
Abstract
The management of localised and advanced gastrointestinal stromal tumours (GISTs) in terms of histological diagnosis, surgery, imaging, medical treatment and molecular biology has rapidly changed since introduction of imatinib mesylate for molecularly targeted therapy in 2000. In this minireview, we briefly summarise and discuss the current data relevant to the increasing role of molecular characterisation of GISTs in the diagnosis, risk assessment and effective targeted therapy.
Similar content being viewed by others
Main
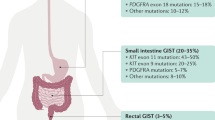
Gastrointestinal stromal tumours (GISTs), the most common mesenchymal tumours of the gastrointestinal tract, hypothetically evolve from a progenitor related to the interstitial cells of Cajal. Population-based studies reported GIST annual incidence rates ranging from 6.5 to 14.5 per million (Joensuu, 2006). The most common sites of origin for GIST are stomach (39–70%) and small intestine (31–45%), but GISTs may arise anywhere along the gastrointestinal tract or within the abdomen as extragastrointestinal tumours (Demetri et al, 2007). Frequent clinical symptoms include bloating, gastrointestinal tract bleeding, fatigue and anaemia. Gastrointestinal stromal tumours are highly resistant to traditional chemotherapeutic agents. The usefulness of radiotherapy is also very limited in the therapeutic setting mostly due to toxicity to the surrounding structures (Joensuu et al, 2002). Surgery has been the basis of treatment for GISTs. The 5-year survival rate is only 54% after complete resection of a localised primary GIST (Dematteo et al, 2002). The median survival for patients with high risk and overtly malignant tumours is <2 years and <4 years, respectively. Independent prognostic factors that predict the tumour-free survival are tumour size and proliferative index (Nilssen et al, 2005). The common metastatic sites for GIST include the liver and omentum. Due to recognition of the disease as a separate entity, the diagnosis of GIST has dramatically enhanced since 1998, and survival has greatly improved since introduction of imatinib mesylate in 2000.
Pathogenesis, differential diagnosis and risk assessment
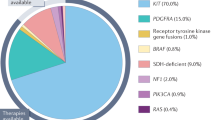
Identification of KIT-activating mutations as a key factor in the pathogenesis of GIST has substantially altered the diagnosis and treatment of GIST (Hirota et al, 1998; Corless et al, 2004; Miettinen and Lasota, 2006). Oncogenic KIT mutations are detectable in 75–85% of all GISTs, even in small, incidentally discovered lesions. These mutations most frequently involve the intracellular juxtamembrane domain of the receptor encoded by exon 11 (57–70%), followed by the extracellular domain encoded by exon 9 (5–18%). The KIT exon 11 mutations are quite heterogeneous, encompassing mainly in-frame deletions of variable sizes, basic amino acid substitutions, or more complex deletions–insertions. The KIT exon 9 mutations represent mainly in-frame tandem duplication, AY502-503dup. KIT mutations in the split kinase domains I and II (encoded by exon 13 and exon 17) appear to be rather uncommon, accounting for 0.6–1.4% of all mutations. Alternatively, a subset of GISTs (5–10%) carries constitutively activating mutations in the gene encoding platelet-derived growth factor receptor α (PDGFRA), a receptor tyrosine kinase homologous to KIT (Heinrich et al, 2003b). The mutations reported within PDGFRA involve exon 12, 14 and 18, being homologous to KIT exon 11, 13 and 17, respectively. In >90% of cases, PDGFRA mutations target codons 842–849 (exon 18), with the D842V substitution being most common. Gastrointestinal stromal tumours that harbour different KIT or PDGFRA mutations have different molecular signatures at the level of gene expression, which further contributes to the complexity of GIST biology and variable response to treatment (Antonescu et al, 2004).
Approximately 10–15% of GISTs that arise in adults lack detectable mutations of KIT or PDGFRA (referred to as wild-type GISTs). Notably, wild-type genotype is a characteristic feature of the vast majority of GISTs, which are diagnosed in children and adolescents and GISTs associated with familial syndromes such as neurofibromatosis, Carney–Stratakis syndrome or the Carney Triad (Corless et al, 2004; Miettinen and Lasota, 2006).
