
Abstract
Background:
Although inhibition of SGK1 has been shown to delay cancer progression, the underlying mechanisms have not yet been elucidated.
Methods:
We investigated the cellular responses to GSK650394 treatment and SGK1 silencing (or overexpression) in human prostate cancer (PCa) cell lines and PC3 xenografts by flow cytometry, western blotting, immunofluorescence, transmission electron microscopy and immunohistochemistry.
Results:
In the present study, we demonstrated that SGK1 inhibition, mediated by either GSK650394 or SGK1 shRNA, induced G2/M arrest, apoptosis and autophagy. Furthermore, 3MA-mediated autophagy inhibition attenuated SGK1 inhibition-induced apoptosis, suggesting that induction of autophagy precedes apoptosis. Moreover, ectopic expression of SGK1 significantly attenuated the GSK650394-induced effects. Suppression of mTOR and Foxo3a phosphorylation is critical for blockade of SGK1-induced autophagy and apoptosis, at least partially via pFoxo3a (S253)-LC3 and pFoxo3a (S253)-p27 interactions. Dual inhibition of mTOR and SGK1 enhances autophagy activation and leads to synergistic cytocidal effects in PCa cells.
Conclusions:
In summary, our findings show that SGK1 inhibition exhibits significant antitumour effects against PCa in vitro and in vivo. This study uncovered a novel mechanism of SGK1 inhibition in PCa, which is mediated, at least in part, by inducing autophagy-dependent apoptosis via the mTOR-Foxo3a pathway.
Similar content being viewed by others

Main
Prostate cancer (PCa) remains the most common urological malignancy and the second leading cause of male cancer-related deaths in many developed countries (Siegel et al, 2016). Although the improvements in PCa diagnostic strategies and in multiple treatments have led to a significant decrease in PCa-related deaths in the past three decades, most men initially diagnosed with early-stage cancer eventually develop metastatic disease (Gundem et al, 2015). Thus, at this point, it remains practically incurable, and the aims of therapy are the improvement of overall survival time and quality of life. It is important to develop novel therapeutic strategies in order to suppress tumour progression and restore sensitivity to therapy.
Serum and glucocorticoid-induced protein kinase 1 (SGK1) is a member of the ‘AGC’ subfamily of protein kinases, including protein kinases A, G and C. Particularly, SGK1 shares ∼54% identity of its catalytic domain with protein kinase B (PKB, also called Akt), indicating analogous functions (Kobayashi et al, 1999). SGK1 is ubiquitously expressed, and it covers a wide variety of physiological functions (e.g., transport, hormone release, neuroexcitability, cell proliferation and apoptosis) and contributes to a multitude of pathophysiological conditions (e.g., tumour growth, neurodegeneration, fibrosing disease and the sequelae of ischaemia). Most notably, extensive research has shown that SGK1 is closely related to the development and progression of multiple types of tumours (Lang et al, 2006). Increased expression of SGK1 has been observed in several tumours, including gastric cancer (Yao et al, 2016), ovarian cancer (Melhem et al, 2009) and PCa (Szmulewitz et al, 2012). Particularly, SGK1-overexpressing PCa xenografts displayed accelerated castrate-resistant tumour initiation, supporting a role for SGK1-mediated CRPC progression (Isikbay et al, 2014). However, the underlying mechanism is not fully understood.
In this study, our data showed that SGK1 inhibition, mediated by either GSK650394 or SGK1 shRNA, induced G2/M arrest and apoptosis. Furthermore, the most interesting finding was a novel response to GSK650394 in human PCa cells involving the formation of acidic vesicular organelles, and autophagosomes were observed. Given the dynamic role of autophagy in tumourigenesis and that most cancer therapies induce autophagy, adjuvant autophagy inhibition has become an area of research interest (Vyas et al, 2013; Nguyen et al, 2014). Hence, we initially hypothesised that GSK650394-induced autophagy is a cytoprotective response that PCa cells used to evade the antiproliferative effects of GSK650394. In contrast to our initial hypothesis, our further results indicated that GSK650394-induced autophagy was cytotoxic, as evidenced by the restoration of cell viability in GSK650394-treated PCa cells after treatment with autophagy inhibitors. However, an understanding of the mechanism by which SGK1 modulates apoptosis and autophagy is pivotal for its further development as a clinically useful therapeutic target. Furthermore, this knowledge could shed new light on the molecular mechanisms underlying the development and progression of PCa and contribute in developing more effective therapies in future clinical trials.
Materials and methods
Cell lines and cell culture
PC3, LNCaP, DU145 and CWR22-Rv1 PCa cell lines were purchased from American Type Culture Collection (Manassas, VA, USA), cells were cultured in RPMI-1640 media supplemented with 10% foetal bovine serum in 37 °C and 5% CO2 atmosphere.
Reagents and antibodies
GSK650394, chloroquine (CQ), 3-methyladenine (3MA), broad-spectrum caspase inhibitor (z-VAD-fmk) and rapamycin were purchased from Selleck (Shanghai, China). Acridine orange (AO), monodansylcadervarine (MDC), DAPI (4’6-diamid ino-2-phe nylindole) and Hoechst 33342 were obtained from Beyotime (Haimen, China). Antibodies against Bax>Bcl-2, Bim, caspase-3, caspase-8, caspase-9, poly (ADPribose) polymerase (PARP), Fas, FasL, LC3B, p27, p21, mTOR, pmTOR, pSGK1 (S78), Foxo3a, pFoxo3a, p53, Beclin 1, GRP78 and GAPDH were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against SGK1, pSGK1 (S422) and p62 were purchased from Abcam (Cambridge, MA, USA). Secondary antibodies (HRP-conjugated sheep anti-rabbit antibodies or HRP-conjugated sheep anti-mouse antibodies) for western blot were obtained from Millipore, Billerica, MA, USA. DyLight 488 AffiniPure Goat Anti-Rabbit IgG (H+L) for immunofluorescence were obtained from Abbkine (Redlands, CA, USA).
Immunohistochemistry and evaluation of immunostaining
Briefly, 4-μm-thick sections mounted on glass slides were processed for immunohistochemistry (IHC). All slides were dewaxed in xylene and dehydrated in an alcohol gradient, and then, endogenous peroxidase activity was quenched with 3% hydrogen peroxide for 10 min. Antigen retrieval was achieved by heating slides covered with citrate buffer (pH=6.0) at 95 °C for 10 min. Then, 10% goat serum albumin was used to block nonspecific binding by incubating sections for 2 h at room temperature while gently tilting the sections without washing them, followed by incubation with SGK1, Foxo3a, p-Foxo3a, mTOR, pmTOR and LC3B antibodies at 4 °C overnight in a moist chamber. After being washed three times with phosphate-buffered saline (PBS), the sections were incubated with a secondary antibody at room temperature for 1 h and rinsed in PBS. Diaminobenzidine (DAB) was used as a chromogen, and sections were counterstained with haematoxylin. Negative controls were obtained by incubating specimens with PBS instead of primary antibody. Brown particles present in the cytoplasm and/or nuclei were considered positive signals.
The CCK-8 assay
Cell suspension of 100 μl was dispensed (4–6 × 103 cells/well) in 96-well plates. The plates were pre-incubated for 24 h, followed by the treatments of either DMSO (control) or various concentrations of GSK650394 for 24, 48 and 72 h. Cell proliferation was measured using a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Kumamoto, Japan) following the manufacturer’s instructions. The plate was incubated for 2 h in the incubator. The absorbance was measured at 450 nm using a micro-plate spectrophotometer (BIO-RAD xMark, Hercules, CA, USA).
Vital staining with AO or MDC
PCa cells were seeded in a 24-well culture plate and after subsequent treatments, cells were stained with AO (1 μg ml−1) or MDC (0.05 mmol l−1) directly for 10 min at 37 °C, and then observed under the fluorescence microscope (Olympus, Tokyo, Japan).
Transmission electron microscopy
Briefly, cells were harvested after treatment with DMSO (control) or GSK650394, fixed with ice-cold 2.5% glutaraldehyde in PBS (pH 7.3) for 2 h, post-fixed in 1% osmium tetroxide, dehydrated in a graded series of ethanol (50–100%) and acetone, and embedded in Epon. Representative areas were chosen for ultrathin sectioning and examined with a transmission electron microscope (TEM).
