
Abstract
Background:
We investigated the predictive value of dihydropyrimidine dehydrogenase (DPD) phenotype, measured as pretreatment serum uracil and dihydrouracil concentrations, for severe as well as fatal fluoropyrimidine-associated toxicity in 550 patients treated previously with fluoropyrimidines during a prospective multicenter study.
Methods:
Pretreatment serum concentrations of uracil and dihydrouracil were measured using a validated LC-MS/MS method. The primary endpoint of this analysis was global (any) severe fluoropyrimidine-associated toxicity, that is, grade ⩾3 toxicity according to the NCI CTC-AE v3.0, occurring during the first cycle of treatment. The predictive value of uracil and the uracil/dihydrouracil ratio for early severe fluoropyrimidine-associated toxicity were compared. Pharmacogenetic variants in DPYD (c.2846A>T, c.1679T>G, c.1129-5923C>G, and c.1601G>A) and TYMS (TYMS 5′-UTR VNTR and TYMS 3′-UTR 6-bp ins/del) were measured and tested for associations with severe fluoropyrimidine-associated toxicity to compare predictive value with DPD phenotype. The Benjamini-Hochberg false discovery rate method was used to control for type I errors at level q<0.050 (corresponding to P<0.010).
Results:
Uracil was superior to the dihydrouracil/uracil ratio as a predictor of severe toxicity. High pretreatment uracil concentrations (>16 ng ml−1) were strongly associated with global severe toxicity (OR 5.3, P=0.009), severe gastrointestinal toxicity (OR 33.7, P<0.0001), toxicity-related hospitalisation (OR 16.9, P<0.0001), as well as fatal treatment-related toxicity (OR 44.8, P=0.001). None of the DPYD variants alone, or TYMS variants alone, were associated with severe toxicity.
Conclusions:
High pretreatment uracil concentration was strongly predictive of severe, including fatal, fluoropyrimidine-associated toxicity, and is a highly promising phenotypic marker to identify patients at risk of severe fluoropyrimidine-associated toxicity.
Similar content being viewed by others

Main
Chemotherapy with fluoropyrimidines is used by over two million cancer patients each year (Scrip’s Cancer Chemotherapy Report, Scrip's World Pharmaceutical News, 2002). Of the patients treated, 10–30% experience severe treatment-related toxicity, which is lethal in 0.5–1% of the patients (Mikhail et al, 2010). Identifying biomarkers that are predictive of patients’ risk of fluoropyrimidine-associated toxicity has the potential to greatly improve the safety of large numbers of patients.
The most well-known biochemical cause of intolerance to fluoropyrimidines is deficiency of the key 5-fluorouracil (5-FU) metabolic enzyme, dihydropyrimidine dehydrogenase (DPD) (van Kuilenburg, 2004). The fact that decreased DPD activity in peripheral blood mononuclear cells (PBMCs) is found in 39–61% of the patients who experience severe toxicity, demonstrates the critical relationship between DPD activity and fluoropyrimidine-associated toxicity (Milano et al, 1999; van Kuilenburg et al, 2000, 2002; Johnson and Diasio, 2001). Polymorphisms in DPYD, the gene encoding DPD, have received wide-spread attention as predictors of fluoropyrimidine-associated toxicity, and dose adaptation based on several of these DPYD variants is now recommended by the Clinical Pharmacogenetics Implementation Consortium (CPIC) (Caudle et al, 2013). Upfront screening for the most well-known variant, IVS14+1G>A (DPYD*2A), and dose-individualization in DPYD*2A allele carriers has recently been shown to improve the safety of these patients (Caudle et al, 2013; Deenen et al, 2016).
Importantly, however, DPYD genotyping inherently has suboptimal sensitivity and positive predictive value (PPV), as a result of the fact that activity of DPD is regulated not only at the level of DPYD, but also to a relevant extent at the transcriptional level (e.g., by transcription factors SP1 and SP3) and the post-transcriptional level (e.g., by microRNA 27-a and 27-b; van Kuilenburg, 2004; Zhang et al, 2006; Offer et al, 2014; Amstutz et al, 2015; Meulendijks et al, 2016). Genetic variants in TYMS, the gene encoding 5-FU’s target thymidylate synthase (TS), have also been associated with risk of fluoropyrimidine-associated toxicity (Rosmarin et al, 2014), but in contrast to DPYD variants there is currently insufficient evidence for clinical validity of TYMS variants.
Measuring the DPD phenotype has the potential to greatly improve the performance of an upfront test to identify patients at risk of severe and potentially fatal fluoropyrimidine-associated toxicity. However, measuring DPD activity upfront on a routine basis is technically and logistically challenging, laborious, and expensive (Van Kuilenburg et al, 2000).
DPD converts its endogenous substrate uracil (U) into dihydrouracil (DHU), and the pretreatment ratio of serum concentrations of DHU to U – the DHU/U ratio – has been investigated as a phenotypic measure of systemic DPD activity. Several studies have shown that the DHU/U ratio correlates with clearance of 5-FU and with patients’ risk of toxicity (Gamelin et al, 1999; Ciccolini et al, 2004; Jiang et al, 2004; Boisdron-Celle et al, 2007; Zhou et al, 2007; Kristensen et al, 2010; Wettergren et al, 2012; Mueller et al, 2013; Galarza et al, 2016). However, the clinical applicability of the DHU/U ratio has thus far been limited, mainly due to lack of robust evidence on clinical validity. Importantly, bioanalytical issues in previous studies that mainly used HPLC-UV techniques for quantification of U and DHU might have contributed to inconsistent results, as recently emphasised (Sistonen et al, 2014). Importantly, it is unclear whether the DHU/U ratio or U concentrations alone best correlates with systemic DPD activity and risk of fluoropyrimidine-associated toxicity (Boisdron-Celle et al, 2007; Sistonen et al, 2014). While most available studies have correlated the DHU/U ratio to fluoropyrimidine-associated toxicity, a large study by Boisdron-Celle et al in 252 patients showed that while U concentration correlated with 5-FU plasma clearance (r=−0.221, P=0.006 for 5-FU de Gramont regimen and r=−0.219, P=0.05 for weekly 4 h 5-FU), the DHU/U ratio did not correlate with 5-FU clearance (Boisdron-Celle et al, 2007).
