
Abstract
Background:
In the Phase III LUX-Lung 3/6 (LL3/LL6) trials in epidermal growth factor receptor (EGFR) mutation-positive lung adenocarcinoma patients, we evaluated feasibility of EGFR mutation detection using circulating cell-free DNA (cfDNA) and prognostic and predictive utility of cfDNA positivity (cfDNA+).
Methods:
Paired tumour and blood samples were prospectively collected from randomised patients. Mutations were detected using cfDNA from serum (LL3) or plasma (LL6) by a validated allele-specific quantitative real-time PCR kit.
Results:
EGFR mutation detection rates in cfDNA were 28.6% (serum) and 60.5% (plasma). Mutation detection in blood was associated with advanced disease characteristics, including higher performance score, number of metastatic sites and bone/liver metastases, and poorer prognosis. In patients with common EGFR mutations, afatinib improved progression-free survival vs chemotherapy in cfDNA+ (LL3: HR, 0.35; P=0.0009; LL6: HR, 0.25; P<0.0001) and cfDNA− (LL3: HR, 0.46; P<0.0001; LL6: HR, 0.12; P<0.0001) cohorts. A trend towards overall survival benefit with afatinib was observed in cfDNA+ patients.
Conclusions:
Plasma cfDNA is a promising alternative to biopsy for EGFR testing. Detectable mutation in blood was associated with more advanced disease and poorer prognosis. Afatinib improved outcomes in EGFR mutation-positive patients regardless of blood mutation status.
Similar content being viewed by others


Main
Traditionally, treatment of non-small cell lung cancer (NSCLC) has consisted of platinum-based chemotherapy. However, over the past decade, the discovery of frequent molecular alterations in NSCLC, particularly epidermal growth factor receptor (EGFR) mutations, has led to a new treatment paradigm that includes targeted agents (Novello et al, 2016). The reversible EGFR tyrosine kinase inhibitors (TKIs) erlotinib and gefitinib, as well as the irreversible ErbB family blocker, afatinib, are approved for the treatment of advanced EGFR mutation-positive NSCLC. In this era of targeted therapies, identification of patients (e.g., via EGFR mutation detection) who may derive benefit from these agents is a key factor for successful treatment (Olsen and Jorgensen, 2014).
In clinical practice, EGFR mutations are routinely detected using DNA extracted from primary or metastatic tumour tissue obtained during tumour biopsy or resection, which is typically formalin fixed and paraffin embedded (FFPE) (Ellison et al, 2013; Jung, 2013). However, efficient and robust mutation testing using these tissue samples can be challenging owing to small biopsy size or lack of sufficient tumour cells, inherent issues associated with FFPE archival tissue (e.g., poor DNA quality and tissue necrosis), and reluctance or inability of patients to undergo invasive biopsy (Ellison et al, 2013; Jung, 2013; Weber et al, 2014). As such, the development of simpler, minimally invasive assays to detect gene mutations may provide an attractive alternative to tumour biopsy for the identification of patients eligible for targeted treatments.
An emerging methodology to this end is the detection of mutations in circulating cell-free DNA (cfDNA) isolated from blood (i.e., liquid biopsy). Clinical studies in patients with NSCLC have explored the feasibility of EGFR mutation detection using cfDNA from serum and/or plasma samples, as well as the prognostic value and potential utility of EGFR mutation positivity by cfDNA to predict clinical outcomes to EGFR-targeted therapies (Kimura et al, 2006, 2007; Bai et al, 2009; He et al, 2009; Brevet et al, 2011; Lee et al, 2011; Goto et al, 2012; Mok et al, 2013; Li et al, 2014; Douillard et al, 2014a, 2014b; Mok et al, 2015). These studies suggest that detection of EGFR mutation in the blood is feasible and may be useful in the absence of available tumour biopsy. However, there is variability in the detection rates of EGFR mutation in the blood compared with the standard methodology using tumour tissue, and the correlation of blood-derived EGFR mutation positivity with specific patient characteristics or clinical outcomes remains uncertain.
This article describes the findings of two large, randomised Phase III trials (LUX-Lung 3 (LL3) and LUX-Lung 6 (LL6)), which compared the ErbB family blocker afatinib with standard platinum-doublet chemotherapy (cisplatin+pemetrexed in LL3; cisplatin+gemcitabine in LL6) in treatment-naive patients with advanced NSCLC harbouring EGFR mutations in their tumours (Sequist et al, 2013; Wu et al, 2014; Yang et al, 2015). Improvements in clinical outcomes with afatinib vs chemotherapy in both LL3 and LL6, particularly in patients with tumours harbouring common EGFR mutations (Del19/L858R), have been previously reported (Sequist et al, 2013; Wu et al, 2014; Yang et al, 2015). Both trials prospectively collected paired tumour and blood samples from patients for analysis of EGFR mutations. The current analysis evaluates the technical feasibility of detecting EGFR mutations in cfDNA from either serum (LL3) or plasma (LL6) and explores the association of clinical characteristics and outcomes with cfDNA-positive (cfDNA+) or -negative (cfDNA−) status in EGFR mutation-positive patients.