Cell cycle analysis
Briefly, PCa cells were seeded at a density of 5 × 105 cells per well in a six-well culture plate. The following day the medium was replaced with serum-free medium, 24 h later cells were stimulated to growth by adding 10% serum, after subsequent treatments, PCa cells were collected and fixed with 70% ethanol in PBS at −20 °C overnight. Cell cycle analysis was performed using PI/RNase staining buffer (BD Biosciences, San Diego, CA, USA) according to the manufacturer’s specification, and cell cycle distribution was analysed by flow cytometry and ModFit LT software (FACSCalibur, BD, San Jose, CA, USA).
Detection of cell death
Apoptosis induction was quantified using an Annexin V-PE/7-AAD kit (BD Biosciences) according to the manufacturer’s instructions. Briefly, PCa cells were seeded at a density of 5 × 105 cells per well in a six-well culture plate, and after subsequent treatments, cells were harvested following incubation with PE-conjugated annexin V and 7-AAD for 15 min at room temperature in the dark. Then, cells were washed with ice-cold PBS and resuspended with 300 μl of PBS. Samples were analysed with a flow cytometer (FACSCalibur) and CELLQuest software (FACSCalibur). In addition, following treatment with the indicated concentrations of GSK650394, apoptotic nuclei were detected by DAPI (4’6-diamidino-2-phenylindole) or Hoechst 33342 (Beyotime) staining.
Clone formation assay
Cells were seeded at 500 cells/well in six-well plates and pipetted several times, so that most cells were in single-cell forms. After 3 days of incubation, the cells were treated with DMSO (vehicle control) or GSK650394 at various concentrations (2.5–10 μ M), and the covering medium was changed every 2 or 3 days during culture. After 10–14 days, when the cells grew to visible colonies, the medium was removed, and the cells were washed with PBS twice. After being fixed with 4% paraformaldehyde for 15 min and washed with PBS, the cells were stained with 0.1% crystal violet for 10 min. Finally, the excess dye was washed away with PBS, and then, cells were visualised using an ordinary optical microscope (with × 4 objective). The plates were prepared in triplicate, and 10 fields for each well were chosen randomly. Then, the number of colonies in each field was counted, only colonies containing more than approximately 50 cells were counted, and the size of the colonies was measured.
Immunofluorescence
Cells were grown on glass coverslips, so that they reached 30–50% confluence by the day of the experiment. Then, they were treated with GSK650394 for 6, 24 and 48 h, washed twice with PBS, and fixed for 10 min in 4% paraformaldehyde. Cells were permeabilised in 0.5% Triton X-100, washed, and blocked for 2 h in 5% normal goat serum. After blocking, the cells were incubated with primary antibody (1 : 200) for 24 h at 4 °C, washed with PBS and incubated with fluorescently labelled secondary antibody (1 : 200) and DAPI (5 μg ml−1) for 2 h at room temperature. Finally, coverslips were washed and mounted with Vectashield (Solarbio). Immunofluorescence staining was imaged using a fluorescence microscope (Olympus). Five representative fields were captured at × 200 magnification, and the numbers of cells expressing target proteins in the cytoplasm and the nucleus (overlapping with DAPI) cells were counted.
Transfection with lentiviral particles
To generate SGK1-silencing and SGK1-overexpressing stable PCa cell lines, PCa cells were infected with lentiviral particles. The lentiviral expression vectors LV2-Control, LV2-shSGK1, LV6-Control and LV6-SGK1 (SGK1 overexpression) were purchased from Shanghai Gene Pharma Company (Shanghai, China). In brief, the cells were seeded at 2 × 105 cells/well in a 6-well plate before lentiviral particle infection and incubated with 2 ml of complete medium for 24 h. Then, cells were infected with lentiviral particles, and after 12 h, the virus-containing medium of infected cells was substituted with fresh complete medium, and infected cells were selected with 4 μg ml−1 puromycin for 96 h. Empty lentiviral vectors were used as a control.
Transfection with DNA and siRNAs
For transfection, Lipofectamine 2000 (Invitrogen Life Technologies, Shanghai, China) was used for PC3 and LNCaP cells, according to the manufacturer’s user guides. GFP-LC3 plasmid was generously provided by the laboratory of Ye Zhaoming. The siRNAs used in the study were Foxo3a, 5′-GCUGUCUCCAUGGACAAUATT-3′; mTOR, 5′-CCACCCGAAUUGGCAGAUUTT-3′; and a negative control, 5′-UUCUCCGAACGUGUCACGUTT-3′.
Immunoprecipitation
Cells were lysed in cell lysis buffer containing proteinase and phosphatase inhibitors (Beyotime) for western blotting and immunoprecipitation, and 500 μg of soluble protein was first incubated with primary antibodies or immunoglobulin G (IgG), as a negative control, overnight at 4 °C. The immune complexes were recovered with 30 μl of protein A/G magnetic beads (Bio-Rad) for 6 h at 4 °C. The immunoprecipitated proteins were properly washed and then boiled with sample buffer for 5 min, followed by separation by SDS–PAGE (SDS–polyacrylamide gel electrophoresis) and western blotting analysis.
Western blot analysis
PCa cells were seeded at a density of 5 × 105 cells per well in a six-well culture plate and harvested after subsequent treatments, whole-protein lysates were collected cells using RIPA cell lysis reagent containing proteinase and phosphatase inhibitors (Solarbio, Beijing, China) at 4 °C for 30 min. Cell lysates were centrifuged at 12 000 g for 20 min at 4 °C, and protein concentrations of the supernatants were determined using the BCA protein assay reagent kit (Beyotime). Then, the supernatants containing total proteins were mixed with corresponding volume of 5 × SDS loading buffer and heated at 95 °C for 5 min, and 40 μg of total protein from each sample were concentrated on 5% Tris–glycine SDS gels, separated on 8% or 12% Tris–glycine SDS gels, and transferred to 0.22 μm polyvinylidene fluoride membranes. The membranes were blocked with 5% non-fat dry milk in TBST, incubated overnight with the appropriate primary antibody. After being washed three times for 30 min with TBST, the membrane was incubated with HRP-conjugated secondary antibodies for 2 h at room temperature. The bound antibodies were visualised using an enhanced chemiluminescence kit (Millipore).
In vivo tumour biology
Animal studies were conducted in accordance with institutional ethical guidelines for the care and use of experimental animals. Briefly, 4-week-old female BALB/c-nu mice were purchased from Shanghai Laboratory Animal Center of the Chinese Academy of Sciences. They were maintained under specific pathogen-free conditions and supplied with sterilised food and water. For in vivo xenograft studies, ten mice were randomly selected and divided into two groups. On day 0, 2 × 106 PC3LV2-Ctrl cells or 2 × 106 PC3shSGK1 cells suspended in 0.2 ml of PBS were inoculated subcutaneously in the right flank of each mouse (five mice in each group). Tumour sizes were measured daily to observe dynamic changes in tumour growth and calculated by a standard formula, length × width × depth × 0.5236. Tumour formation was defined as the time from inoculation until tumours measured 100 mm3. Subsequently, tumour volume measurements were performed twice weekly, and when the tumours of the PC3LV2 group reached ∼500 mm3, all mice were killed. Tumours were dissected and stored in liquid nitrogen or fixed in formalin for further analysis. All treatment protocols were approved by the Animal Care and Use Committee of Zhejiang University, China.
Statistical analysis
The values are shown as the means±s.d. for triplicate experiments, and significant differences were calculated using one-way ANOVA with Dunnett’s test or Newman–Keuls test and Student’s two-tailed t-test. Levels of statistical significance were set at P<0.05 or 0.01 as indicated.
Results
GSK650394 inhibits cell viability in prostate cancer cells
To investigate the cytotoxic effect of GSK650394 on PCa cells, we treated PCa cells with different concentrations of GSK650394 (0, 10, 20, 40, 80, 100, 120, 160 μ M) for various time points (12, 24, 48 and 72 h). GSK650394 inhibited cell viability in a time- and dose-dependent manner (Figure 1A). While the treatment with less than 40 μ M GSK650394 had little effect, treatments with more than 80 μ M GSK650394 for 48 and 72 h significantly inhibited cell viability. The IC50 values of GSK650394 for 48 h were ∼145.1 μ M for PC3, 81.5 μ M for LNCaP, 113.4 μ M for DU145 and 103.8 μ M for CWR22-Rv1 cells. In addition, clone formation in cells treated with GSK650394 was markedly reduced in a dose-dependent manner compared with control cells, and their proliferation potential was also dramatically impaired based on the size of the colonies as shown in Figure 1B. These results show that GSK650394 could effectively inhibit the proliferation of PCa cells.