We assessed, in 550 patients treated with fluoropyrimidine-based chemotherapy during a previous prospective multicenter study, the association between pretreatment U and DHU concentrations and early severe and fatal fluoropyrimidine-associated toxicity (Deenen et al, 2016). In addition, we investigated correlations of the DHU/U ratio and U concentrations with PBMC DPD activity. Genotyping of DPYD and TYMS were also performed, to compare predictive value for severe toxicity. In this largest study to date investigating pretreatment serum U and DHU concentrations in relation to fluoropyrimidine-associated toxicity, we show that pretreatment U concentration may be of great value as a clinical predictor of severe and fatal fluoropyrimidine-associated toxicity which may complement current genotyping strategies.
Materials and methods
Patients and study design
The primary objective of this study was to assess the performance of pretreatment DPD phenotype, measured as pretreatment U concentrations or the DHU/U ratio, to identify patients at risk of severe fluoropyrimidine-associated toxicity. Genotyping of pharmacogenetic variants in DPYD and TYMS was performed to compare predictive value for severe toxicity.
A prospective multicenter study of DPYD*2A genotype-guided dosing of fluoropyrimidines (NCT00838370) in which 1631 patients were enroled and treated with standard fluoropyrimidine-based regimens (as monotherapy or in combination with other chemotherapy or radiotherapy) was the basis for this analysis (Deenen et al, 2016). The primary endpoint of NCT00838370 was severe toxicity (grade ⩾3) according to the NCI CTC-AE v3.0 (Bethesda, MD, USA).
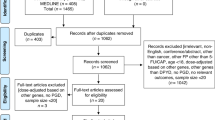
For the current analysis, a subset of patients from NCT00838370 was selected to investigate the performance of pretreatment DPD phenotype to identify patients at risk of severe and fatal toxicity (Figure 1). Pretreatment serum was only collected at the main centre at which NCT00838370 was performed (The Netherlands Cancer Institute) and these samples were used for determination of U and DHU concentrations. All patients of whom a serum sample was available were included in this analysis (see consent procedures below). Patients of whom no serum sample was available were excluded. Patients treated with fluoropyrimidine-based chemoradiotherapy regimens were excluded, to avoid interference by radiotherapy-related toxicities which are not related to DPD deficiency. Further inclusion and exclusion criteria for enrolment in NCT00838370 have been published previously (Deenen et al, 2016). Eighteen patients carrying the DPYD*2A allele were treated in NCT00838370 with an a priori reduced dose of fluoropyrimidines, and these patients were therefore also excluded from the analysis (Figure 1).

Selection of study population for analysis. DPD=dihydropyrimidine dehydrogenase; DPYD=dihydropyrimidine dehydrogenase (gene); TYMS=thymidylate synthase (gene).
This analysis was performed on patient material collected previously for diagnostic purposes (secondary use). The patient material was used in accordance with the Dutch ‘Code of conduct for responsible use of human tissue for medical research’, drawn up by the Federation of Dutch Medical Scientific Societies (FEDERA, www.federa.org). In accordance with the code, Institutional Review Board (IRB) approval was obtained for the study, and anonymized patient material was used. All patients were informed in writing about the secondary use of their tissue, and in line with the code an opt-out procedure was in place (patients could object against the use of their material at any time). Only patients who did not object to the use of their tissue were included in this study. No additional informed consent was obtained from individual patients.
Determination of pretreatment DPD phenotype and associations with fluoropyrimidine-associated toxicity
Pretreatment serum U and DHU concentrations were measured using a validated LC-MS/MS method ((Jacobs et al, 2016b) and Supplementary Material). We first determined whether U or the DHU/U ratio best correlated with DPD activity as measured in PBMCs (which is considered the gold standard for measurement of DPD phenotype) of healthy volunteers. This was done in an independent dataset of measurements in healthy volunteers (n=20, Jacobs et al, (2016a)). Akaike information criterion (AIC) was used to determine model performance, and r was calculated to assess correlations. Subsequently, it was tested in 100 bootstrap samples whether U or the DHU/U ratio resulted in the best model (lowest AIC).
It was then determined whether U or the DHU/U ratio provided the best model describing risk of global severe toxicity in the main dataset of 550 patients treated with fluoropyrimidine-based chemotherapy. This was done by comparing the AIC of a logistic regression model to predict global severe toxicity which included either U or the DHU/U ratio (as continuous variables), with adjustment for age, gender, and treatment regimen (i.e., concomitant chemotherapy; details below). This was done in the original dataset and, subsequently, in 1000 bootstrap samples to assess internal validity.
No cutoff for pretreatment DPD phenotype has yet been defined to classify patients at increased risk of fluoropyrimidine-associated toxicity. Based on the previously estimated frequency of DPD deficiency of at least 3%, we hypothesised that the patients in the lowest 3 percentiles of DPD phenotypes would be at clinically relevant increased risk, corresponding to the highest 3 percentiles of pretreatment U concentrations, percentiles 98–100 (Etienne et al, 1994; Johnson and Diasio, 2001; Van Kuilenburg et al, 2002; Mattison et al, 2006). We investigated the risk of toxicity for this group of patients. Because several studies estimated a much higher frequency of DPD deficiency (up to 61% of the patients who experience severe toxicity (van Kuilenburg, 2004)), we defined two additional groups of patients in which we investigated risk of toxicity, corresponding to percentiles 95–97 and percentiles 92–94 of pretreatment U concentrations. The remaining patients (in percentiles 1–91) were considered the reference group for tests of associations with severe toxicity.