Materials and Methods
Study design and patients
Details of the LL3 and LL6 study designs and patient eligibility criteria have been previously published (Sequist et al, 2013; Wu et al, 2014). In brief, eligible patients in LL3 (n=345; recruited globally) and LL6 (n=364; recruited in China, South Korea, and Thailand) had pathologically confirmed and previously untreated stage IIIB/IV lung adenocarcinoma, an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (Eisenhauer et al, 2009), and adequate organ function. Tumours were confirmed as EGFR mutation positive at screening based on central laboratory analysis of biopsy tissue using a validated test kit (Therascreen EGFR 29; Qiagen, Manchester, UK), as described in detail below.
In each study, patients were randomised (2 : 1) to receive oral afatinib (40 mg day−1) or up to six cycles of intravenous pemetrexed (500 mg m−2) plus cisplatin (75 mg m−2) once every 21 days in LL3 or gemcitabine (1000 mg m−2; days 1 and 8) plus cisplatin (75 mg m−2; day 1) every 21 days in LL6 (Sequist et al, 2013; Wu et al, 2014). Stratification parameters included EGFR mutation type (Del19/L858R/other) and race (Asian/non-Asian; LL3 only). Patients were treated until disease progression, death, unacceptable adverse events or withdrawal of consent for any reason.
The primary end point of each study was progression-free survival (PFS; by independent blinded review) (Sequist et al, 2013; Wu et al, 2014). Key secondary end points for both studies included objective response (complete response (CR) or partial response (PR)), disease control (CR, PR or stable disease), and overall survival (OS). Other secondary end points included patient-reported outcomes of disease symptoms and health-related quality of life and safety, which were previously published (Sequist et al, 2013; Wu et al, 2014).
Study conduct
Each study was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonisation good clinical practice, local laws, and applicable regulatory requirements and was approved by the institutional review board or independent ethics committee of each centre. An independent Data Monitoring Committee monitored conduct. All patients provided written, informed consent for participation in the studies and provision of tumour and blood samples. Trial registration IDs are: ClinicalTrials.gov NCT00949650 (LL3) and NCT01121393 (LL6).
Blood/tumour sample collection and EGFR mutation detection
Tumour tissue from each patient was obtained at an initial diagnostic procedure for NSCLC and was paraffin embedded. Tumour samples for EGFR mutation detection consisted of at least five 10 μm unstained sections mounted on a non-charged microscopic slide and containing at least 20% tumour tissue. In addition to the tumour tissue sample, a mandatory 9-ml blood sample was collected from each patient at the start of treatment (day 1, course 1) in both trials; however, different DNA extraction methodologies were used (see below for further details). The selection of the DNA extraction kit was based on the recommendation from the manufacturer at the time of each study conduct.
In both tumour tissue and paired serum/plasma samples, genotyping of EGFR mutations was conducted at a central laboratory using a validated allele-specific quantitative real-time PCR kit (Therascreen EGFR 29; Qiagen) designed to detect 29 EGFR mutations (19 deletions in exon 19 (collectively termed Del19), L858R, three insertions in exon 20 (collectively termed Ins20), L861Q, G719S, G719A, G719C (or G719X), T790M, and S768I) against a background of wild-type genomic DNA. Although a formal assessment of the amount of serum/plasma used for each sample was not conducted, it is important to note that the Therascreen EGFR 29 assay includes DNA-loading controls, which indicate when a DNA sample is too dilute or is non-amplifiable, and provides a means of performing quality control of the DNA during the assay.
Extraction of DNA from serum
Following phlebotomy (9 ml of venous blood collected over ethylenediaminetetraacetic acid (EDTA)), serum was prepared and frozen prior to shipment to the central laboratory. DNA preparation from frozen serum samples (3 ml) was performed using the QIAamp DNA Blood Mini Kit (Qiagen) according to the manufacturer’s instructions.
Extraction of DNA from plasma
Following phlebotomy (9 ml of venous blood collected over EDTA), plasma was prepared and frozen prior to shipment to the central laboratory. The EDTA plasma sample was thawed at room temperature and then transferred to a 15-ml tube and centrifuged for 10 min at 16 000 × g in a fixed-angle rotor at+4 °C. Proteinase K solution (400 μl) was added to a 50-ml tube. The clarified plasma supernatant was carefully separated from the sediment layer, and a maximum of 4 ml was added to the Proteinase K solution. The sample volume was then adjusted to 4 ml as required using phosphate-buffered saline. Buffer ACL (3.2 ml, containing carrier RNA) was added to the 50-ml tube and the solution was incubated at 60 °C for 30 min. The Proteinase K-digested sample was then further processed as per the QIAamp Circulating Nucleic Acid Kit manufacturer’s instructions (Qiagen), and the final DNA samples were eluted in 100 ml of buffer AVE.
Tumour assessments
Tumour assessments were performed by computed tomography or magnetic resonance imaging every 6 weeks for the first 48 weeks and then every 12 weeks thereafter until disease progression or start of new anticancer therapy (Sequist et al, 2013; Wu et al, 2014). Scans were reviewed by an independent central imaging group consisting of both radiologists and oncologists who were blinded to treatment assignments. Number of metastatic sites was determined by the count of unique sites of metastases across all organs with metastases. The sum of the longest diameters of lesions is the sum of the baseline target lesions identified by an independent review according to the procedures defined in RECIST version 1.1 (Eisenhauer et al, 2009).