GSK650394 inhibits cell viability in prostate cancer cells. (A) Cell viability analysis using a CCK8 assay of prostate cancer cell lines after treatment with various concentrations of GSK650394 or vehicle alone for the indicated time periods. Data are the means of triplicate experiments. The results are expressed as the percentage of viable control cells treated with DMSO alone (vehicle). (B) Colony formation assay of PC3 and LNCaP cells treated with DMSO or GSK650394. Cells were treated with GSK650394 at varying concentrations (2.5–10 μ M) for approximately 10 days for PC3 cells and 14 days for LNCaP cells. (left) The quantification of the number of colonies and the size of the colonies. Significant differences between the treatment groups were found using a t-test (P<0.05). Values represent the means±s.d.
GSK650394 causes G2/M cell cycle arrest and switches autophagy to apoptosis in prostate cancer cells
To investigate the way in which GSK650394 inhibits cell viability, we used flow cytometry to assess whether GSK650394 affected cell cycle progression and apoptosis. GSK650394 blocked cell cycle progression in PC3 (Figure 2A) and LNCaP cells (data not shown). In PC3 cells, a significant increase in the proportion of the cell population in G2/M phase was observed after 24 and 48 h of treatment with GSK650394, concomitant with a dramatic increase in the percentage of cells in sub G1 phase after 48 h treatment with GSK650394 (Figure 2A), which indicated that apoptosis was induced by GSK650394. An annexin V-PE/7-AAD assay further confirmed that GSK650394 did not significantly induce apoptosis in PC3 cells at 6 and 24 h but significantly induced apoptosis at 48 h (Figure 2B).

GSK650394 induces G2/M cell cycle arrest, autophagy and apoptosis. (A) PC3 cells were treated with DMSO or 160 μ M GSK650394 for 24 and 48 h. Cell cycle distribution was analysed by flow cytometry, and results are presented as the mean±s.d. from three independent experiments. (B–D) PC3 cells were treated with DMSO or 160 μ M GSK650394 for various time points (6, 24 and 48 h). Cell apoptosis was analysed by flow cytometry with annexin V-PE/7-AAD, and the apoptotic rate is shown for triplicate experiments (B). Autophagy activity was determined with acridine orange (AO) and monodansylcadaverine (MDC) staining and is quantified in the right panel (C) or immunofluorescence staining for LC3 puncta, which is quantified in the lower panel (D). (E) Transmission electron microscope images show the ultrastructure of PC3 cells treated with DMSO or 160 μ M GSK650394 for 48 h. Red arrows indicate autophagosomes that include residual digested material. (F) PC3 cells were treated as in b; LNCaP cells were treated with DMSO or 80 μ M GSK650394 (GSK) for various time points (6, 24 and 48 h). Total protein lysates were collected for western blot against p21, LC3B, p62, PARP and cleaved (CL) PARP; GAPDH was used as a loading control. *P<0.05 vs control.
Interestingly, PCa cells treated with GSK650394 showed morphological features of cytoplasmic vacuole accumulation that were not observed in DMSO-treated cells (Supplementary Figure 1). GSK650394 induced cytoplasmic vacuolation in a time-dependent manner, and treatments with equal concentrations of GSK650394 for 24 h and 48 h induced more cytoplasmic vacuolation in PCa cells compared to 6 h of treatment (Supplementary Figure 1a). In addition, GSK650394 at concentrations of 80 and 160 μ M induced more cytoplasmic vacuolation than 40 μ M GSK650394 (Supplementary Figure 1b). On the basis of previous research on Rottlerin-induced autophagic vacuoles (Kumar et al, 2014) and the response of cancer treatment-induced autophagy in PCa (Farrow et al, 2014), we preliminarily speculated that a portion of the GSK650394-induced cytoplasmic vacuoles were very likely autophagic vacuoles. To further demonstrate our speculation, GSK650394-induced vacuolation was assessed by immunofluorescence and TEM. We found that PC3 cells exhibited a stark increase in discrete acidic vesicles after 6 h of treatment with 160 μ M GSK650394, especially at 24 and 48 h treatment time points, at which greater than 80% of the cells were MDC-positive, and an average of more than 15 AO puncta were dispersed throughout the cytoplasm in GSK650394-treated PC3 cells (Figure 2C). Autophagy activity was further validated by visualising GSK650394-induced LC3 puncta in PC3 cells using immunofluorescence microscopy (Figure 2D). Higher magnification of autophagosomes revealed a smooth double-membrane containing cellular material, and intense electron-dense lysosomal structures were clearly observed in GSK650394-treated PC3 cells compared to control cells (Figure 2E). Thus, these results collectively demonstrated stimulation of autophagic flux upon GSK650394 treatment.
Next, the expression of cell cycle-, autophagy- and apoptosis-related proteins in PC3 and LNCaP cells was measured. After 6, 24 and 48 h of incubation, GSK650394 upregulated the expression of p21 (Figure 2F), which is also known as a cyclin-dependent kinase (CDK) inhibitor that can inhibit the formation of the cyclin-CDK complex, which leads to G2/M cell cycle arrest (Xiong et al, 1993). In addition, after treatment, immunoblotting revealed steady conversion of cytosolic MAP1LC3B-I/LC3B-I (microtubule-associated protein 1 light chain 3 β-I) to autophagosome-associated MAP1LC3B-II/LC3B-II (microtubule-associated protein 1 light chain 3 β-II) (Figure 2F), a well-known marker of autophagosome assembly (Kaufmann et al, 2014). Moreover, to detect the effect of GSK650394 on autophagic flux, the expression of SQSTM1 (sequestosome 1), also known as p62, a selective substrate of autophagy (Moscat and Diazmeco, 2009), was measured. The level of p62 reduced gradually in a time-dependent manner when the cells were treated with GSK650394, suggesting an enhanced autophagic flux (Figure 2F). In contrast, the level of cleaved PARP increased gradually in a time-dependent manner, but PARP cleavage was not induced at the 6 h treatment time point. All in all, based on the above results, we demonstrated that GSK650394 simultaneously induced G2/M cell cycle arrest, autophagy and apoptosis. Furthermore, autophagy precedes apoptosis in GSK650394-treated PCa cells, but the relationship between GSK650394-induced autophagy and apoptosis needs to be further investigated.
GSK650394-induced autophagy contributes to caspase-dependent apoptosis through intrinsic pathways
A complicated interplay exists between autophagy and apoptosis that is highly dependent on the cellular environment or the stimulus (Roos et al, 2016). To discern the relationship between GSK650394-induced autophagy and apoptosis, a vitality assay was performed with PC3 and LNCaP cells pretreated with 5 mM 3MA, 10 μ M CQ, 20 μ M z-VAD-fmk or vehicle alone before the addition of GSK650394. Treatment with 3MA significantly decreased GSK650394-induced AO puncta (Figure 3B and C) and GSK650394-dependent LC3-I to LC3-II conversion, as expected (Figure 3F). Moreover, the inhibition of autophagy by 3MA or CQ antagonised the inhibitory effects of GSK650394 on cell viability (Figure 3A), thus suggesting that, at least in PC3 and LNCaP cells, GSK650394-dependent SGK1 inhibition activated cytotoxic autophagy, which can be inhibited to restore cell viability. To further demonstrate whether GSK650394-triggered autophagy inhibits or promotes apoptosis, PC3 cells in which autophagy was suppressed by 3MA with or without GSK650394 treatment underwent a flow cytometry apoptosis assay, as shown in Figure 3D and E, and 3MA-mediated autophagy inhibition moderately abrogated GSK650394-induced apoptosis, which was consistent with the reduced cleaved caspase-3 and PARP cleavage (Figure 3F). Subsequently, the effect of inhibition of apoptosis on autophagy was determined. As seen in Figure 3B and C, z-VAD-fmk had a minimal effect on the number of AO-staining red acidic vesicles induced by GSK650394, in accordance with the expression of LC3-II (Figure 3F). However, after pretreatment with z-VAD-fmk, a portion of the cell vitality was successfully restored (Figure 3A), and apoptosis was nearly abrogated in GSK650394-treated PCa cells as expected (Figure 3D and E). Reduced caspase-3 and PARP cleavage further demonstrated that z-VAD-fmk completely diminished GSK650394-mediated apoptotic activity (Figure 3F), indicating that GSK650394-induced apoptosis is caspase-dependent.

