
Abstract
In prostate carcinoma (PCa) increased DNA methylation (‘hypermethylation’) occurs at specific genes such as GSTP1. Nevertheless, overall methylation can be decreased (‘hypomethylation’) because methylation of repetitive sequences like LINE-1 retrotransposons is diminished. We analysed DNA from 113 PCa and 36 noncancerous prostate tissues for LINE-1 hypomethylation by a sensitive Southern technique and for hypermethylation at eight loci by methylation-specific PCR. Hypermethylation frequencies for GSTP1, RARB2, RASSF1A, and APC in carcinoma tissues were each >70%, strongly correlating with each other (P<10−6). Hypermethylation at each locus was significantly different between tumour and normal tissues (10−11<P<103), although hypermethylation, particularly of RASSF1A, was also observed in noncarcinoma tissues. ASC1 hypermethylation was observed in a subgroup of PCa with concurrent hypermethylation. Hypermethylation of CDH1, CDKN2A, and SFRP1 was rare. LINE-1 hypomethylation was detected in 49% PCa, all with hypermethylation at several loci. It correlated significantly with tumour stage, while hypermethylation was neither related to tumour stage nor Gleason score. Coordinate hypermethylation of several genes may occur early in PCa, with additional hypermethylation events and LINE-1 hypomethylation associated with progression. Hypermethylation allows detection of >82% of PCas. PCa may fall into three classes, that is, with few DNA methylation changes, with frequent hypermethylation, or with additional LINE-1 hypomethylation.
Similar content being viewed by others


Main
In many human tumours, two different types of alterations of DNA methylation are found, termed ‘hypermethylation’ and ‘hypomethylation’. At specific sites in the genome methylation is increased, typically in CpG islands around the transcriptional start sites of genes silenced in tumours (Baylin and Jones, 2002). Although hypermethylation occurs to some extent during ageing and in preneoplastic tissues, dense methylation of CpG islands is largely tumour-specific and can be exploited for tumour detection. Hypermethylation is frequent in prostate carcinoma (PCa). For instance, hypermethylation of the GSTP1 gene occurs in >70% of PCa and has been shown to be useful for its detection (Lee et al, 1994; Esteller et al, 1998; Millar et al, 1999; Santourlidis et al, 1999; Goessl et al, 2001; Jeronimo et al, 2001; Nakayama et al, 2004). Hypermethylation of this gene is established during the initial stages of PCa development (Brooks et al, 1998). Several other genes, such as APC, CDH1, CDKN2A, RASSF1A, and RARB2 (Graff et al, 1995; Jarrard et al, 1997; Nakayama et al, 2001; Liu et al, 2002; Maruyama et al, 2002; Yamanaka et al, 2003), have been reported to be hypermethylated in prostate cancer with various frequencies (reviewed in Schulz and Seifert, 2003). Further genes have been reported as hypermethylated in other human cancers, but have not been studied in PCa. They include the WNT signalling modulator gene SFRP1 (Suzuki et al, 2002) at 8p12, a common region of chromosome loss in PCa (Dong, 2001), and the putative apoptosis regulator gene ASC1/TMS1 (Conway et al, 2000) at 16p12.
An important question is how multiple hypermethylation events relate to tumour progression. One possibility is that genes become successively hypermethylated with carcinoma progression, resulting in an increase in the number of methylation changes during tumour development. A second possibility is that PCa may display patterns of methylation changes related to their biological behaviour, implicating that patterns of DNA methylation changes might be associated with different clinical courses. Two recent studies on PCa have yielded discrepant results on these issues (Maruyama et al, 2002; Yamanaka et al, 2003). A third possibility is that carcinomas fall into different subclasses, one of which is characterised by a high frequency of methylation alterations. A precedent is the CIMP+subclass of colon cancers (Toyota et al, 2000).
In spite of hypermethylation at specific sites, the overall methylcytosine content in human tumour cells is often decreased (‘global’ or ‘genome-wide’ hypomethylation) because methylation of repetitive sequences such as retroelements and CpG-rich satellites, which contain most of the methylcytosine in normal somatic cells, is diminished (Robertson, 2001; Baylin and Jones, 2002; Ehrlich, 2002). In particular, LINE-1 retrotransposons comprise 17% of the human genome and contain a disproportionate fraction of methylcytosine. The methylation status of LINE-1 sequences therefore provides a good indicator of global hypomethylation in tumour cells. Moreover, LINE-1 hypomethylation may lead to re-expression of individual elements and promote genomic instability (Florl et al, 1999; Schulz et al, 2002). In PCa, overall DNA methylation and LINE-1 methylation are decreased most consistently in metastatic cases (Bedford and van Helden, 1987). The precise relationship between specific hypermethylation events and global hypomethylation in PCa has not been studied.