Statistical analyses
To formally define the performance of the EGFR Therascreen 29 assay, test results from a mixture of EGFR mutation-positive and -negative samples would be required to calculate the sensitivity, specificity, negative predictive value (NPV), and positive predictive value (PPV) of the assay. Per eligibility criteria in LL3 and LL6, only patients with EGFR mutation-positive tumours were included (i.e., all patients had tumour tissue that was positive for one or more EGFR mutation type). As such, calculations for sensitivity, specificity, PPV, and NPV in this analysis make use of test results for all seven mutation types within each individual patient.
For each study, logistic regression models were used to test for any association between baseline demographic and disease characteristics and the EGFR mutation test result from blood (cfDNA+ or cfDNA−). Separate efficacy analyses were performed on patients who were cfDNA+ or cfDNA− in each study. Comparisons of PFS and OS between treatment arms were made via a log-rank test. Cox proportional-hazard models were used to derive hazard ratios (HRs) and 95% confidence intervals (CIs) comparing the two treatment arms. Kaplan–Meier estimates were used to construct survival curves and calculate median PFS and OS. Logistic regression models were used to compare the rates of objective response and disease control between treatment groups. Additional Cox proportional-hazard models were used to assess whether the blood test result (cfDNA+ or cfDNA−) was prognostic for PFS or OS after adjusting for any effects of treatment. Further Cox proportional-hazard models were used, including the treatment-by-blood test result interaction term, in order to evaluate any evidence of heterogeneity. Efficacy analyses were restricted to those patients with an EGFR mutation blood test result whose tumour harboured a common EGFR mutation (Del19/L858R). Analyses were performed on all such patients and also for patients split by the type of mutation (Del19 or L858R) in the tumour. Efficacy data presented here are from the time point of the main OS analysis, when it was estimated that OS would be sufficiently mature (Yang et al, 2015). All analyses were post hoc and exploratory in nature.
Results
EGFR mutations in blood cfDNA and matched tumour tissue
In LL3 and LL6, 287 (serum) and 334 (plasma) blood samples were analysed for the presence of EGFR mutations (Figure 1). The detection rates of EGFR mutation using cfDNA from blood samples compared with paired tumour samples were 28.6% (82 out of 287) in LL3 and 60.5% (202 out of 334) in LL6. Detection rates for individual EGFR mutations, including the common mutations Del19 and L858R, as well as various uncommon mutations, are shown in Table 1.

Patient disposition in LUX-Lung 3 and LUX-Lung 6. aPatients who were EGFR mutation positive based on tissue biopsy at screening. Abbreviations: cfDNA=cell-free DNA; EGFR=epidermal growth factor receptor.
Characteristics of patients with or without detectable EGFR mutation in the blood
Descriptive summaries supported by statistical tests of association showed that, in both trials, patients who were cfDNA+ for EGFR exhibited characteristics associated with more advanced disease compared with cfDNA− patients. cfDNA+ patients more frequently had a higher ECOG performance status (LL3: P=0.0029; LL6: P=0.0022), greater tumour burden (measured as the sum of the longest diameter of target lesions; approaching significance in LL3: P=0.061; LL6: P=0.0001), more metastatic sites (LL3: P=0.0003; LL6: P<0.0001), and metastases to the bone (LL3 and LL6: P<0.0001) and liver (LL3: P<0.0001; LL6: P=0.0003; Table 2). In terms of laboratory parameters, the cfDNA+ group also exhibited a general trend in higher laboratory values for alkaline phosphatase (ALP), lactate dehydrogenase (LDH), and white blood cell count (approaching significance for LL6), which are associated with metastatic spread to the bone and liver (2015). In addition, there was evidence in LL6 that female patients and those with a poorly/undifferentiated tumour grade were more likely to be cfDNA+ than cfDNA−. No notable differences were observed based on age, race, smoking status, disease stage, pleural metastases, brain metastases, or original EGFR mutation type in the tumour in either study.
Efficacy analysis by cfDNA EGFR mutation detection status
Efficacy analyses were performed in NSCLC patients whose tumours harboured common EGFR mutations (Del19 or L885R) confirmed by tissue biopsy. Within this group of patients, comparisons between treatment arms (afatinib vs chemotherapy) were performed in those who were cfDNA+ or cfDNA− for any EGFR mutation based on their blood test result.
Survival outcomes
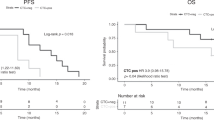
Afatinib significantly improved PFS vs chemotherapy in patients with common EGFR mutations in both the cfDNA+ (LL3: 8.3 vs 3.3 months; P=0.0009; LL6: 9.7 vs 4.6 months; P<0.0001) and the cfDNA−group (LL3: 13.7 vs 6.9 months; P<0.0001; LL6: 16.6 vs 5.8 months; P<0.0001; Figure 2). The PFS benefit of afatinib over chemotherapy was more pronounced in patients with Del19 vs L858R mutation-positive tumours (Table 3).