Sensitivity analyses to determine associations between pretreatment DPD phenotype and fluoropyrimidine-associated toxicity
Sensitivity analyses were performed to investigate the robustness of observed associations between pretreatment DPD phenotype and risk of global severe toxicity. First, at each possible cutoff within the observed range of serum concentrations of U, an OR for severe toxicity and its 95% confidence interval (CI) was determined using a logistic regression model that included a factor with two levels (U above cutoff vs U below cutoff), with adjustment for age, gender, and treatment regimen. This was done first in the original dataset, and ORs and their 95% CIs were plotted against the range of cutoffs. In addition, this analysis was repeated in a bootstrap analysis in which at each cutoff 1000 bootstrap samples were drawn to estimate the risk of severe toxicity for patients with pretreatment U above the cutoff vs patients below the cutoff. For this bootstrap analysis, median ORs and their bias-corrected 95% CIs were plotted against the range of cutoffs.
Pharmacogenetic variants in DPYD and TYMS and associations with fluoropyrimidine-associated toxicity
Candidate pharmacogenetic variants in DPYD and TYMS were identified and selected based on a systematic literature search, to determine their clinical validity (Further details are available in the Supplementary Material). The selected variants were: DPYD c.2846A>T (rs67376798), DPYD c.1679T>G (rs55886062), DPYD c.1129-5923C>G (rs75017182), DPYD c.1601G>A (rs1801158), TYMS 5’-UTR VNTR (variable number of 28-bp tandem repeats, rs34743033/rs45445694), and TYMS 3’-UTR 6-bp ins/del (rs11280056). DPYD*2A was not included, as patients carrying this allele were already excluded from this analysis in view of the dose-adapted treatment they received in NCT00838370. The PCR methods used to determine genotypes are detailed in the Supplementary Material.
All variants were tested for deviation from Hardy–Weinberg equilibrium using the exact test (Wigginton et al, 2005). Associations between pharmacogenetic variants in DPYD and severe toxicity were analysed under dominant models. For the TYMS variants, log-additive, recessive, and dominant models were investigated. In addition, the TYMS risk score (=the total number of risk alleles present for the TYMS 5′-UTR VNTR and TYMS 3′-UTR 6-bp ins/del polymorphisms, as proposed by (Rosmarin et al, 2014)), was investigated. The pharmacogenetic analysis was performed in the same patients as selected for analysis of DPD phenotype (Figure 1). Because DNA samples were available for all 1613 patients who were found to be DPYD*2A wild type in study NCT00838370, a secondary pharmacogenetic analysis was performed in this entire cohort, in order to further characterise the predictive value of these genotypes for severe toxicity.
Endpoints and data analysis
The primary endpoint of this analysis was global (any) severe fluoropyrimidine-associated toxicity, that is, grade ⩾3 toxicity according to the NCI CTC-AE v3.0, occurring during the first cycle of treatment. Global (any) severe toxicity and individual types of severe toxicity, gastrointestinal toxicity, haematological toxicity, hand-foot syndrome, and cardiological toxicity were dichotomized as absent to moderate (grade 0–2) vs severe (grade 3–5). Considering only cycle one for severe toxicity assessment was considered most adequate, because in an analysis of the entire treatment duration using logistic regression there are large differences in treatment duration and consequent fluoropyrimidine-exposure between patients which are not corrected for. This potentially biases the results, for example, as a result of attrition bias or treatment modifications during the course of therapy, such as dose reductions. Also, with increasing treatment duration the cumulative incidence of severe toxicity increases, thereby reducing sensitivity to estimate differences in risk for severe toxicity between groups, due to the fact that the OR will gradually trends towards 1.0 with increasing cumulative incidence (further detailed in Supplementary Material of Meulendijks et al, (2015)).
Associations of the DPD phenotype and of the pharmacogenetic variants with severe toxicity were tested in logistic regression models, with adjustment for age (continuous), gender, and treatment regimen (categorised as capecitabine monotherapy, capecitabine plus platinum, capecitabine plus taxane, capecitabine-based triplet combination, capecitabine plus other drug, or 5-FU-based chemotherapy). The starting dose of capecitabine was highly collinear with type of regimen and was not predictive of toxicity after adjustment for treatment regimen; it was therefore not included in the models. Associations with toxicity-related hospitalisation during the first cycle, and with fatal fluoropyrimidine-associated toxicity were assessed in separate analyses, using the same covariables. For testing associations with fatal fluoropyrimidine-associated toxicity, the whole treatment duration was taken into account, in view of the low number of events of fatal toxicity.
In order to control for type I errors as a result of testing associations with toxicity for four variants in DPYD, two variants in TYMS, and three groups based on low DPD phenotype, the Benjamini-Hochberg false discovery rate (FDR) method was used (Benjamini and Hochberg, 1995). A FDR rate of q<0.050 was used, which corresponded to P<0.010 for testing associations with global severe toxicity. The same threshold was applied for the individual types of toxicity and toxicity-related outcomes, because they were assumed to be dependent on global toxicity. Statistical tests resulting in P<0.010 can therefore be considered formally significant and those that achieved P<0.050 as nominally significant. Sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and F1 score (the harmonic mean of sensitivity and PPV) for predicting severe toxicity were calculated for DPYD variants and the DPD phenotype.