Here, we have investigated 113 PCas for hypermethylation at eight loci by methylation-specific PCR (MS-PCR) and for LINE-1 methylation as an indicator of global hypomethylation by Southern blot hybridisation. The results suggest that with regard to DNA methylation alterations PCas may fall into different subclasses.
Materials and methods
Tissues
PCa specimens were obtained between 1993 and 2002, almost all by radical prostatectomy. Carcinoma and morphologically normal areas of the prostate were identified, and specimens collected by a pathologist, rapidly frozen in liquid nitrogen and stored at −80°C. Since several micrograms of high molecular weight DNA were required for LINE-1 hypomethylation analysis and multiple, repeated MS-PCR assays, no microdissection was performed. Immediately after surgical removal, prostates were sectioned by an experienced pathologist. Tumour and matched tumour-free specimens were only collected (i) when tumours were grossly apparent in the peripheral zone and could be unequivocally identified by their characteristic yellow or orange-yellow colour and (ii) when the transition zone was macroscopically free of tumour burden. Representative samples of 3 mm maximal diameter of tumour and tumour-free tissue specimens were collected, immediately snap frozen in liquid nitrogen and stored at −80°C. Noncancerous tissue samples were taken from areas of the transition zone as far away as possible from the grossly apparent tumour (i.e. in general from the transition zone of the contralateral lobe). Macroscopic separation between tumour and nontumorous tissues was histologically verified by analysing tissue specimens immediately adjacent to the specimens collected for methylation analysis. TNM classification was performed according to the guidelines of the International Union Against Cancer (UICC) from 1997. Of 113 PCa tissues, 48 carcinomas were staged as pT2, 59 as pT3, and five as pT4, one tumour was staged as pT1. Lymph node metastases were present in 21 patients, and distal metastases were detected in one patient. Four tumours had Gleason scores <5, 77 of 5–7, and 32 of >7. The patients' mean age was 66 years, ranging from 46 to 79 years. Overall, 36 carcinoma-free tissues were investigated, of which seven were obtained by adenomectomy or cystoprostatectomy for bladder cancer. The study was approved by the ethics committee of the Heinrich-Heine University medical faculty.
Cell lines
The bladder carcinoma cell line T24 was used as a positive control for methylation of CDH1, CDKN2A, and SFRP1. The PCa cell line LNCaP was used as a positive control for methylation of APC, ASC1, GSTP1, RARB2, and RASSF1A.
DNA extraction
High molecular weight genomic DNA from tissue, cell lines, and whole blood was isolated using the blood and cell culture DNA kit (QIAGEN, Hilden, Germany). Frozen tissues were crushed to a fine powder with a mortar and pestle before extraction.
Methylation-specific PCR
In all, 1 μg of DNA from each sample was bisulphite-treated using the CpGenome™ DNA Modification Kit (Oncor, Heidelberg, Germany) and 80 ng each were used in separate PCR reactions with primer pairs specific for methylated or unmethylated DNA (Supplementary Table). PCR reactions were performed in a volume of 50 μl containing 1.5 mM MgCl2, 150 μ M dNTPs, 0.6 μ M of each primer, and 1.25 U HotStar Taq polymerase (QIAGEN). The initial denaturing step at 95°C for 15 min was followed by 35–38 cycles each consisting of a denaturing step at 94°C for 30 s, primer annealing at 53–65°C (Supplementary Table) for 30 and a 45 s elongation step at 72°C. The final 72°C period was extended to 8 min. PCR products were separated on 2% agarose gels and visualised by ethidium bromide. Results were scored by two independent observers. All assays were repeated at least twice with independent bisulphite treatments and concordant results.
For bisulphite sequencing PCR products were subcloned into the PCR-4-TOPO vector (Invitrogen, Groningen, NL, USA) and several clones each were sequenced by standard methods.