Kaplan–Meier curves of PFS. Kaplan–Meier curves of PFS according to the presence of an EGFR mutation by cfDNA analysisa in patients with common EGFR mutations (Del19 or L858R; based on tumour biopsy) in (A) LUX-Lung 3 and (B) LUX-Lung 6. aPatients who were EGFR Del19 or L858R mutation positive based on tissue biopsy were grouped according to whether any EGFR mutation was detected by cfDNA analysis (cfDNA+ or cfDNA−). Abbreviations: cfDNA=cell-free DNA; cis=cisplatin; EGFR=epidermal growth factor receptor; gem=gemcitabine; pem=pemetrexed; PFS=progression-free survival.
A trend towards improvement in OS with afatinib vs chemotherapy was observed in patients who were cfDNA+ (LL3: 22.1 vs 14.7 months; LL6: 20.0 vs 17.8 months) or cfDNA− (LL3: 33.6 vs 28.6 months; LL6: 35.6 vs 27.0 months; Figure 3). In Del19 patients, a statistically significant improvement in OS with afatinib vs chemotherapy was observed in cfDNA+ patients in LL3 (29.4 vs 14.3 months; P=0.0372) and in cfDNA− patients in both trials (LL3: 33.3 vs 28.2 months; P=0.0247; LL6: 39.4 vs 21.1 months; P=0.0063; Table 3). OS was numerically improved in Del19 patients with a detectable EGFR mutation in plasma in LL6 (23.6 vs 14.6 months). No difference in OS with afatinib vs chemotherapy was observed in the L858R group for those with or without a detectable EGFR mutation in the blood (Table 3).

Kaplan–Meier curves of OS. Kaplan–Meier curves of OS according to the presence of an EGFR mutation by cfDNA analysisa in patients with common EGFR mutations (Del19 or L858R; based on tumour biopsy) in (A) LUX-Lung 3 and (B) LUX-Lung 6. aPatients who were EGFR Del19 or L858R mutation positive based on tissue biopsy were grouped according to whether any EGFR mutation was detected by cfDNA analysis (cfDNA+ or cfDNA−). Abbreviations: cfDNA=cell-free DNA; cis=cisplatin; EGFR=epidermal growth factor receptor; gem=gemcitabine; OS=overall survival; pem=pemetrexed.
cfDNA+ patients had generally shorter PFS and OS in both treatment arms than cfDNA− patients (Supplementary Figures S1 and S2). Additional analyses confirmed the prognostic effect of a detectable EGFR mutation in the blood on PFS and OS; after adjusting for any effects of treatment, a consistent, statistically significant effect was observed (Table 3). There was no evidence to suggest that the treatment effect (afatinib vs chemotherapy) was different for cfDNA+ or cfDNA− patients, as observed by the non-significant interaction term P values (Table 3).
Tumour response
In both cfDNA+ and cfDNA− patients, higher objective response rates (ORRs) based on independent review were observed in those treated with afatinib vs chemotherapy (cfDNA+; LL3: 62.0% vs 20.0%; LL6: 69.6% vs 30.6%; cfDNA−; LL3: 62.8% vs 24.2%; LL6: 69.4% vs 19.0%). Higher ORRs with afatinib vs chemotherapy in cfDNA+ patients (LL3: 78.0% vs 40.0%; LL6: 76.3% vs 34.7%) and cfDNA− patients (LL3: 76.9% vs 45.2%; LL6: 75.0% vs 33.3%) were also observed based on investigator assessment of tumour response.
Discussion
The development of minimally invasive assays to detect gene mutations may provide a promising alternative to tumour biopsy, in the absence of available tissue samples, for identifying patients who would benefit from genotype-directed therapy such as EGFR-targeted agents. In the current analysis of NSCLC patients enrolled in the Phase III LL3 and LL6 trials, the EGFR mutation detection rates between paired tumour and blood samples suggest that the isolation of cfDNA from plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) could be a potential alternative to tumour tissue for EGFR mutation testing in the absence of available tumour samples. Whether the difference in EGFR mutation detection rates between LL3 (28.6%) and LL6 (60.5%) in this analysis was attributable to the different DNA extraction kits used, the difference in DNA source (serum or plasma), or both is currently unclear. However, previous studies suggest that plasma may be a more reliable source than serum for tumour DNA owing to higher background levels of non-tumour wild-type DNA found in serum (Steinman, 1975; Board et al, 2008). In a recent analysis of BRAF mutation testing using amplification refractory mutation system (ARMS) methodology on cfDNA isolated from either serum or plasma collected from advanced melanoma patients, a greater amount of total cfDNA was isolated from serum vs plasma (using the QIAamp Circulating Nucleic Acid Kit (Qiagen) for each blood sample type); however, the proportion of tumour-derived mutant cfDNA was significantly greater in plasma than in serum (approximately 2 × higher) (Aung et al, 2014). Although total cfDNA isolated from serum vs plasma was not investigated in the current analysis, it is possible that the higher EGFR mutation detection rate observed in plasma vs serum may reflect a higher proportion of tumour-derived mutant cfDNA present in plasma.