GSK650394-induced autophagy contributes to caspase-dependent apoptosis through intrinsic pathways. (A) PC3 and LNCaP cells were incubated with or without 160 μ M GSK650394 (G 160) for 48 h after 2 h of pretreatment with 5 mM 3MA (an inhibitor of early autophagy), 10 μ M chloroquine (CQ, an inhibitor of acidification of lysosomes and autophagosomes) or 20 μ M z-VAD-fmk (an irreversible pan-caspase inhibitor). Then, cell viability was analysed by a CCK8 assay. (B–F) PC3 cells were treated as in a, except no cells were treated with CQ. Autophagy activity was determined with AO staining (B) and quantified; *P<0.05 vs G 160 treatment (C). Cell apoptosis was analysed by flow cytometry (D) and quantified (E). Whole-cell lysates were immunoblotted and probed with LC3-I/II, cleaved caspase-3 (Casp.3), PARP, PARP (CL) and GAPDH, as the loading control (F). (G) PC3 cells were treated with G 160 or DMSO for 48 h, and western blot analysis was performed to measure the expression of Fas, FasL, Bax, Bcl-2, cleaved caspase-8, cleaved caspase-9 and GAPDH. The results are expressed as the mean±s.d. from three independent experiments. *P<0.05.
Apoptosis can be triggered either by extrinsic stimuli through cell surface death receptors or by intrinsic stimuli through the mitochondrial signalling pathway. Thus, which pathway was involved in GSK650394-induced apoptosis was determined. As shown in Figure 3F and G, GSK650394 markedly activated caspase-3, -8 and -9 and led to Bax upregulation, Bcl-2 downregulation and PARP cleavage but had almost no effect on Fas and FasL (Figure 3G). Thus, these data imply that GSK650394 triggers caspase-dependent apoptosis by activating the intrinsic pathway rather than the extrinsic pathway.
SGK1 silencing causes G2/M arrest and activates autophagy-dependent apoptosis
Given that the effectiveness of GSK650394 as a growth inhibitor has nonspecific inhibition of other kinases, particularly other members of the SGK protein family, SGK2 and SGK3 (Sherk et al, 2008). Hence, lentiviral shRNA-mediated SGK1 silencing was performed to further demonstrate GSK650394-induced anti-tumour effects by impairing SGK1 activity. We first investigated SGK1 expression in several commonly studied PCa cell lines at baseline, as shown in Figure 4A, baseline SGK1 expression was high in AR-dependent cell line LNCaP (mutated AR), while steady-state levels were low in AR-negative cell lines PC3 and DU145, AR-positive but castration-resistant cell line CWR22-Rv1 is demonstrated middle baseline SGK1 expression. Thus, SGK1 was silenced in LNCaP and PC3 cells, as confirmed by immunoblotting (Figure 4G), and eliminating SGK1 expression impaired cell viability (Figure 4B), caused G2/M arrest with p21 protein upregulation (Figure 4C, D and G), promoted PARP and caspase-3 cleavage and induced apoptosis both in LNCaP and PC3 cells (Figure 4C and D). However, the growth inhibition observed with the SGK1 shRNA (shSGK1) seems to be less dramatic than that observed following treatment with GSK650394. Autophagy activity was also evaluated after SGK1 elimination, shSGK1 significantly increased LC3II protein levels both in LNCaP and PC3 cells (Figure 4G), which was further confirmed by observing LC3 redistribution and LC3 puncta (autophagosomes) formation via transfecting LNCaP cells with pEGFP-LC3 fusion protein (Figure 4F). Whereas, once shSGK1-triggered autophagy was suppressed by 3 MA (Figure 4F and G), shSGK1-induced impaired cell viability was partially restored (Figure 4B), and shSGK1-induced apoptotic cell death was almost completely rescued (Figure 4E). Western blot results further showed that 3 MA blocked shSGK1-induced PARP cleavage but had no effect on SGK1 and p21 protein expression. Collectively, these results suggest that SGK1 silencing causes G2/M arrest and activates autophagy-dependent apoptosis.

SGK1 silencing causes G2/M arrest and activates autophagy-dependent apoptosis. (A) Western blot of SGK1 expression in the PCa cell lines PC3, LNCaP, DU145 and CWR-22Rv1. (B–D) PC3 and LNCaP cells were infected with empty lentivirus control (LV2-Ctrl) or with the lentivirus SGK1-shRNA (LV2-shSGK1) and subsequently incubated for 48 h before pretreatment with 5 mM 3MA or PBS (control). Then, cell viability was analysed with a CCK8 assay (B). Cell cycle distribution was analysed by flow cytometry, and results are presented as the mean±s.d. from three independent experiments (C, D). (E) LNCaP cells were treated as in A, and cell apoptosis was analysed by flow cytometry with annexin V-PE/7-AAD. The apoptotic rate is shown from triplicate experiments. (F) Redistribution of pEGFP-LC3. LNCaP cells were stably transfected with a pEGFP-LC3 fusion construct and then treated as in a. Cells were visualised under a fluorescence microscope to examine the expression of LC3-II. Quantification of punctate dots per cell is based on the number of punctuate dots in pEGFP-LC3-positive cells. Quantification represents at least 100 counted cells and is scored per treatment. (G) PC3 cells were stably infected with the empty lentivirus control or with the lentivirus SGK1-shRNA, and LNCaP cells were treated as in A. Western blots were probed for levels of SGK1, p21, LC3-I, LC3-II, PARP and PARP (CL), and GAPDH was used as a loading control. All results are representative of three experiments and are expressed as the mean±s.d.; *P<0.05.
Ectopic expression of SGK1 attenuates GSK650394-induced autophagy and apoptosis
Although GSK650394 was shown to induce both autophagy and apoptosis, no direct evidence was presented to verify whether the GSK650394-induced events occurred by targeting SGK1. Therefore, we overexpressed SGK1 in PC3 and LNCaP cells. Cells infected with lentivirus containing an SGK1 (LV6-SGK1) expression plasmid were shown to robustly increase the protein level of SGK1 compared to an empty vector (LV6-Ctrl) (Figure 5A), which resulted in significant enhancement of cell viability both in PC3 and LNCaP cells (Figure 5B). Next, PC3 cells stably overexpressing SGK1 or vector alone were treated with 160 μ M GSK650394 for 48 h. Interestingly, overexpression of SGK1 was found to attenuate both the autophagy and apoptosis induced by GSK650394, as shown by the levels of LC3 conversion, p21, Bax, Bcl-2, cleaved caspase-3 and PARP cleavage (Figure 5E). Furthermore, ectopic expression of SGK1 was also shown to reduce the number of MDC-positive cells and apoptotic cells induced by GSK650394 (Figure 5C and D), suggesting that SGK1 promotes cell survival, in part by attenuating cytotoxic autophagy activation. Therefore, a direct role of SGK1 in the regulation of autophagy was demonstrated.

Ectopic expression of SGK1 attenuates GSK650394-induced autophagy and apoptosis. (A and B) PC3 and LNCaP cells were infected with lentivirus containing an SGK1 (LV6-SGK1) expression plasmid or an empty vector (LV6-Ctrl) that confers resistance to puromycin. PC3 and LNCaP cells stably overexpressing SGK1 or vector alone were assessed using a western blot against SGK1 (A), and cell viability was determined using a CCK8 assay (B). (B–D) PC3 cells stably overexpressing SGK1 or vector alone were treated for 48 h with 160 μ M GSK650394. Autophagy activity was assessed using microscopy to observe MDC-positive cells (C) and quantified. (D) Western blot against LC3. (E) Apoptosis was determined using Hoechst 33342 staining. Red arrows indicate chromatin condensation and DNA fragmentation (C), and the apoptotic rate shown is from triplicate experiments (D). Western blots were probed for levels of p21, Bax, Bcl-2, cleaved caspase-3, PARP, PARP (CL), with GAPDH used as a loading control (E). All results are representative of three experiments and are expressed as the mean±s.d.; *P<0.05.