LINE-1 hypomethylation analysis
Aliquots from the same DNA were employed to detect LINE-1 hypomethylation. In all, 1 μg of high molecular weight DNA each was extensively digested with the methylation-sensitive restriction enzyme HpaII or its methylation non-sensitive isoschizomer MspI, separated on agarose gels, blotted and hybridised with a 32P-labelled specific LINE-1 probe. After HpaII digestion, decreased methylation of LINE-1 sequences results in the appearance of new bands in the 1.0–4.0 kb range on Southern blots whose intensities relative to the MspI signals, after correction for unequal loading of the two lanes, can be used to quantitate hypomethylation. This method detects as little as 1% hypomethylation (Florl et al, 1999; Santourlidis et al, 1999). For categorisation, 0–4% hypomethylation was considered unchanged (0), 5–12% as moderate (1) and >12% as strong (2), slightly modified from a previous report (Schulz et al, 2002).
Mathematical procedures
Statistical analyses were performed using S-Plus 4.5. Professional, Release 2, MathSoft Inc., and MatLab, version 6.1.0.450 (R12.1), The Math Works, Inc. For CART analysis no weights were used. Hierarchical clustering was calculated based on Euclidean distance and average linkage.
Results
Hypermethylation was investigated at eight loci in 113 PCa specimens using published validated MS-PCR methods (Supplementary Table). Six of these loci had previously been reported to be hypermethylated in prostate cancers, whereas ASC1 and SFRP1 had been reported as frequently hypermethylated in other cancers, but not been studied in prostate cancer before. They were chosen to be located on different chromosomes (with the exception of RARB2 and RASSF1A), and to represent a wide range of established or presumed functions. In each MS-PCR assay controls were carried through the procedure, that is, DNA from a cell line with hypermethylation of the respective gene as a positive control and DNA from normal blood leukocytes as a negative control (Figure 1).

Methylation specific PCR. Representative examples of MS-PCR analyses for the methylated (M) and the unmethylated (U) forms of eight genes in prostate cancer. Selected tumour samples with decreasing frequency of hypermethylation events from left to right. The bladder cancer cell line T24 was used as a positive control for CDH1, CDKN2A, and SFRP1 hypermethylation. The prostate carcinoma cell line LNCaP was used for ASC1, APC, GSTP1, RARB2, and RASSF1A hypermethylation (M control). Leukocyte DNA from a female without cancer was used as a negative control (U control).
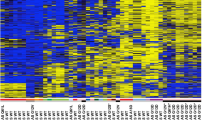
The eight loci fell into three groups with regard to hypermethylation in PCa tissues. First, for APC, GSTP1, RARB2, and RASSF1A hypermethylation was frequent and found in 78, 79, 70, and 78% of the cases, respectively. Methylation at each of the four loci strongly correlated highly significantly with that at any of the other loci. Thus, 68 (60%) PCa specimens exhibited methylation at all four loci, while in 10 (9%) cases none of the loci was hypermethylated (Figure 2A). In addition, 10 PCa specimens displayed methylation in only one gene, most often in RASSF1A, followed by APC. This distribution is highly unlikely to arise by chance (P<10−6) and suggests a coordinate process. Secondly hypermethylation of CDH1, CDKN2A, and SFRP1, was rare in PCa tissues (<5% of the specimens), although they were hypermethylated in cancer cell line controls (Figure 1). This result was not due to lack of sensitivity, since dilution experiments with T24 cell line DNA showed that the MS-PCR method detected ≈1% hypermethylated alleles for each gene (data not shown). Moreover, the data for SFRP1 were confirmed by bisulphite sequencing in several positive and negative samples (data not shown). Thirdly, hypermethylation of ASC1 was found in 40% of PCa. Interestingly, these also displayed hypermethylation in at least three of the genes APC, GSTP1, RARB2, and RASSF1A. Thus, the PCa specimens with ASC1 hypermethylation formed a precise subgroup of those with concomitant hypermethylation of these four genes (Figure 2C).

Hypermethylation in prostate cancer and noncancerous tissues. (A) Number of cases with hypermethylation detected in the indicated number of genes (APC, GSTP1, RARB2, and RASSF1A) in PCa tissues. (B) Number of cases with hypermethylation detected in the indicated number of genes (APC, GSTP1, RARB2, and RASSF1A) in prostate tissues morphologically free of carcinoma. (C) Cluster analysis of APC, ASC1, GSTP1, RARB2, RASSF1, and SFRP1 hypermethylation in PCa tissues. Black: hypermethylated; grey: unmethylated. LINE-1 hypomethylation is also shown. Black: hypomethylation >4%; grey: normal methylation.
