documented ANC less than 500 cells/mm3 on at least three occasions during the 3 months prior to registration; age older than 3 months (unless there is a positive family history); and compatible bone marrow smear examination. The parents or patients were required to give informed consent. SCN was diagnosed on the basis of a median ANC value below 500/mm3 and no cyclic variations. ELA2 gene mutations were identified as reported previously.4 Overall, 101 patients with SCN were included, and a complete report of the survey is available elsewhere.13 Four of these patients were considered fully or partially refractory to G-CSF therapy (no ANC increase with 50 μg/kg/day G-CSF). One patient had bone marrow failure with profound thrombocytopenia and a decreased megakaryocyte count on the bone marrow smear. This patient also had unilateral lower limb hypertrophy and lymphangiectasia (Klippel Trenaunay syndrome) and mild mental retardation. Six patients had MDS/leukemia. The first five patients and four of the six patients with MDS/leukemia underwent HSCT. The two patients with MDS/leukemia who only received chemotherapy died from malignant disease progression.
Statistical methods
Stata® version 7 software was used for all statistical analyses. For survival analyses, the end points were death and MDS/leukemia onset. The period taken into account was the interval between HSCT and either death or the last examination when no event occurred. The Kaplan–Meier method was used to estimate survival rates. The cutoff date for this analysis was 29 February 2004. Median follow-up was 3.1 years (range 1.8–10.9 years). The date of the first HSCT was used in the two cases in which two HSCT procedures were performed.
Description of the patients prior to HSCT (Table 1)
The patients were six girls and three boys. ELA2 mutations were detected in four of the six patients tested. Median age at transplantation was 10 years (3–19 years). Polychemotherapy was administrated to two of the four patients with MDS/leukemia, and resulted in complete remission. In one case, the patient had acute lymphoblastic leukemia, treated on the very high-risk group FRALLE protocol (French acute lymphoblastic leukemia group),14 and the second, with MDS, received an AML-like regimen, including high-dose Aracytine, Mitoxantrone, VP16, and amsacrine.15 In the other two cases, no attempt was made to induce remission prior to HSCT. The median time between leukemic transformation and HSCT was 6 months.
HSCT characteristics (Table 2)
None of the donors were neutropenic. Two patients received marrow from an HLA-identical sibling. Seven patients received HSCT from the following unrelated donors: two HLA 10/10 compatible donors (bone marrow), one one-antigen-mismatch donor (bone marrow), and four unrelated cord blood (UCB) donors with at least four matches among the six HLA A, B, and DRB1 antigens.
All patients received preparatory myeloablation. One patient received a nonmyeloablative regimen based on a combination of fludarabine and ATG before a second HSCT procedure.
Seven patients received a combination of busulfan (16 mg/kg) and cyclophosphamide (200 mg/kg). Three patients received irradiation, combined with cyclophosphamide (120 mg/kg) and etoposide in one case; cyclophosphamide (120 mg/kg) in one case (conditioning for a second HSCT; patient 4); and cyclophosphamide (120 mg/kg) and thiotepa (10 mg/kg) in one case.
Two patients received two HSCT procedures, in one case with the same sibling donor and in the other case with a new, unrelated donor. The indications for these second HSCT procedures were nonengraftment in one case and AML relapse in the other. The latter patient received an AML-based chemotherapy regimen, resulting in complete remission prior to the second HSCT procedure, and was prepared with a total body irradiation (TBI)-based conditioning regimen.
The total number of infused nucleated cells ranged from 0.15 × 108 to 8 × 108/kg among patients who received cord blood cells (median 5.6 × 108/kg) and from 0.31 × 108 to 8 × 108/kg among patients who received marrow cells (median 4.2 × 108/kg). CD34+ cell selection was performed in one case (patient 7).
Graft-versus-host disease (GVHD) prophylaxis
GVHD prophylaxis consisted of cyclosporine (CSA) and short-course methotrexate (MTX) in six patients, and CSA plus methylprednisone in three patients. Seven patients with unrelated donors received ATG in their conditioning regimen.
Results (Table 2)
Hematological recovery after HSCT
Engraftment failed in one case, while full hematological recovery occurred in the other cases. The median time for the neutrophil count to reach 0.5 × 109/l was 38 days (range 18–71 days). The platelet count reached 20 × 109/l after 15–150 days (median 46 days), without further transfusions.
Engraftment
Engraftment failed in one patient. Chimerism analysis was performed on seven patients by using polymerase chain reaction (PCR) amplification of microsatellites. Complete chimerism was found in six patients tested 1–2 months after HSCT, and mixed chimerism was detected in one patient (patient 8) with 84% of donor cells.
GVHD
Eight patients had grade 0–1 acute GVHD and one patient, who had received UCB, had fatal acute grade IV GVHD. One extensive chronic GVHD was observed.