Histopathologically, GISTs are a heterogeneous group of tumours, featuring spindle cell, epithelioid or mixed type morphology, and showing a wide clinical spectrum from benign to frankly malignant sarcomas (Fletcher et al, 2002; Miettinen and Lasota, 2006). Gastrointestinal stromal tumours typically show expression of CD117/KIT (95%) and frequently CD34 (70%) antigens by immunostaining, yet a small fraction of GISTs lack both diagnostic markers (Hornick and Fletcher, 2007). KIT-negative GISTs have a predilection for the stomach and omentum and are most commonly epithelioid or mixed cell in type. Most KIT-negative GISTs harbour PDGFRA mutations (80%) (Heinrich et al, 2003b; Corless et al, 2005); the others contain KIT mutations. The diagnosis of KIT-negative GIST can be problematic. Yet, some of the mutations detected in KIT-negative GISTs (including some PDGFRA mutations) are known to be imatinib sensitive; therefore, the precise diagnosis is of utmost importance and lack of immunoreactivity for KIT in a GIST should not be used as justification to deny patients therapy with imatinib. In this context, mutational analysis is mandatory to confirm the diagnosis of GIST.
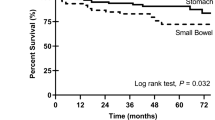
The most important prognostic features of GIST in regard to the malignant potential and prognosis are tumour size (diameter of the largest mass) and mitotic index per high-powered field (Fletcher et al, 2002). In addition, Miettinen and Lasota (2006) confirmed the results of earlier studies indicating that the anatomic location affects the risk of disease recurrence, that is, the gastric tumours showing better prognosis than small intestinal tumours. Yet, even low risk tumours still may have a malignant potential and the prediction of the biological behaviour of GISTs is difficult based on pathomorphological criteria alone. Recent studies indicate that the type of tumour mutation may be an additional prognostic risk factor for GISTs. Thus, the deletions in KIT exon 11, particularly those involving codons 557–558, are associated with high metastatic risk and poor prognosis, whereas KIT exon 11 missense mutations are correlated with longer progression-free survival (PFS) and better overall survival (Singer et al, 2002; Martin et al, 2005; Andersson et al, 2006; Miettinen and Lasota, 2006; Steigen et al, 2007). In addition, duplications in the distal part of KIT exon 11 lead to a less aggressive phenotype. Conversely, the presence of homo-/hemizygous KIT exon 11 mutations is associated with an increased risk for metastasis and with an adverse clinical course (Lasota et al, 2007). Gastrointestinal stromal tumours that are defined by a PDGFRA mutations occur almost exclusively in the gastric location, show epithelioid morphology and usually have a low mitotic count. In most cases, PDGFRA-mutated GISTs are characterised by a low malignant potential (Lasota et al, 2004, 2006). Therefore, detection of a PDGFRA mutation in gastric GISTs may represent an additional prognostic marker of more benign tumour behaviour.
Treatment – response according to molecular subtype
The KIT or PDGFRA mutations in GISTs differ in type and affect different receptor domains (Corless et al, 2004). It is well known that sensitivity to imatinib depends on the location of the KIT/PDGFRA gene mutation. Imatinib effectively inhibits KIT with the juxtamembrane-type mutation, but fails to inhibit KIT with certain activation loop (exon 17) mutations, such as D816V in mastocytosis (Ma et al, 2002; Heinrich et al, 2003a, 2003b, 2006a). Similarly, imatinib efficiently inhibits the juxtamembrane-type PDGFRA mutations, while some PDGFRA tyrosine kinase domain II mutations (mainly point mutations involving codon 842, for example, D842V) confer primary resistance to imatinib (Debiec-Rychter et al, 2005; Heinrich et al, 2006a). The phenomenon can be explained by the concept that some tyrosine kinase domain II-type mutations induce stabilisation of the activation loop in an active conformation and/or structural alteration at the imatinib-binding site of KIT, resulting in a decreased affinity for imatinib. Moreover, experimental in vitro studies have shown that different amino-acid substitutions in KIT juxtamembrane domain, even at the same codon, might also cause different structural alteration and lead to different imatinib sensitivities (Nakagomi and Hirota, 2007). These findings should be an important factor to consider while determining the eligibility for adjuvant or neoadjuvant imatinib therapies, as patients with tumours harbouring mutations primarily resistant to imatinib will not be benefited from the treatment. Recently, neoadjuvant approaches to downsize GISTs prior to surgical resection and the adjuvant treatment of intermediate- and high-risk tumours after complete or almost complete surgical resection are evaluated in a few international clinical trials; both approaches may possibly become routine clinical practice in the future.