Many noncarcinoma prostate specimens were free of hypermethylation, but some yielded signals in MS-PCR analyses. In these, the most frequently hypermethylated gene was RASSF1A (53%), followed by APC (36%), while hypermethylation of GSTP1 (19%) and RARB2 (17%) was infrequent. This is the same order as in tumour tissues with hypermethylation of only a single gene. Importantly, concomitant hypermethylation of several genes was much rarer in noncarcinoma than in carcinoma tissues (Figure 2B).
Since RASSF1A was most frequently methylated in noncarcinoma tissues, its methylation pattern was compared between several normal and tumour specimens by bisulphite sequencing (Figure 3). In tumour tissues (T#232 and T#139), individual RASSF1A alleles were often continuously methylated. In the corresponding normal tissues (N#233 and N#140), methylation of individual alleles was less dense. Tumour tissues reacting weakly positive in MS-PCR (T#133 and T#137) showed patchy methylation and methylation was rare in the corresponding normal tissues (N#134 and N#138). One prostate sample without carcinoma from an adenomectomy for benign hyperplasia and one from a cystoprostatectomy for bladder cancer were devoid of methylation (N#234, N#235). A further carcinoma-free prostate specimen (N#229) obtained by cystoprostatectomy for bladder cancer showed patchy methylation in RASSF1A.

Bisulphite methylation analysis of RASSF1A. Methylation status of the RASSF1A promoter CpG island in paired prostate carcinoma (T) and adjacent normal (N) tissue samples (#232/233, #133/134, #137/138, #139/140) as well as carcinoma-free prostate specimens obtained during adenomectomy for benign prostate hyperplasia (AD, #234) or cystoprostatectomy for bladder cancer (Cy, #229 and #235). Each circle depicts an individual CpG site. Black: methylated site, white: unmethylated site. At the top of the figure, the localisation of the 22 investigated sites in the RASSF1A promoter are indicated. Several potential binding sites for Sp1 are marked by asterisks.
Accordingly, among the four frequently hypermethylated genes, hypermethylation of RASSF1A discriminated least well between normal and tumour tissues. However, even for this gene the frequency of hypermethylation was significantly different between carcinoma and noncarcinoma tissues (p(χ2)=10−3). The significance levels for other loci were 3 × 10−5 (APC), 1.2 × 10−6 (GSTP1), and 1.2 × 10−10 (RARB2). In a CART analysis, optimal separation between tumour and normal tissues was achieved using RARB2 plus GSTP1 hypermethylation. This procedure classified 82% of all cancers correctly, while inclusion of further loci improved detection marginally to 83% (i.e. by one case). The specificity of the combination RARB2 plus GSTP1 hypermethylation was 83%, which was identical to that of RARB2 hypermethylation alone, similar to that of GSTP1 (81%), and considerably higher than those of APC (61%) and RASSF1A (47%).
Hypomethylation of LINE-1 sequences (Figure 4) was observed in 49% of PCa (Figure 5A). In 23 specimens pronounced hypomethylation (defined as >12%) was observed, while 33 displayed moderate hypomethylation (defined as 5–12%). LINE-1 hypomethylation correlated with hypermethylation of each of the genes RARB2, RASSF1A, GSTP1, and APC (1.7 × 10−6 <p(χ2) <3.2 × 10−5). Nevertheless, hypermethylation and hypomethylation were not entirely concordant, because many cases with hypermethylation in three or four genes lacked LINE-1 hypomethylation (Figure 5A). Thus, like ASC1 hypermethylation, LINE-1 hypomethylation was essentially restricted to a subgroup of the cases with concomitant hypermethylation of the four genes (Figure 2C). ASC1 hypermethylation and LINE-1 hypomethylation correlated closely with each other (p(χ2)=6.0 × 10−4), but the subgroups were not identical. These relationships are illustrated by principal component analysis (Figure 5B), which yielded almost identical vectors for hypermethylation of each of the four genes, but separation of the vectors representing ASC1 hypermethylation in the first component and LINE-1 hypomethylation in the second component. No significant LINE-1 hypomethylation was seen in any noncancer prostate specimen analysed (Figure 4).

Southern blot analysis of LINE-1 hypomethylation in prostate and prostate carcinoma tissues. DNA isolated from prostates carrying no carcinoma (left), carrying carcinoma (center), and from prostate carcinomas (right) was analysed for LINE-1 hypomethylation as described in the Materials and Methods section. H: HpaII digestion; M: MspI digestion. Hypomethylation is detectable by the appearance of bands in the 1.4–3.5 kb range in the H lane. Results of densitometric quantification (see Materials and Methods for details) are depicted at the bottom of the figure.