In previous analyses, the detection rates for EGFR mutation in blood (plasma or serum) cfDNA compared with paired tumour tissue samples have ranged from 31% to 97% (Kimura et al, 2006, 2007; Moran et al, 2007; Bai et al, 2009; He et al, 2009; Kuang et al, 2009; Yung et al, 2009; Brevet et al, 2011; Goto et al, 2012; Liu et al, 2013; Zhao et al, 2013; Douillard et al, 2014b; Mok et al, 2015). This wide variation could be due to multiple factors, including differences in sample media (e.g., plasma vs serum), differences in DNA extraction and mutation detection methodologies (e.g., ARMS, digital PCR, or direct sequencing, among others), disease stage of patients (e.g., high cfDNA levels have been associated with more advanced disease; Lee et al, 2011), and the inclusion of patients with wild-type EGFR. Indeed, it is easier to establish the absence of an EGFR mutation in the blood than confirm its presence, suggesting that the inclusion of patients with wild-type EGFR may result in higher overall concordance rates with paired tissue samples.
In LL3 and LL6, EGFR mutation-positive patients who were cfDNA+ tended to have more advanced disease characteristics compared with cfDNA− patients, including higher ECOG performance scores, greater tumour burden, a higher number of metastatic sites, metastases to the bone and liver, and higher laboratory values (e.g., ALP and LDH), which are associated with metastatic spread to the bone/liver. This association may be a result of increased escape of tumour cells and DNA into the blood in patients with greater tumour burden and more metastatic sites. Furthermore, the more advanced baseline disease characteristics of EGFR cfDNA+ patients are likely to contribute to the poorer prognosis of these patients, irrespective of treatment with afatinib or chemotherapy. The correlation between the presence of mutant EGFR in the blood and the increased number of metastatic sites has been reported in a previous analysis (Lee et al, 2011). The higher proportion of females among cfDNA+ patients observed in LL6 is also consistent with some previous reports (Kimura et al, 2007; Jiang et al, 2011). Although some studies have demonstrated a correlation between cfDNA+ status and tumour histology, poor differentiation status, and non-smoking status (Kimura et al, 2007; Bai et al, 2009; Jiang et al, 2011; Zhao et al, 2013), such correlations were not observed in this report.
The therapeutic sensitivity of EGFR mutation-positive NSCLC to EGFR TKIs is well established (Yap and Popat, 2014). In this context, previous studies have demonstrated improved clinical outcomes in gefitinib-treated NSCLC patients with detectable EGFR mutations in serum (Kimura et al, 2006, 2007) and plasma (Bai et al, 2009) vs patients with NSCLC harbouring wild-type EGFR. The current study examined clinical outcomes in EGFR mutation-positive patients diagnosed by tumour biopsy according to their EGFR mutation status by cfDNA. In this analysis, clinical benefit was observed with afatinib- vs platinum-based chemotherapy whether or not the mutation could be found in the blood. Interestingly, cfDNA+ patients displayed less favourable PFS and OS, irrespective of treatment, compared with cfDNA− patients, despite similar ORRs in cfDNA+ and cfDNA− patients. Similar findings were observed in a recent study by Li et al (2014), in which shorter PFS with EGFR TKI treatment was reported in cfDNA+ vs cfDNA− NSCLC patients with EGFR mutation-positive tumours by biopsy. Subgroup analyses in the Phase III EURTAC trial in EGFR mutation-positive patients also demonstrated shorter PFS with erlotinib in cfDNA+ vs cfDNA− patients (Rosell et al, 2012). More recent analyses from the EURTAC trial demonstrated that, in patients with L858R mutations in the tumour, median OS was shorter for cfDNA+ patients compared with those who were cfDNA− (Karachaliou et al, 2015); these findings are consistent with results from our analysis. In contrast to our results, patients with tumour Del19 mutations who were also cfDNA+ had longer median OS than patients who were cfDNA−, and analysis of baseline disease characteristics did not show any significant differences between cfDNA+ and cfDNA− patients (Karachaliou et al, 2015). In recent analyses of the Phase III FASTACT-2 study, improved PFS with erlotinib over placebo was noted in patients who were cfDNA+ vs cfDNA− for EGFR mutations at baseline. However, for the baseline cfDNA+ subgroup, both PFS and OS were significantly worse for those who were cfDNA+ at cycle 3 of treatment compared with those who were cfDNA− at cycle 3 (Mok et al, 2015). In the Phase III First-SIGNAL study, high EGFR mutation positivity in plasma was associated with significantly shorter survival in gefitinib-treated patients with lung adenocarcinoma vs low EGFR mutation positivity (P=0.03) (Lee et al, 2011). Of note, high blood EGFR mutation positivity in the First-SIGNAL study was also associated with more aggressive baseline tumour characteristics (e.g., greater number of metastatic sites); Mok et al (2015) also hypothesised that the changes in outcomes with erlotinib may have been linked to increases in tumour burden or metastases.