The effects of DPYD variants on DPD phenotype were assessed. Kruskal–Wallis and Mann–Whitney U-tests were used to test for differences in DPD phenotype between patients according to DPYD variants. The threshold for significance for the latter analysis was P<0.050. All statistical analyses were performed using R v3.1.0 (Vienna, Austria).
Results
Patients
The process of patient selection for analysis is shown in Figure 1. Patient and treatment characteristics, and frequencies of adverse events are summarised in Table 1.
Pretreatment U concentration as a predictor of severe and fatal fluoropyrimidine-associated toxicity
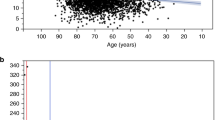
It was first analysed whether U or the DHU/U ratio best correlated with PBMC DPD activity. This analysis showed that pretreatment U was superior to the DHU/U ratio (Supplementary Material and Figure 2A).

Correlation between pretreatment serum uracil concentrations and DPD activity in healthy volunteers and distribution of uracil concentrations in patients. Correlation between dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells and uracil plasma levels (r=−0.51, P=0.023) in 20 healthy volunteers (A, Figure adapted from Jacobs et al (2016a) with permission). Distribution of pretreatment serum uracil concentrations in the entire cohort of 550 patients treated with fluoropyrimidine-based chemotherapy (B). U=uracil; DPD=dihydropyrimidine dehydrogenase.
Subsequently, the association between pretreatment U concentration and severe toxicity was analysed. As expected based on correlations with PBMC DPD activity, also in relation to predicting severe toxicity U was superior to the DHU/U ratio based on AIC (AIC 363.5 for basic clinical model with age, gender, and treatment regimen as covariables, AIC 357.7 for basic clinical model plus U concentration, and AIC 362.0 for basic clinical model plus the DHU/U ratio). Also in bootstrap analysis U concentration was the superior predictor (Supplementary Material).
Uracil as a continuous variable was strongly predictive of global severe toxicity (OR 2.75 per 10 ng ml−l, 95% CI 1.39–5.44, P=0.004), gastrointestinal toxicity (OR 5.58, 95% CI 2.08–14.9, P=0.0006), toxicity-related hospitalisation (OR 2.53, 95% CI 1.23–5.19, P=0.011), and fatal treatment-related toxicity (OR 5.11, 95% CI 1.56–16.7, P=0.007), but not significantly associated with haematological toxicity (OR 1.53, 95% CI 0.59–3.96, P=0.383).
Pretreatment U concentrations were between 3.2 and 38.2 ng ml−l (Figure 2B). Based on the observed distribution for pretreatment U, the upper three percentiles of pretreatment U concentrations corresponded to U>16 ng ml−l (n=17). The next two groups were 13.9–16 ng ml−1 (n=17) and 13–13.8 ng ml−1 (n=16), respectively (Figure 2B).
As shown in Figure 3, patients with pretreatment U>16 ng ml−l, as well as patients with U⩾13.9–16 ng ml−1, were at significantly increased risk of global severe toxicity compared to patients with low pretreatment U (<13 ng ml−1), with OR 8.2 (P=0.0004) and OR 5.3 (P=0.0087) for groups U⩾13.9–16 ng ml−1 and U>16 ng ml−1, respectively. Pretreatment U concentration was also strongly associated with risk of gastrointestinal toxicity and toxicity-related hospitalisation in patients with pretreatment U⩾13.9 ng ml−1. For patients with pretreatment U concentrations 13–13.8 ng ml−1, risks of toxicity outcomes were not significantly increased.

Associations of pretreatment serum uracil concentrations with toxicity outcomes. Associations of pretreatment serum uracil concentrations with toxicity outcomes in the entire population of 550 patients. * 2/17 patients (12%) in the uracil >16 ng ml−l group had fatal treatment-related toxicity, compared to 2/500 patients (0.4%) among patients with uracil concentrations <13 ng ml−1. Fatal treatment-related toxicity did not occur among patients with pretreatment U concentrations of 13–13.8 or 13.9–16 ng ml−1. Associations with fatal toxicity were determined with adjustment for age and gender but not treatment regimen (due to the low number of events). OR=odds ratio; CI=confidence interval.
While fatal treatment-related toxicity was rare in the group of patients with normal pretreatment U (<13 ng ml−1), with 2/500 patients (0.4%) suffering fatal treatment-related toxicity, in the group of patients with pretreatment U concentrations>16 ng/ml, two out of 18 patients (11%) suffered fatal toxicity (P=0.0011, Figure 3).
Figure 4 depicts the results of the sensitivity analysis in which at each possible cutoff between serum U 6–16 ng ml−l an OR for severe toxicity with its corresponding 95% CI were determined using logistic regression. The OR reflects risk of severe toxicity for patients with serum U above the cutoff depicted on the x-axis vs patients who are below the cutoff with adjustment for age, gender, and treatment regimen. Risk of global severe toxicity (Figure 4A) and severe gastrointestinal toxicity (Figure 4B) were found to increase proportionally with increasing pretreatment U concentration. Bootstrap analysis confirmed these results (Supplementary Material), and when pretreatment U was plotted against the log odds for severe toxicity using spline regression, a linear increase in the risk of severe toxicity was observed over the range of serum U concentration of 8–20 ng ml−1 (Supplementary Material).

Risk of global severe toxicity and severe gastrointestinal toxicity at varying cutoff levels for pretreatment serum uracil. Results from the analysis to estimate the risk of global severe (grade ⩾3) toxicity and severe gastrointestinal toxicity at varying cutoffs for pretreatment uracil concentration in the original dataset, adjusted for age, gender, and treatment regimen. The solid line depicts the estimated odds ratio for risk of severe toxicity for patients with pretreatment uracil concentrations above the cutoff, vs patients with uracil concentrations below the cutoff. The dashed lines represent 95% confidence intervals. Odds ratios and 95% confidence intervals are shown on a log scale.