Relationship of LINE-1 hypomethylation to gene hypermethylation in prostate carcinoma samples. (A) Number of cases with hypermethylation detected in the indicated number of genes in prostate carcinoma tissues (APC, GSTP1, RARB2, and RASSF1A) and lack of hypomethylation (black bars), moderate hypomethylation (grey bars), and pronounced hypomethylation (striped bars). (B) Principal component analysis of the tumour methylation data set for the factors LINE-1 hypomethylation (L1-HYPO) as well as APC, ASC1, GSTP1, RARB2, RASSF1A, and SFRP1 hypermethylation. Plot of the first two components from principal component analysis. The first component explains 66.2% of the variance, the second 26.6%. Asterisks represent the factors, while arrows demonstrate the direction from the origin.
None of the more frequent hypermethylation events, including ASC1 hypermethylation, were significantly related to tumour stage or Gleason score. All six cases with SFRP1 hypermethylation were staged as pT2 and this rare change was therefore significantly related to tumour stage (P=0.02). The number of hypermethylation events was unrelated to clinical parameters (Table 1). LINE-1 hypomethylation was significantly more frequent in higher stage carcinomas (P=0.04) and tended to be more pronounced in lymph node-positive PCa (P=0.08).
Discussion
Hypermethylation of GSTP1 is a promising marker for detection of PCa (Lee et al, 1994; Esteller et al, 1998; Millar et al, 1999; Santourlidis et al, 1999; Goessl et al, 2001; Jeronimo et al, 2001). Different studies have reported somewhat different proportions of positive cancers, likely due to differences in methodology as well as to differences between the patient populations investigated. Here, using a sensitive MS-PCR method, we found 70% of carcinoma tissues from a Central European population to display GSTP1 hypermethylation with rare methylation in adjacent normal tissues, supporting its value as a marker. In the same specimens, three further genes, that is APC, RARB2, and RASSF1A, proved to be hypermethylated at similarly high frequencies, and also discriminated well between tumour and normal tissue. A combination of several genes may be optimal for discrimination and could be used as described here or in combination with quantitative methylation analyses (Jeronimo et al, 2001). In our data set, optimal separation between carcinoma and normal tissues was achieved by GSTP1 and RARB2 hypermethylation, identifying >80% tumour samples correctly. The specificity of detection by this combination was 83% and thus not exceedingly high. However, as the noncarcinoma specimens were mostly taken from prostates that harboured cancers, this level of specificity is likely an underestimate (see discussion below).
The fact that two of these four genes are sufficient to detect all carcinomas with hypermethylation follows from the strong correlation between hypermethylation of these individual genes. If each hypermethylation event occurred independently at the observed frequencies (70–79%), most carcinomas should display hypermethylation in two or three genes and <1/113 should lack hypermethylation. Therefore, we observed more tumours with simultaneous hypermethylation of all four genes as well as more tumours without hypermethylation at any of the loci than expected from a stochastic process (cf Figure 2). This suggests that hypermethylation occurs in a coordinate manner in most cases and, conversely, that a smaller subset of PCas is not prone to hypermethylation. It may be difficult to distinguish PCa in this subset reliably from normal or ageing prostate by DNA methylation analysis. There is so far little indication for major differences in clinical course between the hypermethylation-prone and rare-hypermethylation groups (Supplementary Table). This fits the emerging consensus that deregulation of DNA methylation can be as effective at driving tumour development and progression as chromosomal instability or base repair deficiency (Baylin and Jones, 2002).
The strongly coordinated hypermethylation of the above four genes is remarkable in view of the low hypermethylation rate detected in three other genes that are often hypermethylated in other cancers. Our data on CDKN2A/p16 are in accordance with other reports on a low percentage of hypermethylation of this gene in PCa (Jarrard et al, 1997; Maruyama et al, 2002). The reported frequencies of CDH1 hypermethylation in PCa differ widely between published studies (Graff et al, 1995; Woodson et al, 2003). Detailed analyses have shown hypermethylation patterns in CDH1 to be very variable (Graff et al, 2000), which may partly explain these differences. At any rate, such a variability would preclude a use in diagnostics. Hypermethylation of SFRP1 was found in other cancers (Suzuki et al, 2002), but had not been studied before in PCa. This gene was of particular interest because of its location in a chromosomal region with a high frequency of allelic loss in PCa (Dong, 2001). In this regard, our data indicate that the gene may become inactivated by hypermethylation in individual cases of PCa. Our data suggest a more significant involvement of ASC1. This gene has previously been identified as being silenced by hypermethylation in breast carcinoma and is thought to be involved in the regulation of apoptosis (Conway et al, 2000). Its precise role in PCa certainly deserves further investigation.