To our knowledge, the current analysis of the LL3 and LL6 trials provides the largest cohort of data to date assessing both the feasibility of detecting EGFR mutation in the blood and the correlation of clinical characteristics and outcomes with cfDNA+ status. In this context, there are some limitations of the analyses that should be considered when interpreting the findings. First, blood samples were not collected from all screened patients in these studies, and the proportions of missing samples, while generally low, were different between LL3 and LL6, which may be a confounding factor in these analyses. Second, quantitative measurement of the extracted DNA was not performed; thus we were unable to determine whether there was a relationship between DNA quantity and test positivity. In addition, because all patients included in LL3 and LL6 had tumour tissue that was positive for one or more EGFR mutation, we were unable to formally determine the sensitivity and specificity of the methods to detect these mutations, as compared with wild-type EGFR, in serum or plasma. Further, the different sample media used in each study (plasma vs serum) prevented combining the study data. Although combination of the LL3 and LL6 data would have potentially improved the overall statistical power of the outcomes analyses, the separate analysis of plasma and serum did provide a preliminary assessment of which media may be better for the detection of EGFR mutations. As described in the Methods section, all analyses were post hoc and therefore considered exploratory. No adjustment for multiplicity was made to the P values presented, resulting in an increased chance of false-positive results; however, the results were generally consistent and of sufficient magnitude to suggest a true effect rather than a chance finding. Finally, serial postbaseline blood sampling was not conducted in LL3 and LL6; thus potential changes in blood mutation status during treatment and at the time of progression could not be assessed.
In summary, our findings suggest that detection of EGFR mutation in cfDNA from plasma appears to be a promising and minimally invasive alternative to tumour biopsy for patients without available tissue samples. Patients with EGFR mutations detected in the blood (irrespective of mutation type) displayed more advanced disease characteristics and had worse prognosis compared with blood EGFR mutation-negative patients. Importantly, clinical benefit with afatinib over platinum-based chemotherapy was consistently observed in both cfDNA+ and cfDNA− populations. Improvements in the methodology of cfDNA testing for EGFR mutations are needed to increase sensitivity and to enable this to be offered to all patients considered for EGFR-targeted therapy, with the goal of reducing the overall burden of performing invasive biopsy procedures. Future prospective studies in patients with paired tumour tissue and blood samples will be important to further develop the utility of this methodology with regard to patient prognosis and predicting clinical outcomes.
References
Aung KL, Donald E, Ellison G, Bujac S, Fletcher L, Cantarini M, Brady G, Orr M, Clack G, Ranson M, Dive C, Hughes A (2014) Analytical validation of BRAF mutation testing from circulating free DNA using the amplification refractory mutation testing system. J Mol Diagn 16 (3): 343–349.
Bai H, Mao L, Wang HS, Zhao J, Yang L, An TT, Wang X, Duan CJ, Wu NM, Guo ZQ, Liu YX, Liu HN, Wang YY, Wang J (2009) Epidermal growth factor receptor mutations in plasma DNA samples predict tumor response in Chinese patients with stages IIIB to IV non-small-cell lung cancer. J Clin Oncol 27 (16): 2653–2659.
Board RE, Williams VS, Knight L, Shaw J, Greystoke A, Ranson M, Dive C, Blackhall FH, Hughes A (2008) Isolation and extraction of circulating tumor DNA from patients with small cell lung cancer. Ann NY Acad Sci 1137: 98–107.
Brevet M, Johnson ML, Azzoli CG, Ladanyi M (2011) Detection of EGFR mutations in plasma DNA from lung cancer patients by mass spectrometry genotyping is predictive of tumor EGFR status and response to EGFR inhibitors. Lung Cancer 73 (1): 96–102.
Douillard JY, Ostoros G, Cobo M, Ciuleanu T, Cole R, McWalter G, Walker J, Dearden S, Webster A, Milenkova T, McCormack R (2014a) Gefitinib treatment in EGFR mutated caucasian NSCLC: circulating-free tumor DNA as a surrogate for determination of EGFR status. J Thorac Oncol 9 (9): 1345–1353.
Douillard JY, Ostoros G, Cobo M, Ciuleanu T, McCormack R, Webster A, Milenkova T (2014b) First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer 110 (1): 55–62.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45 (2): 228–247.
Ellison G, Zhu G, Moulis A, Dearden S, Speake G, McCormack R (2013) EGFR mutation testing in lung cancer: a review of available methods and their use for analysis of tumour tissue and cytology samples. J Clin Pathol 66 (2): 79–89.
Goto K, Ichinose Y, Ohe Y, Yamamoto N, Negoro S, Nishio K, Itoh Y, Jiang H, Duffield E, McCormack R, Saijo N, Mok T, Fukuoka M (2012) Epidermal growth factor receptor mutation status in circulating free DNA in serum: from IPASS, a phase III study of gefitinib or carboplatin/paclitaxel in non-small cell lung cancer. J Thorac Oncol 7 (1): 115–121.
He C, Liu M, Zhou C, Zhang J, Ouyang M, Zhong N, Xu J (2009) Detection of epidermal growth factor receptor mutations in plasma by mutant-enriched PCR assay for prediction of the response to gefitinib in patients with non-small-cell lung cancer. Int J Cancer 125 (10): 2393–2399.