Associations between pharmacogenetic variants in DPYD and TYMS and fluoropyrimidine-associated toxicity
All pharmacogenetic variants were in Hardy–Weinberg equilibrium (P<0.05). None of the individual DPYD variants were found to be significantly associated with global severe toxicity (Figure 5A). With regard to individual subtypes of toxicity outcomes, associations were found between DPYD c.1129-5923C>G and toxicity-related hospitalisation (OR 3.2, P=0.047), DPYD c.1601G>A and gastrointestinal toxicity (OR 5.0, P=0.026), and DPYD c.1601G>A and toxicity-related hospitalisation (OR 3.1, P=0.018). When the DPYD variants were combined, they were found to be associated with gastrointestinal toxicity (OR 4.1, P=0.017) and associated with toxicity-related hospitalisation (OR 3.3, P=0.002). None of the TYMS variants were associated with toxicity outcomes (Figure 5B).

Results of the pharmacogenetic analysis. Results of the pharmacogenetic analysis investigating associations between DPYD variants (A) and TYMS variants (B) in the primary cohort of 550 patients. The frequencies of early hand-foot syndrome and cardiological toxicity were too low in the population to investigate associations with these individual types of toxicity. The associations mentioned as ‘not estimable’ could not be estimated due to too few events of severe toxicity. For the TYMS variants, the results are shown for the log-additive pharmacogenetic model. The other models (dominant, recessive, or risk score) resulted in similar, non-significant, associations between TYMS genotypes and toxicity outcomes (details not shown). Results of the pharmacogenetic analysis in the larger cohort of 1613 patients are shown in C. OR=odds ratio; CI=confidence interval.
When the entire cohort of 1613 patients was genotyped, none of the individual DPYD variants were found to be associated with global severe toxicity, nor were the TYMS variants (Figure 5C). For c.2846A>T and c.1679T>G combined, there was evidence for an association with global severe toxicity (OR 3.0, 95% CI 1.05–8.77, P=0.040). In addition, DPYD c.1679T>G alone was associated with haematological toxicity (OR 24.9, 95% CI 1.74–354, P=0.018). The four DPYD variants, combined, were associated with toxicity-related hospitalisation (OR 2.1, 95% CI 1.20–3.63, P=0.009). There were no significant associations between DPYD variants and fatal treatment-related toxicity (OR 3.0, P=0.202, for all DPYD variants combined, not shown in Figure 5C).
Associations between DPYD variants and increased pretreatment U concentration
Figure 6 shows the relationship between DPYD variants and pretreatment U concentration. Overall, the DPYD variants (combined) were significantly associated with U concentrations (P=0.009). Both c.2846A>T and c.1679T>G were individually associated with increased U concentrations (P<0.001 and P=0.024, respectively). In contrast, c.1129-5923C>G and c.1601G>A were not associated with pretreatment U (P=0.105 and P=0.431, respectively).

Relationships between DPYD variants and pretreatment uracil concentrations. The figure shows pretreatment serum uracil concentrations by DPYD variant. The horizontal lines represent median concentrations. Overall, DPYD variants were associated with an increase of pretreatment uracil concentrations of 12% (P=0.003). The c.2846A>T and c.1679T>G variants were associated with significantly higher uracil concentrations (+82%, P<0.001 and +41%, P=0.024, respectively). In contrast, c.1129-5923C>G and c.1601G>A were not significantly associated with pretreatment uracil concentrations (+12%, P=0.105 and −1%, P=0.431, respectively). * wild type for DPYD*2A, c.2846A>T, c.1679T>G, c.1129-5923C>G, and c.1601G>A. ** the cohort of 550 patients contained 2 patients with the c.1679T>G variant. In view of the low frequency of this variant, the third patient who carried c.1679T>G from the entire cohort of 1613 patients was phenotyped solely for the purpose of this analysis investigating the association between DPYD variants and pretreatment serum uracil concentration (this patient received chemoradiotherapy and was therefore excluded from the main analysis).
Performance of DPYD variants and pretreatment U to predict early severe toxicity
Sensitivity, specificity, PPV, NPV, and F1 score for first cycle global toxicity were calculated for DPYD genotyping and pretreatment phenotyping, as well as for combination strategies. To assess diagnostic accuracy of genotyping, we combined the DPYD variants which were clinically validated in a recent meta-analysis which was published while preparing the current manuscript, that is, c.2846A>T, c.1679T>G, and c.1129-5923C>G (Meulendijks et al, 2015). The variant DPYD c.1601G>A was not included is these calculations as this variant could not be clinically validated in the meta-analysis (Meulendijks et al, 2015).
For combined genotyping of c.2846A>T, c.1679T>G, and c.1129-5923C>G, sensitivity was 6%, PPV 13%, specificity 95%, and NPV 88%. For pretreatment U (at a cutoff of ⩾13.9 ng ml−1), sensitivity was 18%, PPV 35%, specificity 95%, and NPV 90%. Since sensitivity and PPV are considered the most relevant parameters in this context, we also calculated the F1 score, the harmonic mean of sensitivity and PPV. F1 score was 8% for genotyping of variants c.2846A>T, c.1679T>G, and c.1129-5923C>G, while it was 24% for phenotyping alone (cutoff for U concentration ⩾13.9 ng ml−1). For genotyping and phenotyping combined, sensitivity was 22%, PPV 24%, specificity 91%, NPV 90%, and F1 score 23%. Further data on diagnostic accuracy are detailed in the Supplementary Material.