Suppression of Foxo3a phosphorylation is critical for blockade of SGK1-induced apoptosis
To decipher the molecular mechanisms by which blockade of SGK1 induces events in PCa, we investigated changes in signalling molecules downstream of the SGK1 pathway. It was found that forkhead-transcription-factor 3 (Foxo3a), an important downstream target of SGK1 (You et al, 2004) and a potential tumour suppressor (Hu et al, 2004), was one of the molecules that was not changed in PC3 cells treated with GSK650394 nor in LNCaP cells with stable knockdown of SGK1 (Figure 6A). However, interestingly, SGK1 inhibition significantly reduced pSGK1 (S78) and suppressed phosphorylation of endogenous Foxo3a at Ser-253 and Thr-32 (Figure 6A), and it dramatically increased p27 and Bim levels according to western blot analysis, which was consistent with our previous results (3.2 and 3.4) indicating that blockade of SGK1-induced G2/M arrest and apoptosis. We next determined whether SGK1 also induces phosphorylation of Foxo3a, and our results showed that SGK1 overexpression efficiently upregulated pSGK1 (S78), which contributed to the phosphorylation of endogenous Foxo3a at Ser-253 and Thr-32 (Figure 6B) in PC3 cells, but the level of Foxo3a in these cells was almost comparable with that in LV2-Ctrl-PC3 cells. It appears that the level of Foxo3a phosphorylation is critical for SGK1-mediated effects. The activity of Foxo3a can be affected by post-translational modifications, such as phosphorylation and acetylation, which inactivate Foxo3a protein and cause it to translocate from the nucleus to the cytoplasm, resulting in the promotion of cell proliferation by preventing Foxo3a from inducing cell cycle arrest and apoptosis (Brunet et al, 2001). Therefore, we further investigated Foxo3a localisation in PC3 cells with stable knockdown or overexpression of SGK1 using an immunofluorescence analysis. As shown in Figure 6C, SGK1 silencing induced nuclear accumulation of Foxo3a, whereas ectopic expression of SGK1 triggered translocation of Foxo3a from the nucleus into the cytoplasm. In summary, suppression of Foxo3a phosphorylation is critical for blockade of SGK1-induced apoptosis.

Suppression of Foxo3a phosphorylation is critical for blockade of SGK1-induced apoptosis. (A) PC3 cells were incubated with DMSO or 160 μ M GSK650394 (G 160) for 48 h. LNCaP cells were infected with LV2-Ctrl or LV2-shSGK1 and selected with puromycin. Total protein lysates were collected for immunoblot analysis, using antibodies to the indicated antigens. (B) Lysates of PC3 cells with stable overexpression of SGK1 or vector alone were assessed using a western blot against the indicated antigens. (C) Immunofluorescence analysis of Foxo3a localisation was performed in PC3 cells with stable knockdown or overexpression of SGK1.
SGK1 inhibition-mediated mTOR dephosphorylation enhances autophagy activity through a p-Foxo3a-LC3 interaction
The molecular mechanisms by which SGK1 affects autophagy regulation are far from being elucidated. Given the importance of mammalian target of rapamycin (mTOR) in autophagy regulation (Kim et al, 2011; Seront et al, 2013), we investigated whether mTOR was involved in SGK1 inhibition-triggered autophagy. Our data showed that SGK1 silencing significantly decreased the phosphorylation of mTOR, although it failed to affect the mTOR protein level (Figure 7A). Our data further showed that SGK1 overexpression stimulated the phosphorylation of mTOR, but the mTOR protein level was again not significantly altered (Figure 7A). All of the above data suggested that Foxo3a and mTOR might be involved in the SGK1-mediated effects in PCa. Therefore, we investigated the roles of Foxo3a and mTOR by transfecting PCa cells with siFoxo3a or simTOR for 48 h. Immunoblotting showed that siFoxo3a and simTOR efficiently silenced Foxo3a and mTOR expression, respectively, in PC3 (Figure 7C) and LNCaP cells (data not shown). Then, the viability of PCa cells was assessed using CCK8. As shown in the left panel of Figure 7B, siRNA-mediated Foxo3a downregulation remarkably increased the viability of LNCaP cells. Conversely, simTOR dramatically decreased the viability of LNCaP cells compared to siScr, and similar results were observed in PC3 cells, shown in the right panel of Figure 7B. Our further results indicated that both simTOR and siFoxo3a could induce LC3-I to LC3-II conversion, suggesting that mTOR inhibition or Foxo3a inhibition can trigger autophagy. We further found that Foxo3a silencing suppressed the phosphorylation of Foxo3a at Ser-253 and Thr-32 and reduced the expression of p27, but mTOR, pmTOR, SGK1, pSGK1 (S78), PARP and PARP (CL) were not significantly altered (Figure 7C). Furthermore, interestingly, our results showed that mTOR knockdown in PC3 cells significantly reduced pmTOR, pSGK1 (S78), pFoxo3a (S253) and pFoxo3a (T32) levels, markedly increased p27 levels and moderately induced PARP cleavage but had no significant effect on SGK1 and Foxo3a (Figure 7C). These results suggested that SGK1 inhibition-mediated mTOR dephosphorylation enhances autophagy activity by suppressing the phosphorylation of Foxo3a. Moreover, importantly, these results indicated that a positive feedback loop between pSGK1 (S78) and pmTOR is involved in autophagy and apoptosis modulation in PCa.

SGK1 inhibition-mediated mTOR dephosphorylation enhances autophagy activity through a p-Foxo3a-LC3 interaction. (A) Total protein lysates of PC3 cells with stable knockdown or overexpression of SGK1 were assessed using western blotting against mTOR and pmTOR. (B) LNCaP and PC3 cells were transfected with siFoxo3a, simTOR or siScr alone for 48 h, and cell viability was determined using a CCK8 assay. (C) PC3 cells were transfected with siFoxo3a, simTOR or siScr alone, and total protein lysates were assessed using a western blot against the indicated antigens. (D) Total protein lysates of LNCaP cells were prepared for co-IP using pFoxo3a (S253) antibody. (E) Total protein lysates of LNCaP cells were prepared for co-IP using p27 antibody. (F) Total protein lysates of LNCaP cells were prepared for co-IP using LC3 antibody. (G) PC3 cells were stably infected with the empty lentivirus control or shSGK1 lentivirus, and protein lysates collected for co-IP using pFoxo3a (S253) antibody. All results are representative of three experiments and are expressed as the mean±s.d.; *P<0.05.
In view of the important role of pFoxo3a in SGK1-mediated autophagy and apoptosis modulation, proteins of interest that may interact with SGK1, pSGK1 (S78), pFoxo3a (T32) and pFoxo3a (S253) were detected using co-immunoprecipitation (IP) in LNCaP cells. It was found that three proteins, SGK1, pSGK1 (S78) and pFoxo3a (T32) were unable to interact with each other, and they had no interaction with LC3, p27, p21, mTOR or pmTOR (data not shown). To our excitement, p27 and LC3 were found to interact with pFoxo3a (S253) (Figure 7D), and this was further confirmed using reverse IP with p27 and LC3 antibodies (Figure 7E and F), although pSGK1 (S78), pmTOR and p21 did not interact with pFoxo3a (S253) (Figure 7D). Moreover, SGK1 silencing decreased pFoxo3a (S253)-LC3 and pFoxo3a (S253)-p27 interactions (Figure 7G). Collectively, these data suggest that SGK1 inhibition-mediated mTOR dephosphorylation enhances autophagy activity at least partially through a p-Foxo3a-LC3 interaction, especially via pFoxo3a (S253) rather than pFoxo3a (T32).
Combined suppression of SGK1 and mTOR leads synergistic cytocidal effects on PCa cells
Our results demonstrated that both SGK1 inhibition and mTOR inhibition can induce autophagy and impair cell viability of PCa cells. However, whether the combination of both could lead to more deleterious effect on PCa cell viability needed to be further investigated. As expected, while the impairment of cell viability upon SGK1 silencing or mTOR silencing were modest, the combination of both induced a much more deleterious effect when compared with the control in PC3 cells (Figure 8A). Similar results were also obtained in LNCaP cells; the combination of rapamycin and shSGK1 led to a more pronounced response than either treatment alone (Figure 8B). We also examined the combination of rapamycin and shSGK1 on the induction of autophagy in LNCaP cells using immunofluorescence staining for LC3. As shown in Figure 8C, treatment with either rapamycin or SGK1-silencing shRNA significantly increased LC3 puncta compared to vehicle control, and the combination of both exhibited a synergistic effect, with ∼70% of the cells exhibiting more LC3 dots than were observed with the individual treatments (Figure 8D), which was further verified via LC3-I/LC3-II conversion (Figure 8G). However, this seems to be slightly inconsistent with the apoptosis results showing that treatment with rapamycin alone induced significant autophagy rather than apoptosis (Figure 8E). Interestingly, the combination of rapamycin and SGK1 silencing induced more apoptotic cell death compared with SGK1 silencing alone (Figure 8E and F). Moreover, it was found that rapamycin failed to trigger PARP cleavage, whereas the combination significantly increased cleaved PARP compared to SGK1 silencing alone, indicating that rapamycin-induced autophagy contributes to SGK1 inhibition-induced apoptotic cell death.