Malignant relapse
HSCT was indicated for hematological malignancies in four patients. One patient relapsed 6 months after HSCT. Complete remission was achieved with chemotherapy and was followed by a second HSCT. She is in remission with normal blood counts more than 10 years after initial HSCT.
Survival, causes of death, and complications
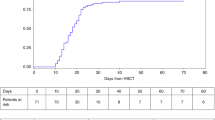
Three patients died (patients 2, 5, and 9). The overall survival rate at 5 years is 61±20% (Figure 1). The median time between HSCT and death was 0.7 years.

Survival after HSCT in nine patients with SCN.
The first patient (patient 2) developed acute grade IV GVHD, chronic extensive GVHD, and died 258 days after HSCT from Gram-negative sepsis. The second patient (patient 5) died 374 days after HSCT from pulmonary aspergillosis following secondary pancytopenia. The third patient (patient 9) developed EBV post-transplant lymphoproliferative disease (PTLD) and died of Gram-negative sepsis 42 days after a second HSCT performed because of primary nonengraftment (2.5 months after the first transplantation).
These three patients had received HSC from unrelated donors (marrow in one case, cord blood in two cases). They were significantly older than the other patients (15 vs 7 years, P=0.03).
Thus, severe infectious complications occurred in four patients and were fatal in three. Two patients developed EBV PTLD: one was cured by anti-CD20 therapy, while the other died from associated sepsis. Two patients developed CMV antigenemia and were treated preemptively with foscavir or ganciclovir.
Two patients had long-term complications. One patient developed bronchial dilatation and cardiac hypokinesia.16 This patient underwent a second HSCT for AML relapse after the first HSCT and received intensive chemotherapy before the second procedure. The second patient (patient 8) had mild neurological abnormalities before transplantation. Neurological manifestations worsened after transplantation, with ataxia, seizures, and language retardation. MRI was normal, and metabolic disorders and viral leukoencephalopathy were excluded. The neurological disorders were probably related to the underlying disease, as similar manifestations have already been reported in three patients of Kostmann's pedigree.17
Discussion
We report the outcome of HSCT in nine patients with SCN and a poor prognosis. Six patients are currently alive and cured of their initial disease. Infectious complications associated with acute GVHD were the main cause of death (three patients). Increased age at HSCT seemed to be associated with poor outcome.
All patients received a myeloablative conditioning regimen prior to the first HSCT, usually with busulfan–cyclophosphamide combination. This regimen was effective, as engraftment failed in only one patient; in addition, full chimerism was obtained in six of the seven patients tested, while the other patient had mixed chimerism. Less toxic, nonmyeloablative conditioning regimens might reduce transplanted-related mortality, but experience is still limited, particularly in nonmalignant settings.11,18
MDS/leukemia in SCN patients carries an extremely poor prognosis, and long-term survival has only been reported after HSCT.19 However, the success rate of HSCT for AML or MDS is low, as 17 of the 24 relevant patients included in the International Severe Chronic Neutropenia Registry died.7 The need for chemotherapy prior to HSCT is controversial.20 It can be useful for patients with overt leukemia, as in patients who have acute lymphoblastic leukemia,13,21,22 but it can be deleterious in other patients as it may contribute to increasing infection risk. In our series, two patients with MDS and one with AML underwent HSCT without initial chemotherapy. Only the patient with AML relapsed after HSCT, but a subsequent course of AML chemotherapy led to complete remission. Cases of successful HSCT without prior chemotherapy have already been described,9 but larger studies are needed to settle this question.
HSCT is therefore a worthwhile option for patients who do not respond to G-CSF therapy or who have only a partial response and continue to have recurrent bacterial infections, as well for patients with bone marrow failure. Previously reported results have been encouraging, as 10 of the 12 patients transplanted for this indication have survived.11,12 However, it should be noted that eight of these patients had HLA-identical sibling donors, whereas two of the four patients with alternative donors died.
These results suggest that HSCT should not be delayed for patients at risk of MDS/leukemia or for those who do not respond to G-CSF therapy, particularly if a HLA-matched sibling donor is available. Myeloablative preparation with the busulfan–cyclophosphamide combination appears adequate. The use of cytoreductive chemotherapy before HSCT is controversial, particularly in patients with MDS.
References
Kostmann R . Infantile genetic agranulocytosis. Acta Paediatr Scand 1956; 45 (Suppl 105): 1–78.
Kostmann R . Infantile genetic agranulocytosis. Acta Paediatr Scand 1975; 64: 362–368.
Ancliff PJ, Gale RE, Liesner R et al. Mutations in the ELA2 gene encoding neutrophil elastase are present in most patients with sporadic severe congenital neutropenia but only in some patients with the familial form of the disease. Blood 2001; 98: 2645–2650.