Discussion
In this study we showed that high pretreatment serum U concentration (>16 ng ml−1) was strongly associated with severe fluoropyrimidine-associated toxicity (OR 5.3, P=0.0087), as well as fatal fluoropyrimidine-associated toxicity (OR 44.8, P=0.0011), and risk of severe toxicity increased proportionally with increasing serum U concentration. Sensitivity to identify patients at risk of early severe toxicity was three times higher for phenotyping (18%), than for genotyping of the established DPYD variants c.2846A>T, c.1679T>G, and c.1129-5923C>G (6%) (Meulendijks et al, 2015). Similarly, PPV of phenotyping was found to be 35 vs 13% for genotyping of the established DPYD variants. Combined genotyping and phenotyping did not further improve diagnostic accuracy compared to phenotyping alone.
In contrast to the strong predictive value of U concentration, known DPYD genotypes appeared only moderately predictive of severe toxicity in the same patient population. The TYMS variants, which were previously found to have a modest association with severe fluoropyrimidine-associated toxicity in a meta-analysis (OR 1.36 for TYMS 5′-VNTR, P<0.001; and OR 1.25 for TYMS 3′-UTR, P=0.02; Rosmarin et al, 2014), were not associated with toxicity in our study. In view of the modest effect size observed for these variants, both in this study and the previous meta-analysis, the value of these pharmacogenetics variants in TYMS in clinical practice should be questioned.
Our results suggest that pretreatment serum U concentration can potentially strongly improve an upfront test to identify patients with DPD deficiency who are at high risk of severe and potentially fatal toxicity. Concentration of U was found to correlate better with PBMC DPD activity (in healthy volunteers) and better predicted toxicity than the DHU/U ratio in patients. These findings may be explained by the metabolism of U and DHU. Uracil is metabolised via three sequential reactions: U is converted into DHU by DPD, which is converted into beta-ureidopropionate by dihydropyrimidinase, which is converted into beta-alanine, ammonia, and CO2 by beta-ureidopropionase. The activity of the first enzyme in this scheme, DPD, is the main determinant of toxicity upon treatment with 5-FU because 5-FU is converted by DPD into the non-cytotoxic compound 5,6-dihydrofluorouracil. Ito et al modelled the catabolism of U, and showed that in the overall cascade of enzymes, dihydropyriminidase (which converts DHU into beta-ureidopropionate) is rate-limiting (Ito et al, 2005). This is reflected by the approximately 10 times higher concentrations of DHU compared to U in plasma, and indicates that concentrations of DHU are mainly determined by dihydropyrimidinase, and not by DPD. This may explain our finding that taking into account concentrations of DHU in addition to U – by incorporating it in the DHU/U ratio – did not lead to better prediction of toxicity than by using U concentration alone. Further supportive data come from a study in which 500 mg m−2 U was administered to a group of individuals with DPD deficiency and a group with normal DPD activity (van Staveren et al, 2011). It was found that while exposure to U (measured as AUC) was 230% in DPD deficient subjects (130% increased) compared to subjects with normal DPD activity, the difference in DHU exposure was much smaller (25% reduction in exposure in DPD deficient subjects compared to DPD proficient subjects).
In our study U and DHU concentrations in relation to fluoropyrimidine-associated toxicity were measured using a validated LC-MS/MS method (Jacobs et al, 2016b). This is of relevance, since previous studies mainly used methods based on UV detection and, as shown recently, there has been large variability in the reported ranges for the DHU/U ratio in these studies, which could indicate that bioanalytical issues may have negatively affected results in previous studies (Sistonen et al, 2014).
Determination of an optimal cutoff for pretreatment U concentration to identify patients at risk of severe toxicity was not a formal aim in our study. Based on our results, however, it appears that it can be safely assumed that there is a clinically relevant increase in risk of severe toxicity above 16 ng/ml, and possibly for patients with pretreatment U⩾13.9–16 ng ml−1.
Dose adaptation in patients with elevated pretreatment U concentration has the potential to increase the safety of DPD deficient patients at high risk of severe and fatal toxicity, and prescreening could be an effective approach to improve patient safety. Determination of the threshold for dose adaptation should therefore be an important objective of future studies.
A limitation of our study is that we did not externally validate the clinical validity of U concentration in an independent cohort of patients treated with fluoropyrimidines. However, we did validate the association between U concentration and PBMC DPD activity in a small independent dataset of healthy volunteers. We also performed different sensitivity analyses, which showed that risk of severe toxicity increased proportionally with increasing pretreatment U concentration, supporting the validity of our findings, which are further backed up by previous smaller studies suggesting the clinical validity of pretreatment DPD phenotype (Boisdron-Celle et al, 2007; Zhou et al, 2007; Kristensen et al, 2010; Wettergren et al, 2012). We are currently undertaking a prospective validation study to replicate our current findings in 1250 patients (NCT02324452).
Our findings should be interpreted in the context of the fact that patients with the DPYD*2A variant were excluded from the analysis (n=18, 1% of a typical Caucasian population). Calculations of diagnostic accuracy could therefore be affected as a result, but it is expected that this will only affect sensitivity and PPV to a minor extent in view of the low frequency of DPYD*2A. Furthermore, we found that DPYD*2A genotype strongly correlates with high pretreatment U concentration (unpublished observations), indicating that pretreatment U concentration is also able to identify these patients.
In conclusion, this study indicates that pretreatment U is a highly promising phenotypic marker with high sensitivity and PPV to identify patients at high risk of fluoropyrimidine-associated toxicity, that could be used alone or in combination with DPYD genotype-based dose-individualization to improve patient safety. The safety of 3–6% of the patients treated with fluoropyrimidines could thereby be improved. Prospective investigations to confirm the clinical validity and, importantly, the clinical utility of pretreatment U concentration are now warranted.