Even more importantly, data from randomised, multicenter North American and EORTC-Australian phase II/III clinical trials for patients with unresectable or metastatic GISTs provided clear evidence that tumour mutational status is associated with outcome of imatinib therapy (Heinrich et al, 2003a, 2003b; Debiec-Rychter et al, 2006). Patients with GISTs harbouring KIT exon 11 mutations have higher response rates and longer PFS than those whose tumours carry KIT exon 9 mutations or with no detectable mutations. Notably, the EORTC-Australian 62005 study demonstrated that patients with KIT exon 9 mutations, but not those with other tumour genotype, had significantly longer PFS when treated with imatinib 800 mg day−1 compared with 400 mg day−1 (Debiec-Rychter et al, 2006). The significant PFS advantage observed with the 800 mg day−1 dose in patients with KIT exon 9 mutations in the EORTC-Australian study was not confirmed in the North American S0033 study. However, it remained significant (P=0.017) when the pooled data set was examined further in a meta-analysis of 1640 patients who had been followed for a median of 45 months (Van Glabbeke et al, 2007).
Predictive value of mutational analysis for imatinib-resistant GISTs
Resistance of GIST tumours to imatinib treatment is emerging as a clinical challenge.
Early resistance has been reported in 10–20% of cases (Demetri et al, 2007). However, the vast majority of responding patients will eventually develop secondary tumour progression. Development of imatinib resistance can follow several patterns, including progression at the primary tumour site or the development of new metastatic lesions. Available data suggest that many cases of focal progression during imatinib therapy result from clonal evolution (Antonescu et al, 2005; Debiec-Rychter et al, 2005; Heinrich et al, 2006a; Wardelmann et al, 2006). Depending on the series of patients recently published, the PFS under imatinib treatment ranges between 7 and 53 months.
Late imatinib resistance is most commonly associated with acquisition of secondary KIT mutations in the split kinase domains I and II (exon 13, 14 or exon 17). Some of these mutations alter specifically the configuration of the ATP-binding kinase pocket (V654A and T670I), inhibiting imatinib binding. Others stabilise the active conformation of the receptor, which also prevents imatinib binding (D820Y and N822K). Secondary PDGFRA mutations have also been described (Debiec-Rychter et al, 2005; Heinrich et al, 2006a). Importantly, several different types of mutations may occur independently indicating polyclonal resistance (Heinrich et al, 2006a; Wardelmann et al, 2006). It has been shown that GISTs with an underlying primary KIT mutation in exon 11, known to be the subgroup with better response rates than other mutational subtypes, reveal more frequently secondary mutations compared to tumours with an underlying exon 9 mutation (Antonescu et al, 2005; Debiec-Rychter et al, 2005; Wardelmann et al, 2006). This observation suggests that the development of a secondary KIT mutation is an important escape mechanism for tumour cells of which KIT-dependent proliferation is chronically inhibited by imatinib.
Another putative mechanism of imatinib mesylate resistance is KIT gene amplification (Debiec-Rychter et al, 2005). In addition, it is proposed that other oncogenes or tumour suppressor genes may have become important in sustaining the tumorigenic potential, rendering the tumour independent from KIT signalling and thus, making it insensitive to imatinib treatment (Heinrich et al, 2006a). Identifying altered expression of genes, known to be important in tumorigenesis, is an important research area that could lead to validation of interesting targets for specific molecular therapies.