Combined suppression of SGK1 and mTOR leads to a much more deleterious effect on PCa cell viability. (A) PC3 cells were infected with shSGK1, transfected with simTOR, or a combination of both silencing treatments for 48 h, and cell viability was determined using a CCK8 assay. (B) LNCaP cells were treated with DMSO or rapamycin (100 nM) or infected with shSGK1 or a combination of both treatments for 48 h, and cell viability was determined using a CCK8 assay. (C–G) LNCaP cells were treated as in B. Autophagy activity was determined with immunofluorescence staining for LC3 puncta (C) and quantified (D). Apoptosis was analysed by flow cytometry with annexin V-PE/7-AAD (E), and the apoptotic rate is shown from triplicate experiments (F). Total protein lysates of LNCaP cells were assessed using western blotting against the indicated antigens. All results are representative of three experiments and expressed as the mean±s.d.; *P<0.05.
Downregulation of SGK1 inhibits prostate cancer growth in vivo
We next expanded our results in vivo. The in vivo effect of SGK1 inhibition in PCa was determined in a tumour-transplant mouse model. It was found that injection of PC3 cells with stable knockdown of SGK1 caused a 9.4% weight loss in mice 30 days after inoculation (Figure 9A). In addition, it is worth noting that the gap in tumour volume between the two groups gradually became larger (Figure 9B), and there was a significant (80%) reduction in tumour weight in mice inoculated with PC3shSGK1 cells when compared with LV2-Ctrl mice, as shown in Figure 9C. Immunohistochemistry demonstrated that SGK1, pFoxo3a (S253) and pmTOR were downregulated and LC3 was upregulated, whereas mTOR and Foxo3a were not obviously altered in the shSGK1 group compared to the LV2-Ctrl group (Figure 9D). Immunoblotting results further confirmed that shSGK1 resulted in inhibition of SGK1 and LC3-I/LC3-II conversion and an increase in p21, p27 and cleaved caspase-3 in vivo (Figure 9E). Taken together, these results indicate that SGK1 inhibition suppresses PCa growth via activation of both autophagy and apoptosis in vivo.

Downregulation of SGK1 inhibits prostate cancer growth in vivo. Briefly, 2 × 106 PC3LV2-Ctrl cells or 2 × 106 PC3shSGK1 cells suspended in 0.2 ml PBS were inoculated subcutaneously in the right flank of each mouse (five mice in each group). Following inoculation, tumour volume measurements were performed at the indicated days. When the tumours of the PC3LV2 group reached ∼500 mm3, all mice were killed. (A–C) Body weights, tumour sizes and tumour weights were measured. (D) SGK1, Foxo3a, pFoxo3a (S253), mTOR, pmTOR and LC3B levels were determined by IHC. Representative images are presented. (E) The levels of SGK1, LC3B, p21, p27 and cleaved caspase-3 in tumour xenograft tissues were also measured by western blot. *P<0.05.
Discussion
SGK1, a serine/threonine protein kinase, has a pivotal role in several oncogenic signalling cascades that are involved in cell transformation, tumour progression and chemo/radio resistance (Lang et al, 2006; Shanmugam et al, 2007; Sherk et al, 2008; Pau et al, 2016; Talarico et al, 2016). Although SGK1-specific inhibitors, including GSK650394, EMD638683 and SI113 (Ortuso et al, 2014), have been demonstrated to exhibit significant anti-tumour effects by inducing G2/M arrest and apoptosis in vitro and in vivo (Shanmugam et al, 2007; Sherk et al, 2008; Talarico et al, 2016), the mechanism underlying these effects is not fully understood. The present work found that SGK1 inhibition, mediated by either GSK650394 or shSGK1, not only induced G2/M arrest and apoptosis but also enhanced autophagosome biogenesis. We further provide evidence that SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway, and dual inhibition of mTOR and SGK1 enhances autophagy activation resulting in augmented cytocidal effects that restrain PCa progression.
We have confirmed that SGK1 expression (shRNA-mediated downregulation) and activity (GSK650394 studies) are required for both AR-dependent and AR-independent PCa growth (Figures 1 and 4) and not just AR-dependent PCa cell growth (Shanmugam et al, 2007; Sherk et al, 2008), indicating that SGK1 inhibition may be a promising treatment strategy for aggressive androgen-refractory PCa. Interestingly, it was found that the IC50 values of GSK650394 at 48 h were higher in AR-negative cell lines (PC3 and DU145) than in AR-positive cell lines (LNCaP and CWR22-Rv1) (Figure 1), suggesting that AR-positive PCa cell lines are more sensitive to GSK650394, which is consistent with our findings regarding the high expression of SGK1 in AR-positive cell lines but not in AR-negative cell lines. In addition, given the nonspecific inhibitory effects of GSK650394 on other protein kinases, such as SGK2, SGK3 and Akt (Sherk et al, 2008), which we have not investigated in the present study, the possibility that these kinases may also contribute to this difference needs to be further explored. Furthermore, the inhibitory effects observed following treatment with GSK650394 were more dramatic than those observed with SGK1 shRNA (Figures 1 and 4). Similar results were reported by Sherk et al (2008), and this might be ascribed to the residual expression of SGK1 observed in our silencing experiments.
We have demonstrated that SGK1 inhibition suppressed cell viability primarily by inducing G2/M arrest and apoptosis. Although SGK1 inhibition-induced cell cycle arrest was also observed by other research groups (Brunet et al, 2001; Tao et al, 2013), the delay in cell cycle progression at G1 or G2 was not defined. On the one hand, SGK1-mediated p27 T157 phosphorylation and cytoplasmic p27 mislocalisation facilitated the promotion of G1 progression in breast cancer (Hong et al, 2008). On the other hand, knockdown of SGK1 significantly increased p21 levels (Fagerli et al, 2011), a negative regulator of cell cycle progression at G1 (Xiong et al, 1993). Here, our results revealed that SGK1 inhibition caused G2/M arrest accompanied by p21 and p27 elevation, in line with recent investigations in HCC (Talarico et al, 2015, 2016). And similar results were observed in a synchronised HeLa continuous cell line, the inhibition of SGK1 by its dominant negative mutant caused a striking delay in the G2/M-phase entry (Amato et al, 2009).
In addition, extensive studies demonstrated that SGK1 inhibition can significantly trigger apoptosis (Brunet et al, 2001; Sherk et al, 2008; Tao et al, 2013; Talarico et al, 2016). However, apoptosis can be triggered through both intrinsic and extrinsic pathways, and these studies did not show further results examining these pathways. Endo et al (2011) reported that SGK1 knockdown activated the extrinsic apoptosis pathway in the ectoderm. In contrast, our data demonstrated that SGK1 inhibition activated apoptosis through the mitochondrial-mediated intrinsic pathway rather than the extrinsic pathway in PCa. Thus, SGK1 inhibition-induced cell cycle arrest and apoptosis are closely related to cell type and cellular context.
Autophagy (also known as macroautophagy), or cellular self-digestion, is a cellular pathway involved in the degradation of proteins, lipids and organelles, with an astonishing number of connections to human physiology and disease (Mizushima et al, 2008). In some cellular contexts, autophagy serves as a cell survival pathway, preventing tumour cells from apoptosis induced by cancer therapies, and in other contexts, prolonged stress and progressive autophagy can eventually lead to cell death, either in collaboration with apoptosis or as a backup mechanism (White and Dipaola, 2011). Autophagy was induced, as demonstrated by the accumulation of AO puncta, the formation of autophagosomes observed with TEM and increased LC3-I/LC3-II conversion. However, the interplay between autophagy and apoptosis induced by SGK1 has not previously been investigated. We further revealed that SGK1 inhibition-induced cell death was markedly restored by 3MA or z-VAD-fmk, indicating that SGK1 inhibition induced autophagic cell death, which contributes to apoptosis and is caspase-dependent. Recently, cytotoxic autophagy was also observed in human glioblastoma multiforme cells (Talarico et al, 2016) and endometrial cancer cells (Conza et al, 2017) after treatment with SI113, a novel SGK1 inhibitor, could significantly decreased the expression endoplasmic reticulum stress markers GRP78 (glucose-regulated protein 78, also known as BiP or HSPA5) (Talarico et al, 2016). In contrast, SGK1 inhibition dramaticlly increased the expression of GRP78 in PC3 cells (Supplementary Figure 2a), which is in line with a recent report (Conza et al, 2017). Intriguingly, suppression of SGK1-induced autophagy has been used as an effective therapeutic approach to suppressing glycogen accumulation without killing cells in Lafora disease (LD) animal models (Andres-Mateos et al, 2013; Singh et al, 2014).