Jiang B, Liu F, Yang L, Zhang W, Yuan H, Wang J, Huang G (2011) Serum detection of epidermal growth factor receptor gene mutations using mutant-enriched sequencing in Chinese patients with advanced non-small cell lung cancer. J Int Med Res 39 (4): 1392–1401.
Jung CY (2013) Biopsy and mutation detection strategies in non-small cell lung cancer. Tuberc Respir Dis (Seoul) 75 (5): 181–187.
Karachaliou N, Mayo-de las CC, Queralt C, de Aguirre I, Melloni B, Cardenal F, Garcia-Gomez R, Massuti B, Sánchez JM, Porta R, Ponce-Aix S, Moran T, Carcereny E, Felip E, Bover I, Insa A, Reguart N, Isla D, Vergnenegre A, de Marinis F, Gervais R, Corre R, Paz-Ares L, Morales-Espinosa D, Viteri S, Drozdowskyj A, Jordana-Ariza N, Ramirez-Serrano JL, Molina-Vila MA, Rosell R Group. SLC (2015) Association of EGFR L858R mutation in circulating free DNA with survival in the EURTAC trial. JAMA Oncol 1 (2): 149–157.
Kimura H, Kasahara K, Kawaishi M, Kunitoh H, Tamura T, Holloway B, Nishio K (2006) Detection of epidermal growth factor receptor mutations in serum as a predictor of the response to gefitinib in patients with non-small-cell lung cancer. Clin Cancer Res 12 (13): 3915–3921.
Kimura H, Suminoe M, Kasahara K, Sone T, Araya T, Tamori S, Koizumi F, Nishio K, Miyamoto K, Fujimura M, Nakao S (2007) Evaluation of epidermal growth factor receptor mutation status in serum DNA as a predictor of response to gefitinib (IRESSA). Br J Cancer 97 (6): 778–784.
Kuang Y, Rogers A, Yeap BY, Wang L, Makrigiorgos M, Vetrand K, Thiede S, Distel RJ, Janne PA (2009) Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res 15 (8): 2630–2636.
Lee YJ, Yoon KA, Han JY, Kim HT, Yun T, Lee GK, Kim HY, Lee JS (2011) Circulating cell-free DNA in plasma of never smokers with advanced lung adenocarcinoma receiving gefitinib or standard chemotherapy as first-line therapy. Clin Cancer Res 17 (15): 5179–5187.
Li X, Ren R, Ren S, Chen X, Cai W, Zhou F, Zhang Y, Su C, Zhao C, Li J, Cheng N, Zhao M, Zhou C (2014) Peripheral blood for epidermal growth factor receptor mutation detection in non-small cell lung cancer patients. Transl Oncol 7 (3): 341–348.
Liu X, Lu Y, Zhu G, Lei Y, Zheng L, Qin H, Tang C, Ellison G, McCormack R, Ji Q (2013) The diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologies. J Clin Pathol 66 (12): 1065–1069.
Merck Manual (professional version) (2015) Overview of cancer: cancer diagnosis http://www.merckmanuals.com/professional/hematology-and-oncology/overview-of-cancer/cancer-diagnosis.html.
Mok T, Wu YL, Lee JS, Yu CJ, Sriuranpong V, Wen W, Tsai J, Truman M, Klughammer B, Wu L (2013) Detection of EGFR-activating mutations from plasma DNA as a potent predictor of survival outcomes in FASTACT 2: A randomized phase III study on intercalated combination of erlotinib (E) and chemotherapy (C). J Clin Oncol 31 (15 suppl): : abstract 8021.
Mok T, Wu YL, Lee JS, Yu CJ, Sriuranpong V, Sandoval-Tan J, Ladrera G, Thongprasert S, Srimuninnimit V, Liao M, Zhu Y, Zhou C, Fuerte F, Margono B, Wen W, Tsai J, Truman M, Klughammer B, Shames DS, Wu L (2015) Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes in NSCLC patients treated with first-line intercalated erlotinib and chemotherapy. Clin Cancer Res 21 (14): 3196–3203.
Moran T, Paz-Ares L, Isla D, Cobo M, Massuti B, Insa A, Queralt C, Pradas A, Taron M, Rosell R (2007) High correspondence between EGFR mutations in tissue and in circulating DNA from non-small-cell lung cancer (NSCLC) patients (p) with poor performance status (PS). J Clin Oncol 25 (18 suppl): : abstract 7505.
Novello S, Barlesi F, Califano R, Cufer T, Ekman S, Levra MG, Kerr K, Popat S, Reck M, Senan S, Simo GV, Vansteenkiste J, Peters S; ESMO Guidelines Committee (2016) Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment andfollow-up. Ann Oncol 27(suppl 5): v1–v27.