Since secondary resistance of GIST patients to imatinib is a growing clinical problem, multiple novel inhibitors are in development to interfere with kinase signalling using alternative KIT and/or PDGFRs inhibitors or by targeting critical downstream-signalling proteins to regain disease control after failure of imatinib (von Mehren, 2006).
Sunitinib mesylate (SU011248, Sutent®, Pfizer) is approved for imatinib-refractory GIST. Sunitinib is an oral tyrosine kinase inhibitor that targets multiple kinases, including the vascular endothelial growth factor receptors (VEGFR-1, VEGFR-2 and VEGFR-3), PDGFRs, KIT, FLT3 and the receptor encoded by the RET proto-oncogene. It has both antiangiogenic and antiproliferative activities (Goodman et al, 2007). A phase III double-blind trial comparing sunitinib with placebo was conducted in 312 patients with GIST who had documented failure or intolerance of imatinib (Demetri et al, 2006). Only 5% of the patients who had imatinib-resistant GIST showed objective response, but 58% had disease stabilisation. Importantly, patients with imatinib-resistant GISTs with KIT exon 9 mutations may benefit more from sunitinib than those with an exon 11 mutation. In the patient group reported by Heinrich et al (2006b), 37% of the imatinib-resistant GISTs with a primary exon 9 mutation responded to sunitinib, as compared with 5% of cases with a primary exon 11 mutation. PFS and overall survival were also longer for patients with GIST with either a primary KIT exon 9 mutation or with no detectable KIT/PDGFRA mutation, compared with tumours with a KIT exon 11 mutation. The confounding factor might be a higher incidence of secondary KIT mutations in exon 11-mutant tumours, which may reflect longer duration of imatinib therapy and therefore increase chances for selection of mutations producing imatinib resistance in these tumours. Sunitinib shows lower efficacy for secondary KIT mutations in exons 17 and 18 in comparison with secondary mutations in exons 13 and 14 (Prenen et al, 2006; Heinrich et al, 2006a, 2006b). Given that GIST progression is a polyclonal event and a variety of secondary, imatinib-resistant KIT mutations may be present in parallel (Wardelmann et al, 2006), sunitinib may not be efficient to inhibit the proliferation of the different tumour clones present. As it will be impossible to identify all emerging resistant tumour clones, KIT sequencing has only a limited role for predicting clinical benefit of sunitinib, and the same holds true for the other second-line TK inhibitors.
Nilotinib (AMN107, Novartis) is an oral tyrosine kinase inhibitor that inhibits downstream signalling of BCR-ABL, KIT and PDGFRs. Preclinical studies have shown better efficacy of Nilotinib against certain KIT and PDGFRA mutations in comparison with imatinib (Weisberg et al, 2006; Guo et al, 2007).
Dasatinib (BMS-354825, Bristol-Myers Squibb) is a dual SRC/ABL inhibitor. This drug binds to the active conformation of KIT to which imatinib cannot bind. In this perspective, dasatinib could be a valid therapy for GIST patients expressing a secondary KIT mutation that stabilises the receptor in its active conformation form (Schittenhelm et al, 2006).
Sorafenib (BAY 43-9006, Nexavar®, Bayer) is a novel biaryl urea compound that was initially developed as a specific inhibitor of serine–threonine kinase RAF. In addition, sorafenib inhibits multiple receptor tyrosine kinases, including VEGFR-2, VEGFR-3, PDGFRs and KIT. Preclinical data show that sorafenib is more efficient, compared to nilotinib and dasatinib, to inhibit the downstream signalling of KIT receptors containing imatinib-resistant secondary mutations (Guo et al, 2007).