The comparison of the two groups of genes – APC, GSTP1, RARB2, and RASSF1A vs CDH1, CDKN2A, and SFRP1 – suggests no obvious explanation why they should become hypermethylated at such different frequencies. Specifically, GSTP1, RARB2, APC, and RASSF1A are not known to function in a common pathway or – with the exception of RARB2 and RASSF1A – reside on the same chromosome. Our data call for a mechanistic explanation of this unexpected specificity.
In the course of PCa, the origin of GSTP1 hypermethylation has been traced to late preneoplastic stages like high-grade prostatic intraepithelial neoplasia (Brooks et al, 1998; Yamanaka et al, 2003) and proliferative inflammatory atrophy lesions (Nakayama et al, 2003; Yamanaka et al, 2003). If GSTP1 hypermethylation is associated with the onset of prostate carcinogenesis, it cannot be related to tumour stage or Gleason score, as observed here and by others (Yamanaka et al, 2003). Furthermore, because GSTP1 hypermethylation correlated strongly with that of three other genes, their hypermethylation may also occur at early stages of cancer development. Moreover, our data suggest that hypermethylation of RASSF1A might begin in preneoplastic tissue. Hypermethylation of RASSF1A was most frequently found in normal tissue from carcinoma-carrying prostates, and RASSF1A was the locus most often hypermethylated without hypermethylation at other loci. In addition, RASSF1A methylation on individual alleles was denser in carcinoma than in normal prostate tissue (Figure 3). This observation is very reminiscent of findings in the colon. There, some genes are only hypermethylated in colon carcinoma, whereas others start to become hypermethylated in ageing and preneoplastic colon. The patchy methylation in ageing tissue becomes intensified in tumour cells (Issa, 2000). Our data suggest that RASSF1A may belong to this class in prostate tissue.
The findings on RASSF1A raise the question of how to interpret hypermethylation events observed in normal-appearing prostate tissue. In some cases, hypermethylation might be derived from minimal carcinoma infiltrates not detected on adjacent histological sections, particularly, in the few cases where hypermethylation of several genes was observed (Figure 2B). However, there were several differences between hypermethylation detected in normal and carcinoma tissues. First, the order of the frequencies for different genes was different. GSTP1, RARB2, APC, and RASSF1A were each hypermethylated in 70–80% of carcinoma tissues, whereas the order in noncarcinoma tissues was RASSF1A>APC>GSTP1=RARB2. Secondly, concomitant hypermethylation was rare in morphologically normal tissues, but was the rule in carcinomas (Figure 2). Thirdly, the methylation pattern in RASSF1A was always patchy in non-neoplastic tissues, whereas dense methylation was typically seen in carcinomas (Figure 3). Taken together, these results might argue for age-associated hypermethylation in the prostate like that reported for other organs (Issa, 2000). Hypermethylation in the prostate might help to establish a preneoplastic state that disposes to carcinoma development. Indeed, if the silencing of GSTP1 by hypermethylation was to promote the initial development of PCa by inducing an increased sensitivity to electrophilic carcinogens, as postulated by others (Nakayama et al, 2004), one would have to expect methylation changes to actually precede the development of morphological changes.
Our finding of a lack of correlation between DNA hypermethylation and tumour stage and Gleason grade in the prostate is in good agreement with a study of Japanese patients (Yamanaka et al, 2003), but differs from the results in an American cohort (Maruyama et al, 2002) in which the number of methylated genes increased with tumour stage and Gleason score. The reasons for these differences may thus be population-related, or may reflect methodical differences. For instance, the study reporting a good correlation of hypermethylation with stage and grade detected GSTP1 hypermethylation in only 36% of PCa tissues, which is by far the lowest rate reported in the literature.