Olsen D, Jorgensen JT (2014) Companion diagnostics for targeted cancer drugs–clinical and regulatory aspects. Front Oncol 4: 105.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, Longo F, Moran T, Insa A, De Marinis F, Corre R, Bover I, Illiano A, Dansin E, de Castro J, Milella M, Reguart N, Altavilla G, Jimenez U, Provencio M, Moreno MA, Terrasa J, Munoz-Langa J, Valdivia J, Isla D, Domine M, Molinier O, Mazieres J, Baize N, Garcia-Campelo R, Robinet G, Rodriguez-Abreu D, Lopez-Vivanco G, Gebbia V, Ferrera-Delgado L, Bombaron P, Bernabe R, Bearz A, Artal A, Cortesi E, Rolfo C, Sanchez-Ronco M, Drozdowskyj A, Queralt C, de Aguirre I, Ramirez JL, Sanchez JJ, Molina MA, Taron M, Paz-Ares L (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 13 (3): 239–246.
Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, Gorbunova V, Lee KH, Shah R, Massey D, Zazulina V, Shahidi M, Schuler M (2013) Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31 (27): 3327–3334.
Steinman CR (1975) Free DNA in serum and plasma from normal adults. J Clin Invest 56 (2): 512–515.
Weber B, Meldgaard P, Hager H, Wu L, Wei W, Tsai J, Khalil A, Nexo E, Sorensen BS (2014) Detection of EGFR mutations in plasma and biopsies from non-small cell lung cancer patients by allele-specific PCR assays. BMC Cancer 14: 294.
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, Shi Y, Geater SL (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 15 (2): 213–222.
Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, Zhou C, Hu CP, O’Byrne K, Feng J, Lu S, Huang Y, Geater SL, Lee KY, Tsai CM, Gorbunova V, Hirsh V, Bennouna J, Orlov S, Mok T, Boyer M, Su WC, Lee KH, Kato T, Massey D, Shahidi M, Zazulina V, Sequist LV (2015) Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 16 (2): 141–151.
Yap TA, Popat S (2014) Toward precision medicine with next-generation EGFR inhibitors in non-small-cell lung cancer. Pharmgenomics Pers Med 7: 285–295.
Yung TK, Chan KC, Mok TS, Tong J, To KF, Lo YM (2009) Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin Cancer Res 15 (6): 2076–2084.
Zhao X, Han RB, Zhao J, Wang J, Yang F, Zhong W, Zhang L, Li LY, Wang MZ (2013) Comparison of epidermal growth factor receptor mutation statuses in tissue and plasma in stage I-IV non-small cell lung cancer patients. Respiration 85 (2): 119–125.
Acknowledgements
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Katie McClendon, PhD, of GeoMed, an Ashfield business, part of UDG Healthcare plc, during the preparation of this article. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version. This study was supported by Boehringer Ingelheim.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
LVS has participated in non-compensated consulting from Boehringer Ingelheim, Clovis Oncology, AstraZeneca, Novartis, Merrimack, Taiho, and Genentech. MSc has received grants from Novartis Pharma and personal fees from Novartis Pharma, AstraZeneca, Pfizer, GlaxoSmithKline, and Lilly. TM has received personal fees from AstraZeneca, Roche, Lilly, Merck Serono, Eisai, Bristol-Myers Squibb, AVEO, Pfizer, Boehringer Ingelheim, Novartis Pharmaceuticals, GlaxoSmithKline, Clovis Oncology, Amgen, Janssen, BioMarin Pharmaceuticals, and Threshold Pharmaceuticals. KO’B has received personal fees from Boehringer Ingelheim, MSD, Lilly, AstraZeneca, Bristol-Myers Squibb, Pfizer, and Roche. VH has participated on advisory boards for Boehringer Ingelheim. NG is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG. DM is an employee of Boehringer Ingelheim Ltd UK. JC-HY has received advisory board and speaker honoraria from Boehringer Ingelheim, AstraZeneca, Roche/Genentech, Pfizer, Novartis, MSD, Merck Serono, Clovis Oncology, and Bayer. MK is an employee of Boehringer Ingelheim GmbH. All the other authors declare no potential conflicts of interest.
Additional information
Presented, in part, at the American Society for Clinical Pathology Annual Meeting, 8–10 October 2014, Tampa, FL, USA.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wu, YL., Sequist, L., Hu, CP. et al. EGFR mutation detection in circulating cell-free DNA of lung adenocarcinoma patients: analysis of LUX-Lung 3 and 6. Br J Cancer 116, 175–185 (2017). https://doi.org/10.1038/bjc.2016.420
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2016.420
Keywords
This article is cited by
-
Liquid biopsy for detecting epidermal growth factor receptor mutation among patients with non-small cell lung cancer treated with afatinib: a multicenter prospective study
BMC Cancer (2022)
-
The value of cell-free circulating tumour DNA profiling in advanced non-small cell lung cancer (NSCLC) management
Cancer Cell International (2021)
-
Association between oligo-residual disease and patterns of failure during EGFR-TKI treatment in EGFR-mutated non-small cell lung cancer: a retrospective study
BMC Cancer (2021)
-
Routine Molecular Screening of Patients with Advanced Non-SmallCell Lung Cancer in Circulating Cell-Free DNA at Diagnosis and During Progression Using OncoBEAMTM EGFR V2 and NGS Technologies
Molecular Diagnosis & Therapy (2021)
-
Smoking habit and chemo-radiotherapy and/or surgery affect the sensitivity of EGFR plasma test in non-small cell lung cancer
BMC Research Notes (2020)