Heat-shock protein 90 inhibitors prevent heat-shock protein 90 from stabilising client proteins, like KIT. The targeted proteins are then increasingly directed towards the proteasome for degradation. Preclinical data show that these compounds are able to decrease downstream signalling from the receptor that contains primary and secondary imatinib-resistant mutations by increasing KIT degradation (Bauer et al, 2006). Clinical trials in imatinib-resistant GIST patients are ongoing.
Additional molecular characterisation of GISTs becomes increasingly important to identify which of the immense variety of newly developed tyrosine kinase inhibitors might be successful for the treatment of imatinib resistant GISTs. Moreover it has become clear that identifying additional genomic alterations and establishing protein and gene expression profiles in GIST might ultimately help identify new markers of tumour behaviour, prognosis and drug response.
Mutational analysis of gists in the management of the disease
The molecular characterisation of GISTs has become an essential part of the routine management of this disease.
At first diagnosis of resectable disease, mutational analysis mainly serves academic purposes, as the prognostic assessment of individual cases at present is based on solid clinical and morphological variables. However, according to recent NCCN clinical practice guidelines, GIST mutational analysis is strongly recommended for the primary intermediate or high-risk tumours (Demetri et al, 2007). Additionally, in selected cases with atypical histopathological or clinical features, molecular studies are required to help make a clear diagnosis. Molecular techniques are also essential to establish the diagnosis of emerging subtypes of GIST, and to differentiate them from common GIST or other mesenchymal malignancies.
At first diagnosis of disseminated, unresectable disease, mutational analysis should be considered a standard of care, as various studies have clearly shown that the type of mutation of GISTs clearly correlates with key clinical outcome parameters, such as response rate and PFS. While the results of molecular studies might not have an immediate impact on the choice of drug or daily dose, due to non-availability of approved treatment alternatives to imatinib 400 mg in most health care systems, patients with ‘poor response genotype’ might be considered for more intense follow-up, with higher frequency of early imaging assessments, including computer tomography and/or FDG-PET. This helps in predicting and identifying the treatment failure and to switch to a more appropriate treatment as early as possible, and could even reduce overall treatment costs.
In patients progressing during treatment with standard doses of imatinib who potentially qualify for dose escalation, and in patients failing the highest available doses of the imatinib who are potential candidates for treatment with sunitinib, mutational analysis of progressing lesions is of academic purposes. The comparison between the initial genotype with the mutational profile of refractory lesions might contribute to the further understanding of the natural biology and evolution of GIST and other solid tumours.
Patients failing all conventional systemic treatment options at present qualify for clinical trials involving new targeted agents or innovative drug combinations. In such an experimental setting, it is absolutely crucial to gain as much information about the treated tumour as possible, enabling the researcher to interpret the outcome of the study in the most appropriate way. In this situation molecular studies are generally regarded essential. This can involve analysis of historical tumour material, fresh biopsies of progressing lesions prior to study entry or even sequential biopsies during the conduct of the trial. The molecular profile of the tumour can be used at study entry to enrich the patient population for better outcome, which can help to establish the real value of a new drug or combination. The same principle holds true for trials focusing on preoperative (neoadjuvant) or postoperative (adjuvant or additive) use of targeted agents in this malignancy, where molecular studies can have major impact on trial outcome and interpretation of the study data.
A major limitation though is the global nonavailability of cross-validated methodology used for mutational analysis in GIST. It is an absolute requirement to overcome this issue by sharing biological material, co-analysing samples, comparing the results and establish common standards between reference laboratories. Such projects are considered within the framework of various academic and commercial groups, but at present without visible outcome.