In our hands, LINE-1 hypomethylation represents the methylation change best related to histopathological parameters of PCa. In previous studies, global DNA hypomethylation was found to be particularly pronounced in specimens from androgen-refractory carcinomas (Bedford and van Helden, 1987; Schulz et al, 2002). Accordingly, the present study showed LINE-1 hypomethylation to be more prevalent in high stage and lymph-node positive PCas. LINE-1 hypomethylation obviously characterises a set of PCas within the subclass of those with frequent hypermethylation (Figures 2C and 4) and is associated with higher stage disease. The most straightforward explanation for this finding is that LINE-1 hypomethylation in PCa is a secondary event during progression following DNA hypermethylation. As a corollary, the mechanisms causing DNA hypermethylation at specific sites and DNA hypomethylation of repetitive sequences are likely distinct, as also reported for Wilms tumours (Ehrlich et al, 2002). Another change occurring during progression may be ASC1 hypermethylation, which was also found in a precise subgroup of the hypermethylation-prone cancers, but apparently not the same one exhibiting LINE-1 hypomethylation. However, no significant relationship to stage or grade was found for this change.
Our study therefore suggests a distinction of three subclasses of PCa, one with few DNA methylation changes, one with hypermethylation of several genes, and one with additional hypomethylation of repetitive sequences. This last subclass may be over-represented among high-stage PCa (P=0.086 in the present study according to Kruskal–Wallis test). The distinction between these three subclasses might be useful for further development of methylation-based diagnosis of PCa.
Finally, regarding the functional implications of our findings, hypermethylation of promoter sequences is established as a mechanism of silencing of tumour suppressor genes in human cancers (Baylin and Jones, 2002). The GSTP1 gene is frequently hypermethylated in prostate cancers, but is a less than ideal candidate for a tumour suppressor (reviewed by Nakayama et al, 2004). Its loss of function may lead to increased sensitivity against certain electrophilic compounds and perhaps decreased apoptosis, which could promote carcinogenesis in the prostate. The finding in the present study that hypermethylation of GSTP1 occurs in a coordinate manner with that of other genes raises the interesting possibility that GSTP1 hypermethylation might be a bystander effect of the inactivation of a gene more directly involved. Specifically, all three other genes found to be coordinately hypermethylated with GSTP1 in this study, APC, RARB2, and RASSF1A, are established as functionally important in other cancers. Regarding the functional implications of hypomethylation, it has previously been reported to be almost ubiquitous in metastatic PCa (Bedford and van Helden, 1987, Schulz et al 2002), in accord with a significant correlation of LINE-1 hypomethylation with tumour stage in the present study. Moreover, LINE-1 hypomethylation was found to be significantly correlated with chromosomal instability (Schulz et al, 2002). It is not known, however, whether genome-wide hypomethylation causes chromosomal instability, although there is some experimental support for this idea (Eden et al, 2003).
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Baylin SB, Jones PA (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428
Bedford MT, van Helden PD (1987) Hypomethylation of DNA in pathological conditions of the human prostate. Cancer Res 47: 5274–5276
Brooks JD, Weinstein M, Lin X, Sun Y, Pin SS, Bova GS, Epstein JI, Isaacs WB, Nelson WG (1998) CG island methylation changes near the GSTP1 gene in prostatic intraepithelial neoplasia. Cancer Epidemiol Biomarkers Prev 7: 531–536
Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM (2000) TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res 60: 6236–6242
Dong JT (2001) Chromosomal deletions and tumor suppressor genes in prostate cancer. Cancer Metast Rev 20: 173–193
Eden A, Gaudet F, Waghmare A, Jaenisch R (2003) Chromosomal instability and tumors promoted by DNA hypomethylation. Science 300: 455
Ehrlich M (2002) DNA methylation in cancer: too much, but also too little. Oncogene 21: 5400–5413
Ehrlich M, Jiang G, Fiala E, Dome JS, Yu MC, Long TI, Youn B, Sohn OS, Widschwendter M, Tomlinson GE, Chintagumpala M, Champagne M, Parham D, Liang G, Malik K, Laird PW (2002) Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene 21: 6694–6702
Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG (1998) Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res 58: 4515–4518
Florl AR, Loewer R, Schmitz-Dräger BJ, Schulz WA (1999) DNA methylation and expression of L1 LINE and HERV-K provirus sequences in urothelial and renal cell carcinoma. Br J Cancer 80: 1312–1321