Previous investigation indicates that autophagy and apoptosis share the same set of cellular regulatory proteins (Pattingre et al, 2005). For example, the well-known anti-apoptotic proteins Bcl-2 and Bcl-X(L) suppress both mitochondria-mediated apoptosis, through antagonising pro-apoptotic BH3-only proteins, and autophagy, through binding and neutralising Beclin1, which contains a BH3 domain (Shimizu et al, 2004; Maiuri et al, 2007). In this regard, the mechanism of SGK1 inhibition-induced autophagy may be attributable to Bcl-2 reduction (Figure 3G), thereby releasing Beclin1 to trigger autophagy (Supplementary Figure 2a). More importantly, our results showed that ectopic expression of SGK1 significantly attenuated GSK650394-induced autophagy and apoptosis. However, ectopic expression of SGK1 did not completely abrogate GSK650394-induced autophagy and apoptosis, perhaps partially because GSK650394 seems also to downregulate protein activity of ectopic SGK1 in addition to that of the endogenous SGK1, indicating that SGK1 functions as a negative regulator in autophagy and apoptosis modulation. Further studies are needed to better understand the underlying mechanism by which SGK1 inhibition induces autophagy.
Next, we explored the downstream pathways. Forkhead transcription factor FKHRL1, also called Foxo3a, has a pro-apoptotic role by inducing cell cycle arrest and apoptosis (Brunet et al, 2001; Dehner et al, 2008). Post-translational modifications, especially phosphorylation, have an important role in modulating the activity of Foxo3a. Unphosphorylated Foxo3a has been suggested to be its active form, which is primarily located in the nucleus and functions as a transcriptional regulator for a variety of genes (Brunet et al, 2001; Hu et al, 2004; Tao et al, 2013). Our data revealed that SGK1 inhibition significantly suppressed phosphorylation of endogenous Foxo3a at Ser-253 and Thr-32 and dramatically increased p27 and Bim, according to western blot analysis, which was consistent with previous studies (You et al, 2004; Tao et al, 2013). However, ectopic expression of SGK1-induced phosphorylation of endogenous Foxo3a at Ser-253 and Thr-32 and triggered the translocation of Foxo3a from the nucleus into the cytoplasm, which resulted in the promotion of cell proliferation. Brunet et al (2001) also showed that SGK1, similar to Akt, promoted cell survival and that it did so in part by phosphorylating Foxo3a at Ser-253, Ser-315 and Thr-32, thereby inactivating Foxo3a.
Despite these co-associations, molecular mechanisms linking SGK1 and autophagy are undefined. Our data revealed that SGK1 silencing significantly decreased the phosphorylation of mTOR, which contributed to the activation of autophagy, although it failed to affect mTOR, a negative regulator of autophagy (Kim et al, 2011). Our data further suggested that SGK1 inhibition induced autophagy by dephosphorylating Foxo3a, thereby activating Foxo3a and attenuating a p-Foxo3a-LC3 interaction. This is the first time that p-Foxo3a was demonstrated to interact with LC3. Indeed, Foxo3a is required for fasting-induced autophagy (Mammucari et al, 2007; Warr et al, 2013), and Foxo3a activation significantly upregulates Foxo3a-target genes, including autophagy-related gene 5 (Atg5), Atg6/Beclin, Atg12 and Atg7, thereby activating autophagy (Chiacchiera and Simone, 2009). Surprisingly, we also found that pFoxo3a could interact with p27. Therefore, these data suggest that SGK1 inhibition-induced autophagy and apoptosis can be attributed to two mechanisms, one is mediated by Foxo3a activation-induced upregulation of Foxo3a target genes, and the other is mediated by attenuating pFoxo3a-p27/LC3 interactions; however, the latter mechanism needs to be further studied. In addition, we also found that, in PC3 cells, SGK1 inhibition significantly increased the expression level of p53 protein (Supplementary Figure 2a), a tumour suppressor, has an important role in regulation of autophagy (Tasdemir et al, 2008).
Interestingly, a positive feedback loop between pSGK1 (S78) and pmTOR was defined in the present study. Accumulating evidence suggests that mTOR activation facilitates an increase in SGK1 phosphorylation, thereby activating the SGK1 signalling pathway (Garcíamartínez and Alessi, 2008; Hong et al, 2008; Pau et al, 2016), which is consistent with our results showing that siRNA-mediated downregulation of mTOR remarkably decreased the phosphorylation of SGK1. In addition, our data further showed that SGK1 silencing significantly decreased the phosphorylation of mTOR, and SGK1 overexpression stimulated the phosphorylation of mTOR, but modulating SGK1 expression failed to affect the total protein level of mTOR. What’s more, we also found that, in PC3 cells, mTOR inhibition markedly attenuated the expression level of pSGK1 (S422) protein (Supplementary Figure 2b), which is in line with a previous report (Hong et al, 2008). Recently, Pau et al (2016) demonstrated that PDK1-mediated SGK1 activation contributes to the maintenance of residual mTORC1 activity through direct phosphorylation and inhibition of TSC2, which further confirms this positive feedback loop.
Therapies targeting autophagy combined with other therapeutic strategies have been studied extensively (Kondo and Kondo, 2006; Koukourakis et al, 2016). In fact, there are active clinical trials involving patients using both autophagy inhibitors or inducers together with other cancer treatments to achieve better curative effects (Høyerhansen and Jäättelä, 2008). In our study, we showed that mTOR inhibition-induced autophagy synergised with SGK1 inhibition and dramatically amplified the cytocidal effects in PCa cells. In this regard, co-targeting mTOR and SGK1 prevents Foxo3a phosphorylation, and may present a novel combination therapy regimen that will benefit PCa patients due to the amplified cytocidal effects that restrain cancer progression.
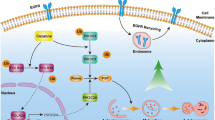
In summary, our findings show that SGK1 inhibition exhibits significant antitumour effects against PCa in vitro and in vivo. This study uncovered a novel mechanism of SGK1 inhibition in PCa, which is mediated, at least in part, by inducing autophagy-dependent apoptosis via the mTOR-Foxo3a pathway (Supplementary Figure 3), and highlights how co-targeting SGK1 and autophagy can amplify cytocidal effects and delay cancer progression.
References
Amato R, D’Antona L, Porciatti G, Agosti V, Menniti M, Rinaldo C, Costa N, Bellacchio E, Mattarocci S, Fuiano G, Soddu S, Paggi MG, Lang F, Perrotti N (2009) Sgk1 activates MDM2-dependent p53 degradation and affects cell proliferation, survival, and differentiation. J Mol Med 87: 1221–1239.
Andres-Mateos E, Brinkmeier H, Burks TN, Mejias R, Files DC, Steinberger M, Soleimani A, Marx R, Simmers JL, Lin B (2013) Activation of serum/glucocorticoid-induced kinase 1 (SGK1) is important to maintain skeletal muscle homeostasis and prevent atrophy. EMBO Mol Med 5: 80–91.
Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME (2001) Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21: 952–965.
Chiacchiera F, Simone C (2009) Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 5: 1030–1033.
Conza D, Mirra P, Cali G, Tortora T, Insabato L, Fiory F, Schenone S, Amato R, Beguinot F, Perrotti N, Ulianich L (2017) The SGK1 inhibitor SI113 induces autophagy, apoptosis, and endoplasmic reticulum stress in endometrial cancer cells. J Cell Physiol 8: 25850.
Dehner M, Hadjihannas M, Weiske J, Huber O, Behrens J (2008) Wnt signaling inhibits Forkhead box O3a-induced transcription and apoptosis through up-regulation of serum- and glucocorticoid-inducible kinase 1. J Biol Chem 283: 19201–19210.
Endo T, Kusakabe M, Sunadome K, Yamamoto T, Nishida E (2011) The kinase SGK1 in the endoderm and mesoderm promotes ectodermal survival by down-regulating components of the death-inducing signaling complex. Sci Signal 4: 131–139.
Fagerli UM, Ullrich K, Stuhmer T, Holien T, Kochert K, Holt RU, Bruland O, Chatterjee M, Nogai H, Lenz G, Shaughnessy JD Jr, Mathas S, Sundan A, Bargou RC, Dorken B, Borset M, Janz M (2011) Serum/glucocorticoid-regulated kinase 1 (SGK1) is a prominent target gene of the transcriptional response to cytokines in multiple myeloma and supports the growth of myeloma cells. Oncogene 30: 3198–3206.