In conclusion, the type of KIT or PDGFRA mutation appears to influence tumour development and response to current therapies and, therefore, might have profound power in assessing the prognosis of the disease. Currently, KIT/PDGFRA mutational analysis is mandatory for KIT-immunonegative GISTs and strongly recommended for the primary intermediate or high-risk tumours. With newly developed small molecule inhibitors or alternative drug strategies, mutational analysis may become indispensable for rationale and effective GIST treatment in the future.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andersson J, Bumming P, Meis-Kindblom JM, Sihto H, Nupponen N, Joensuu H, Odén A, Gustavsson B, Kindblom LG, Nilsson B (2006) Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology 130: 1573–1581
Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP (2005) Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 11: 4182–4190
Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, Maki NDS, DeMatteo RP, Besmer B (2004) Gene expression in gatrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res 10: 3282–3290
Bauer S, Yu LK, Demetri GD, Fletcher JA (2006) Heat-shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res 66: 9153–9161
Corless CL, Fletcher JA, Heinrich MC (2004) Biology of gastrointestinal stromal tumors. J Clin Oncol 22: 3813–3825
Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC (2005) PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 23: 5357–5364
Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, Hagemeijer A, Van Oosterom A, Marynen P (2005) Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 128: 270–279
Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, Zalcberg J, Verweij J, Van Glabbeke M, Hagemeijer A, Judson I (2006) KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 42: 1093–1103
Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G (2002) Clinical management of gastrointestinal tumors: before and after STI-571. Hum Pathol 33: 466–477
Demetri GD, Benjamin RS, Blanke CD, Blay JY, Casali P, Choi H, Corless CL, Debiec-Rychter M, DeMatteo RP, Ettinger DS, Fisher GA, Fletcher CD, Gronchi A, Hohenberger P, Hughes M, Joensuu H, Judson I, Le Cesne A, Maki RG, Morse M, Pappo AS, Pisters PW, Raut CP, Reichardt P, Tyler DS, Van den Abbeele AD, von Mehren M, Wayne JD, Zalcberg J, NCCN Task Force (2007) NCCN Task Force Report: management of patients with gastrointestinal stromal tumor (GIST)-update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw 5: S1–S2
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomized controlled trial. Lancet 368: 1329–1338
Fletcher CDM, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Learry TJ, Remotti H, Rubin BP, Shoookler B, Sobin LH, Weiss SW (2002) Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 33: 459–465
Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JVS, Booth BP, Verbois L, Morse DE, Liang CY, Chidambaram N, Jiang JX, Tang S, Mahjoob K, Justice R, Pazdur R (2007) Approval summary: sunitinib for the treatment of imatinib refactory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res 13: 1367–1373
Guo T, Agaram NP, Wong GC, Hom G, D'Adamo D, Maski R, Schwartz GK, Veach D, Clarkson BD, Singer S, DeMatteo RP, Besmer P, Antonescu CR (2007) Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumors. Clin Cancer Res 13: 4874–4881
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M, Fletcher CDM, Sandau K, McDougall K, Ou W, Chen C, Fletcher JA (2006a) Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 24: 1–11
Heinrich M, Maki RG, Corless C (2006b) Sunitinib (SU) response in imatinib-resistant (IM-R) GIST correlates with KIT and PDGFRA mutation status. J Clin Oncol 25(18S): 520s. Abstract 9502
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA (2003a) Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 21: 4342–4349
Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen C-J, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CDM, Fletcher JA (2003b) PDGFRA-activating mutations in gastrointestinal stromal tumors. Science 299: 708–710
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Masato H, Kurata A, Takeda M, Ghulam M, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y (1998) Gain-of-function of c-kit in human gastrointestinal tumors. Science 279: 577–580
Hornick JL, Fletcher CDM (2007) The role of KIT in the management of patients with gastrointestinal stromal tumors. Hum Pathol 38: 679–687