Change history
23 May 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amstutz U, Offer SM, Sistonen J, Joerger M, Diasio RB, Largiadèr CR (2015) Polymorphisms in MIR27A Associated with Early-Onset Toxicity in Fluoropyrimidine-Based Chemotherapy. Clin Cancer Res 21: 2038–2044.
Benjamini Y, Hochberg Y (1995) Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J R Stat Soc B 57: 289–300.
Boisdron-Celle M, Remaud G, Traore S, Poirier AL, Gamelin L, Morel A, Gamelin E (2007) 5-Fluorouracil-related severe toxicity: a comparison of different methods for the pretherapeutic detection of dihydropyrimidine dehydrogenase deficiency. Cancer Lett 249: 271–282.
Caudle KE, Thorn CF, Klein TE, Swen JJ, McLeod HL, Diasio RB, Schwab M (2013) Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clin Pharmacol Ther 94: 640–645.
Ciccolini J, Mercier C, Blachon M-F, Favre R, Durand A, Lacarelle B (2004) A simple and rapid high-performance liquid chromatographic (HPLC) method for 5-fluorouracil (5-FU) assay in plasma and possible detection of patients with impaired dihydropyrimidine dehydrogenase (DPD) activity. J Clin Pharm Ther 29: 307–315.
Deenen MJ, Meulendijks D, Cats A, Sechterberger MK, Severens JL, Boot H, Smits PHM, Rosing H, Mandigers CMPWPW, Soesan M, Beijnen JH, Schellens JHMM (2016) Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J Clin Oncol 34: 227–234.
Etienne MC, Lagrange JL, Dassonville O, Fleming R, Thyss A, Renée N, Schneider M, Demard F, Milano G (1994) Population study of dihydropyrimidine dehydrogenase in cancer patients. J Clin Oncol 12: 2248–2253.
Galarza AFA, Linden R, Antunes MV, Hahn RZ, Raymundo S, da Silva ACC, Staggemeier R, Spilki FR, Schwartsmann G (2016) Endogenous plasma and salivary uracil to dihydrouracil ratios and DPYD genotyping as predictors of severe fluoropyrimidine toxicity in patients with gastrointestinal malignancies. Clin Biochem 49 (16-17): 1221–1226.
Gamelin E, Boisdron-Celle M, Guérin-Meyer V, Delva R, Lortholary A, Genevieve F, Larra F, Ifrah N, Robert J (1999) Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5-FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: A potential interest for predicting 5-FU toxicity and determining optimal 5-FU dosage. J Clin Oncol 17: 1105.
Ito S, Kawamura T, Inada M, Inoue Y, Hirao Y, Koga T, Kunizaki J, Shimizu T, Sato H (2005) Physiologically based pharmacokinetic modelling of the three-step metabolism of pyrimidine using C-uracil as an in vivo probe. Br J Clin Pharmacol 60: 584–593.
Jacobs BAW, Deenen MJ, Pluim D, van Hasselt JGC, Krähenbühl MD, van Geel RMJM, de Vries N, Rosing H, Meulendijks D, Burylo A, Cats AM, Beijnen JH, Huitema ADR, Schellens JHM (2016a) Pronounced between-subject and circadian variability in thymidylate synthase and dihydropyrimidine dehydrogenase enzyme activity in human volunteers. Br J Clin Pharmacol 82 (3): 706–716.
Jacobs BAW, Rosing H, de Vries N, Meulendijks D, Henricks LM, Schellens JHM, Beijnen JH (2016b) Development and validation of a rapid and sensitive UPLC–MS/MS method for determination of uracil and dihydrouracil in human plasma. J Pharm Biomed Anal 126: 75–82.
Jiang H, Lu J, Jiang J, Hu P (2004) Important role of the dihydrouracil/uracil ratio in marked interpatient variations of fluoropyrimidine pharmacokinetics and pharmacodynamics. J Clin Pharmacol 44: 1260–1272.
Johnson MR, Diasio RB (2001) Importance of dihydropyrimidine dehydrogenase (DPD) deficiency in patients exhibiting toxicity following treatment with 5-fluorouracil. Adv Enzyme Regul 41: 151–157.
Kristensen MH, Pedersen P, Mejer J (2010) The value of dihydrouracil/uracil plasma ratios in predicting 5-fluorouracil-related toxicity in colorectal cancer patients. J Int Med Res 38: 1313–1323.
van Kuilenburg ABP (2004) Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer 40: 939–950.
van Kuilenburg ABP, Haasjes J, Richel DJ, Zoetekouw L, Van Lenthe H, De Abreu RA, Maring JG, Vreken P, van Gennip AH (2000) Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD gene. Clin Cancer Res 6: 4705–4712.
Van Kuilenburg ABP, Van Lenthe H, Tromp A, Veltman PC, Van Gennip AH (2000) Pitfalls in the diagnosis of patients with a partial dihydropyrimidine dehydrogenase deficiency. Clin Chem 46: 9–17.
Van Kuilenburg ABP, Meinsma R, Zoetekouw L, Van Gennip AH (2002) Increased risk of grade IV neutropenia after administration of 5-fluorouracil due to a dihydropyrimidine dehydrogenase deficiency: high prevalence of the IVS14+1g>a mutation. Int J Cancer 101: 253–258.
Mattison LK, Fourie J, Desmond RA, Modak A, Saif MW, Diasio RB (2006) Increased prevalence of dihydropyrimidine dehydrogenase deficiency in African-Americans compared with Caucasians. Clin Cancer Res 12: 5491–5495.