Farrow JM, Yang JC, Evans CP (2014) Autophagy as a modulator and target in prostate cancer. Nat Rev Urol 11: 508–516.
Garcíamartínez JM, Alessi DR (2008) mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J 416: 375–385.
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JM, Papaemmanuil E, Brewer DS, Kallio HM, Hognas G, Annala M, Kivinummi K, Goody V, Latimer C, O’Meara S, Dawson KJ, Isaacs W, Emmert-Buck MR, Nykter M, Foster C, Kote-Jarai Z, Easton D, Whitaker HC, Neal DE, Cooper CS, Eeles RA, Visakorpi T, Campbell PJ, McDermott U, Wedge DC, Bova GS (2015) The evolutionary history of lethal metastatic prostate cancer. Nature 520: 353–357.
Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM (2008) mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell 30: 701–711.
Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC (2004) IkappaB kinase promotes tumourigenesis through inhibition of forkhead FOXO3a. Cell 117: 225–237.
Høyerhansen M, Jäättelä M (2008) Autophagy: an emerging target for cancer therapy. Autophagy 4: 574–580.
Isikbay M, Otto K, Kregel S, Kach J, Yi C, Griend DJV, Conzen SD, Szmulewitz RZ (2014) Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Hormones Cancer 5: 72–89.
Kaufmann A, Beier V, Franquelim HG, Wollert T (2014) Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell 156: 469–481.
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141.
Kobayashi T, Deak M, Morrice N, Cohen P (1999) Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Int J Dermatol 344 (Pt 1): 1193–1194.
Kondo Y, Kondo S (2006) Autophagy and cancer therapy. Autophagy 2: 85–90.
Koukourakis MI, Mitrakas AG, Giatromanolaki A (2016) Therapeutic interactions of autophagy with radiation and temozolomide in glioblastoma: evidence and issues to resolve. Br J Cancer 114: 485–496.
Kumar D, Shankar S, Srivastava RK (2014) Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/Akt/mTOR signaling pathway. Cancer Lett 343: 179–189.
Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V (2006) (Patho) physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 86: 1151–1178.
Maiuri MC, Le TG, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N (2007) Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 26: 2527–2539.
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Piccolo PD, Burden SJ, Lisi RD, Sandri C, Zhao J (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6: 458–471.
Melhem A, Yamada SD, Fleming GF, Delgado B, Brickley DR, Wu W, Kocherginsky M, Conzen SD (2009) Administration of glucocorticoids to ovarian cancer patients is associated with expression of the anti-apoptotic genes SGK1 and MKP1/DUSP1 in ovarian tissues. Clin Cancer Res 15: 3196–3204.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075.
Moscat J, Diazmeco TM (2009) p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137: 1001–1004.
Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki D, Lara PN Jr, DeVere White RW, Gao AC, Evans CP (2014) Targeting autophagy overcomes enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 33: 4521–4530.
Ortuso F, Amato R, Artese A, D’Antona L, Costa G, Talarico C, Gigliotti F, Bianco C, Trapasso F, Schenone S (2014) In silico identification and biological evaluation of novel selective serum/glucocorticoid-inducible kinase 1 inhibitors based on the pyrazolo-pyrimidine scaffold. J Chem Inform Model 54: 1828–1832.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939.
Pau C, Haley E, Ruzica B, Eneda T, Pedram R, Javier CF, Srinivasaraghavan K, Verma CS, Maura D, Sarat C (2016) PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell 30: 229–242.
Roos WP, Thomas AD, Kaina B (2016) DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer 16: 20–33.
Seront E, Boidot R, Bouzin C, Karroum O, Jordan BF, Gallez B, Machiels JP, Feron O (2013) Tumour hypoxia determines the potential of combining mTOR and autophagy inhibitors to treat mammary tumours. Br J Cancer 109: 2597–2606.
Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B (2007) Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ 14: 2085–2094.
Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D (2008) Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res 68: 7475–7483.
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawakobayashi S, Thompson CB, Tsujimoto Y (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6: 1221–1228.
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66: 7–30.
Singh PK, Singh S, Ganesh S (2014) Activation of serum/glucocorticoid-induced kinase 1 (SGK1) underlies increased glycogen levels, mTOR activation, and autophagy defects in Lafora disease. Mol Biol Cell 25: 3776–3786.
Szmulewitz RZ, Chung E, Al-Ahmadie H, Daniel S, Kocherginsky M, Razmaria A, Zagaja GP, Brendler CB, Stadler WM, Conzen SD (2012) Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate 72: 157–164.
Talarico C, D’Antona L, Scumaci D, Barone A, Gigliotti F, Fiumara CV, Dattilo V, Gallo E, Visca P, Ortuso F, Abbruzzese C, Botta L, Schenone S, Cuda G, Alcaro S, Bianco C, Lavia P, Paggi MG, Perrotti N, Amato R (2015) Preclinical model in HCC: the SGK1 kinase inhibitor SI113 blocks tumour progression in vitro and in vivo and synergizes with radiotherapy. Oncotarget 6: 37511–37525.
Talarico C, Dattilo V, D’Antona L, Barone A, Amodio N, Belviso S, Musumeci F, Abbruzzese C, Bianco C, Trapasso F (2016) SI113, a SGK1 inhibitor, potentiates the effects of radiotherapy, modulates the response to oxidative stress and induces cytotoxic autophagy in human glioblastoma multiforme cells. Oncotarget 7: 15868–15884.
Tao GZ, Lehwald N, Jang KY, Baek J, Xu B, Omary MB, Sylvester KG (2013) Wnt/β-catenin signaling protects mouse liver against oxidative stress-induced apoptosis through the inhibition of forkhead transcription factor FoxO3. J Biol Chem 288: 130–133.
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10: 676–687.
Vyas AR, Hahm ER, Arlotti JA, Watkins S, Stolz DB, Desai D, Amin S, Singh SV (2013) Chemoprevention of prostate cancer by d,l-sulforaphane is augmented by pharmacological inhibition of autophagy. Cancer Res 73: 5985–5995.
Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E (2013) FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494: 323–327.
White E, Dipaola RS (2011) The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 15: 5308–5316.
Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366: 701–704.
Yao Y, Jiang Q, Jiang L, Wu J, Zhang Q, Wang J, Feng H, Zang P (2016) Lnc-SGK1 induced byHelicobacter pyloriinfection and highsalt diet promote Th2 and Th17 differentiation in human gastric cancer by SGK1/Jun B signaling. Oncotarget 7: 20549–20560.
You H, Jang Y, Youten AI, Okada H, Liepa J, Wakeham A, Zaugg K, Mak TW (2004) p53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1. Proc Natl Acad Sci USA 101: 14057–14062.
Acknowledgements
We thank Clinical Research Center from the Second Affiliated Hospital of Zhejiang University School of Medicine for essential technical supports. We thank American Journal Experts (AJE) for English language editing. This study was supported by grant from the National Natural Science Foundation of China (Grant nos. 81271917), Natural Science Foundation of Zhejiang province (Grant nos. LY14H200002, LY15H200002 and LY16H160023).
Author contributions
WL and ZT designed the study. WL, XW, ZL, YW, BY, PY and XD performed the experiments. ZL and YC assessed animal tissue histology. CL, XL and YD supervised specific experiments and revised the manuscript. WL and ZT wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Liu, W., Wang, X., Liu, Z. et al. SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway. Br J Cancer 117, 1139–1153 (2017). https://doi.org/10.1038/bjc.2017.293
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.293
Keywords
This article is cited by
-
LncRNA LBX2-AS1 promotes proliferation and migratory capacity of clear cell renal cell carcinoma through mitophagy
European Journal of Medical Research (2024)
-
The role of serum-glucocorticoid regulated kinase 1 in reproductive viability: implications from prenatal programming and senescence
Molecular Biology Reports (2024)
-
Dexmedetomidine promotes autophagy and apoptosis of colon cancer cells by activating endoplasmic reticulum (ER) stress-mediated PERK/eIF2α/ATF4 signaling pathway
Molecular & Cellular Toxicology (2023)
-
Circular RNA COL1A2 Mediates High Glucose-Induced Oxidative Stress and Pyroptosis by Regulating MiR-424-5p/SGK1 in Diabetic Nephropathy
Applied Biochemistry and Biotechnology (2023)
-
Targeting autophagy in prostate cancer: preclinical and clinical evidence for therapeutic response
Journal of Experimental & Clinical Cancer Research (2022)