Joensuu H (2006) Gastrointestinal stromal tumor (GIST). Ann Oncol 17(Suppl 10): 280–286
Joensuu H, Fletcher C, Dimitrijevec S, Silberman S, Roberts P, Demetri G (2002) Management of malignant gastrointestinal stromal tumours. Lancet Oncol 3: 655–664
Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M (2004) A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest 84: 874–883
Lasota J, Stachura J, Miettinen M (2006) GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epitheloid morphology. Lab Invest 86: 94–100
Lasota J, Vel Dobosz AJ, Wasag B, Wozniak A, Kraszewska E, Michej W, Ptaszynski K, Rutkowski P, Sarlomo-Rikala M, Steigen SE, Schneider-Stock R, Stachura J, Chosia M, Ogun G, Ruka W, Siedlecki JA, Miettinen M (2007) Presence of homozygous KIT exon 11 mutations is strongly associated with malignant clinical behavior in gastrointestinal stromal tumors. Lab Invest 87: 1029–1041
Ma Y, Zeng S, Metcalfe DD, Akin C, Dimitrijevic S, Butterfield JH, McMahon G, Longley BJ (2002) The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profile than wild-type kinases and those with regulatory-type mutations. Blood 99: 1741–1744
Martin J, Poveda A, Llombart-Bosch A, López-Guerrero JA, García del Muro J, Maurel J, Calabuig S, Gutierrez A, González de Sande JL, Martínez J, De Juan A, Laínez N, Losa F, Alija V, Escudero P, Casado A, García P, Blanco R, Buesa JM (2005) Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish group for sarcoma research (GEIS). J Clin Oncol 23: 6190–6198
Miettinen M, Lasota J (2006) Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 23: 70–83
Nakagomi N, Hirota S (2007) Juxtamembrane-type c-kit gene mutation found in aggressive systemic mastocytosis induces imatinib-resistant constitutive KIT activation. Lab Invest 87: 365–371
Nilssen B, Bumming P, Meis-Kindblom JM, Oden A, Dortok A, Gustavsson B, Sablinska K, Kindblom L-G (2005) Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era. Cancer 103: 821–829
Prenen H, Cools J, Mentens N, Folens C, Sciot R, Schöffski P, Van Oosterom A, Marynen P, Debiec-Rychter M (2006) Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res 12: 2622–2627
Schittenhelm MM, Shiraga S, Schroeder A, Corbin AS, Griffith D, Lee FY, Bokemeyer C, Deininger MWN, Druker BJ, Heinrich MC (2006) Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res 66: 473–481
Singer S, Rubin BP, Lux ML, Chen CJ, Demetri GD, Fletcher CD, Fletcher JA (2002) Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol 20: 3898–3905
Steigen SE, Eide TJ, Wasag B, Lasota J, Miettinen M (2007) Mutations in gastrointestinal stromal tumors – a population-based study from Northern Norway. APMIS 115: 289–298
Van Glabbeke MM, Owzar K, Rankin C, Simes J, Crowley J (2007) Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors (GIST): a meta-analysis based on 1640 patients. J Clin Oncol 25(18S): 546s. Abstract 10004
von Mehren M (2006) Beyond Imatinib: second generation c-KIT inhibitors for the management of gastrointestinal stromal tumors. Clin Colorectal Cancer 6(Suppl 1): S30–S34
Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus H-U, Reichardt P, Hohenberger P (2006) Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res 12: 1743–1749
Weisberg E, Wright RD, Jiang J, Ray A, Moreno D, Manley PW, Fabro D, Hall-Meyers E, Catley L, Podar K, Kung AL, Griffin JD (2006) Effects of PKC412, nilotinib, and imatinib against GIST-associated PDGFRA mutants with differential imatinib sensitivity. Gastroenterology 131: 1734–1742
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hoeben, A., Schöffski, P. & Debiec-Rychter, M. Clinical implications of mutational analysis in gastrointestinal stromal tumours. Br J Cancer 98, 684–688 (2008). https://doi.org/10.1038/sj.bjc.6604217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604217
Keywords
This article is cited by
-
Potential indicators predict progress after surgical resection of gastrointestinal stromal tumors
Frontiers of Medicine (2012)
-
Cyclin H expression is increased in GIST with very-highrisk of malignancy
BMC Cancer (2010)
-
Evaluation of Self-Reported Progression and Correlation of Imatinib Dose to Survival in Patients with Metastatic Gastrointestinal Stromal Tumors: An Open Cohort Study
Journal of Gastrointestinal Cancer (2010)
-
Gastrointestinal stromal tumor of duodenum: a cause of upper gastrointestinal hemorrhage
The Chinese-German Journal of Clinical Oncology (2010)
-
The benefits of molecular pathology in the diagnosis of musculoskeletal disease
Skeletal Radiology (2010)