Bellanne-Chantelot C, Clauin S, Leblanc T et al. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood 2004; 103: 4119–4125.
Dale DC, Person RE, Bolyard AA et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood 2000; 96: 2317–2322.
Person RE, Li FQ, Duan Z et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet 2003; 34: 308–312.
Dale DC, Cottle TE, Fier CJ et al. Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol 2003; 72: 82–93.
Bonilla MA, Gillio AP, Ruggeiro M et al. Effects of recombinant human granulocyte colony-stimulating factor on neutropenia in patients with congenital agranulocytosis. N Engl J Med 1989; 320: 1574–1580.
Dallorso S, Manzitti C, Dodero P et al. Uneventful outcome of unrelated hematopoietic stem cell transplantation in a patient with leukemic transformation of Kostmann syndrome and long-lasting invasive pulmonary mycosis. Eur J Haematol 2003; 70: 322–325.
Rappeport JM, Parkman R, Newburger P et al. Correction of infantile agranulocytosis (Kostmann's syndrome) by allogeneic bone marrow transplantation. Am J Med 1980; 68: 605–609.
Toyoda H, Azuma E, Hori H et al. Successful unrelated BMT in a patient with Kostmann syndrome complicated by pre-transplant pulmonary ‘bacterial’ abscesses. Bone Marrow Transplant 2001; 28: 413–415.
Zeidler C, Welte K, Barak Y et al. Stem cell transplantation in patients with severe congenital neutropenia without evidence of leukemic transformation. Blood 2000; 95: 1195–1198.
Donadieu J, Leblanc T, Bader-Meunier B et al. Analysis of risk factors for myelodysplasia/leukemia and infectious death among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Hematol J 2004; in press.
Michel G, Landman-Parker J, Auclerc MF et al. Use of recombinant human granulocyte colony-stimulating factor to increase chemotherapy dose-intensity: a randomized trial in very high-risk childhood acute lymphoblastic leukemia. J Clin Oncol 2000; 18: 1517–1524.
Perel Y, Auvrignon A, Leblanc T et al. Maintenance therapy in childhood acute myeloid leukemia. Ann Hematol 2004; 83 (Suppl 1): S116–S119.
Stoll C, Alembik Y, Lutz P . A syndrome of facial dysmorphia, birth defects, myelodysplasia and immunodeficiency in three sibs of consanguineous parents. Genet Counsel 1994; 5: 161–165.
Carlsson G, Fasth A . Infantile genetic agranulocytosis, morbus Kostmann: presentation of six cases from the original ‘Kostmann family’ and a review. Acta Paediatr 2001; 90: 757–764.
Amrolia P, Gaspar HB, Hassan A et al. Nonmyeloablative stem cell transplantation for congenital immunodeficiencies. Blood 2000; 96: 1239–1246.
Freedman MH, Bonilla MA, Fier C et al. Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood 2000; 96: 429–436.
Anderson JE, Gooley TA, Schoch G et al. Stem cell transplantation for secondary acute myeloid leukemia: evaluation of transplantation as initial therapy or following induction chemotherapy. Blood 1997; 89: 2578–2585.
Cassinat B, Bellanne-Chantelot C, Notz-Carrére A et al. Screening for G-CSF receptor mutations in patients with secondary myeloid or lymphoid transformation of severe congenital neutropenia. Leukemia 2004; 18: 1553–1555.
Germeshausen M, Ballmaier M, Schulze H et al. Granulocyte colony-stimulating factor receptor mutations in a patient with acute lymphoblastic leukemia secondary to severe congenital neutropenia. Blood 2001; 97: 829–830.
Acknowledgements
This work was supported by grant RAF0007 from the Inserm AFM network for rare diseases, Société d'Hématologie et Immunologie Pédiatrique, Amgen SA, and Chugai Aventis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ferry, C., Ouachée, M., Leblanc, T. et al. Hematopoietic stem cell transplantation in severe congenital neutropenia: experience of the French SCN register. Bone Marrow Transplant 35, 45–50 (2005). https://doi.org/10.1038/sj.bmt.1704718
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1704718
Keywords
This article is cited by
-
HSCT may lower leukemia risk in ELANE neutropenia: a before–after study from the French Severe Congenital Neutropenia Registry
Bone Marrow Transplantation (2020)
-
Severe congenital neutropenias
Nature Reviews Disease Primers (2017)
-
Allogeneic hematopoietic stem cell transplantation for inherited bone marrow failure syndromes
International Journal of Hematology (2016)
-
Recommendations on hematopoietic stem cell transplantation for inherited bone marrow failure syndromes
Bone Marrow Transplantation (2015)
-
Clinical spectrum and long-term follow-up of 14 cases with G6PC3 mutations from the French severe congenital neutropenia registry
Orphanet Journal of Rare Diseases (2014)