Meulendijks D, Henricks LM, Amstutz U, Froehlich TK, Largiadèr CR, Beijnen JH, de Boer A, Deenen MJ, Cats A, Schellens JHM (2016) Rs895819 in MIR27A improves the predictive value of DPYD variants to identify patients at risk of severe fluoropyrimidine-associated toxicity. Int J Cancer 138: 2752–2761.
Meulendijks D, Henricks LM, Sonke GS, Deenen MJ, Froehlich TK, Amstutz U, Largiadèr CR, Jennings BA, Marinaki AM, Sanderson JD, Kleibl Z, Kleiblova P, Schwab M, Zanger UM, Palles C, Tomlinson I, Gross E, van Kuilenburg ABP, Punt CJA, Koopman M, Beijnen JH, Cats A, Schellens JHM (2015) Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: a systematic review and meta-analysis of individual patient data. Lancet Oncol 16: 1639–1650.
Mikhail SE, Sun JF, Marshall JL (2010) Safety of capecitabine: a review. Expert Opin Drug Saf 9: 831–841.
Milano G, Etienne MC, Pierrefite V, Barberi-Heyob M, Deporte-Fety R, Renée N (1999) Dihydropyrimidine dehydrogenase deficiency and fluorouracil-related toxicity. Br J Cancer 79: 627–630.
Mueller F, Büchel B, Köberle D, Schürch S, Pfister B, Krähenbühl S, Froehlich TK, Largiader CR, Joerger M (2013) Gender-specific elimination of continuous-infusional 5-fluorouracil in patients with gastrointestinal malignancies: results from a prospective population pharmacokinetic study. Cancer Chemother Pharmacol 71: 361–370.
Offer SM, Butterfield GL, Jerde CR, Fossum CC, Wegner NJ, Diasio RB (2014) microRNAs miR-27a and miR-27b directly regulate liver dihydropyrimidine dehydrogenase expression through two conserved binding sites. Mol Cancer Ther 13: 742–751.
Rosmarin D, Palles C, Church D, Domingo E, Jones A, Johnstone E, Wang H, Love S, Julier P, Scudder C, Nicholson G, Gonzalez-Neira A, Martin M, Sargent D, Green E, McLeod H, Zanger UM, Schwab M, Braun M, Seymour M, Thompson L, Lacas B, Boige V, Ribelles N, Afzal S, Enghusen H, Jensen SA, Etienne-Grimaldi M-C, Milano G, Wadelius M, Glimelius B, Garmo H, Gusella M, Lecomte T, Laurent-Puig P, Martinez-Balibrea E, Sharma R, Garcia-Foncillas J, Kleibl Z, Morel A, Pignon J-P, Midgley R, Kerr D, Tomlinson I, Astrup Jensen S (2014) Genetic Markers of Toxicity From Capecitabine and Other Fluorouracil-Based Regimens: Investigation in the QUASAR2 Study, Systematic Review, and Meta-Analysis. J Clin Oncol 32: 1031–1039.
Scrip’s Cancer Chemotherapy Report, Scrip’s World Pharmaceutical News (2002). PJB Publications Ltd: London, UK.
Sistonen J, Büchel B, Froehlich TK, Kummer D, Fontana S, Joerger M, van Kuilenburg AB, Largiadèr CR (2014) Predicting 5-fluorouracil toxicity: DPD genotype and 5,6-dihydrouracil:uracil ratio. Pharmacogenomics 15: 1653–1666.
van Staveren MC, Theeuwes-Oonk B, Guchelaar HJ, van Kuilenburg ABP, Maring JG (2011) Pharmacokinetics of orally administered uracil in healthy volunteers and in DPD-deficient patients, a possible tool for screening of DPD deficiency. Cancer Chemother Pharmacol 68: 1611–1617.
Wettergren Y, Carlsson G, Odin E, Gustavsson B (2012) Pretherapeutic uracil and dihydrouracil levels of colorectal cancer patients are associated with sex and toxic side effects during adjuvant 5-fluorouracil-based chemotherapy. Cancer 118: 2935–2943.
Wigginton JE, Cutler DJ, Abecasis GR (2005) A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 76: 887–893.
Zhang X, Li L, Fourie J, Davie JR, Guarcello V, Diasio RB (2006) The role of Sp1 and Sp3 in the constitutive DPYD gene expression. Biochim Biophys Acta 1759: 247–256.
Zhou ZW, Wang GQ, Wan D, Sen, Lu ZH, Chen YB, Li S, Chen G, Pan ZZ (2007) The dihydrouracil/uracil ratios in plasma and toxicities of 5-fluorouracil-based adjuvant chemotherapy in colorectal cancer patients. Chemotherapy 53: 127–131.
Acknowledgements
The study was funded by The Netherlands Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Meulendijks, D., Henricks, L., Jacobs, B. et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine-associated toxicity. Br J Cancer 116, 1415–1424 (2017). https://doi.org/10.1038/bjc.2017.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.94
Keywords
This article is cited by
-
Integrating rare genetic variants into DPYD pharmacogenetic testing may help preventing fluoropyrimidine-induced toxicity
The Pharmacogenomics Journal (2024)
-
Personalizing adjuvant therapy for patients with colorectal cancer
Nature Reviews Clinical Oncology (2024)
-
Impact of pharmacogenomic DPYD variant guided dosing on toxicity in patients receiving fluoropyrimidines for gastrointestinal cancers in a high-volume tertiary centre
BMC Cancer (2023)
-
Evaluation of UGT1A1 and CYP3A Genotyping and Single-Point Irinotecan and Metabolite Concentrations as Predictors of the Occurrence of Adverse Events in Cancer Treatment
Journal of Gastrointestinal Cancer (2023)
-
Assay performance and stability of uracil and dihydrouracil in clinical practice
Cancer Chemotherapy and Pharmacology (2023)