Goessl C, Muller M, Heicappell R, Krause H, Straub B, Schrader M, Miller K (2001) DNA-based detection of prostate cancer in urine after prostatic massage. Urology 58: 335–338
Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JR (2000) Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem 275: 2727–2732
Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, Baylin SB (1995) E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 44: 5195–5199
Issa JP (2000) CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol 249: 101–118
Jarrard DF, Bova GS, Ewing CM, Pin SS, Nguyen SH, Baylin SB, Cairns P, Sidransky D, Herman JG, Isaacs WB (1997) Deletional, mutational, and methylation analyses of CDKN2 (p16/MTS1) in primary and metastatic prostate cancer. Genes Chromosomes Cancer 19: 90–96
Jeronimo C, Usadel H, Henrique R, Oliveira J, Lopes C, Nelson WG, Sidransky D (2001) Quantitation of GSTP1 methylation in non-neoplastic prostatic tissue and organ-confined prostate adenocarcinoma. J Natl Cancer Inst 93: 1747–1752
Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG (1994) Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci USA 91: 11733–11737
Liu L, Yoon JH, Dammann R, Pfeifer GP (2002) Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene 21: 6835–6840
Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zochbauer-Muller S, Farinas AJ, Minna JD, McConnell J, Frenkel EP, Gazdar AF (2002) Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res 8: 514–519
Millar DS, Ow KK, Paul CL, Russell PJ, Molloy PL, Clark SJ (1999) Detailed methylation analysis of the glutathione S-transferase pi (GSTP1) gene in prostate cancer. Oncogene 18: 1313–1324
Nakayama M, Bennett CJ, Hicks JL, Epstein JI, Platz EA, Nelson WG, de Marzo AM (2003) Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture microdissection. Am J Pathol 163: 923–933
Nakayama M, Gonzalgo ML, Yegnasubramanian S, Lin X, de Marzo AM, Nelson WG (2004) J Cell Biochem 91: 540–552
Nakayama T, Watanabe M, Yamanaka M, Hirokawa Y, Suzuki H, Ito H, Yatani R, Shiraishi T (2001) The role of epigenetic modifications in retinoic acid receptor beta2 gene expression in human prostate cancer. Lab Invest 81: 1049–1057
Robertson KD (2001) DNA methylation, methyltransferases, and cancer. Oncogene 20: 3139–3155
Santourlidis S, Florl AR, Ackermann R, Wirtz HC, Schulz WA (1999) High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate 39: 166–174
Schulz WA, Elo JP, Florl AR, Pennanen S, Santourlidis S, Engers R, Burchardt M, Seifert H-H, Visakorpi T (2002) Genome-wide DNA hypomethylation is associated with alterations on chromosome 8 in prostate carcinoma. Genes Chromosomes Cancer 35: 58–65
Schulz WA, Seifert H-H (2003) DNA methylation in urological cancers. In Szyf M (ed) DNA methylation and cancer therapy, Landes Biosciences, http://www.landesbioscience.com/
Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, Herman JG, Baylin SB (2002) A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 31: 141–149
Toyota M, Ohe-Toyota M, Ahuja N, Issa JP (2000) Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA 97: 710–715
Woodson K, Hayes R, Wideroff L, Villaruz L, Tangrea J (2003) Hypermethylation of GSTP1, CD44, and E-cadherin genes in prostate cancer among US Blacks and Whites. Prostate 55: 199–205
Yamanaka M, Watanabe M, Yamada Y, Takagi A, Murata T, Takahashi H, Suzuki H, Ito H, Tsukino H, Katoh T, Sugimura Y, Shiraishi T (2003) Altered methylation of multiple genes in carcinogenesis of the prostate. Int J Cancer 106: 382–387
Acknowledgements
Helpful discussions with Dr MV Cronauer are gratefully acknowledged. The study was financially supported by the Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary information accompanies the paper on British Journal of Cancer website (http://www.nature.com/bjc).
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Florl, A., Steinhoff, C., Müller, M. et al. Coordinate hypermethylation at specific genes in prostate carcinoma precedes LINE-1 hypomethylation. Br J Cancer 91, 985–994 (2004). https://doi.org/10.1038/sj.bjc.6602030
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6602030
Keywords
This article is cited by
-
DNA methylation profiles of bronchoscopic biopsies for the diagnosis of lung cancer
Clinical Epigenetics (2021)
-
Genome-wide tracts of homozygosity and exome analyses reveal repetitive elements with Barrets esophagus/esophageal adenocarcinoma risk
BMC Bioinformatics (2019)
-
Epigenetic modulators as therapeutic targets in prostate cancer
Clinical Epigenetics (2016)
-
Inter-locus as well as intra-locus heterogeneity in LINE-1 promoter methylation in common human cancers suggests selective demethylation pressure at specific CpGs
Clinical Epigenetics (2015)
-
Global DNA hypomethylation in prostate cancer development and progression: a systematic review
Prostate Cancer and Prostatic Diseases (2015)
