
Abstract
Oncolytic viruses can be found at the confluence of virology, genetic engineering and pharmacology where versatile platforms for molecularly targeted anticancer agents can be designed and optimised. Oncolytic viruses offer several important advantages over traditional approaches, including the following. (1) Amplification of the active agent (infectious virus particles) within the tumour. This avoids unnecessary exposure to normal tissues experienced during delivery of traditional stoichiometric chemotherapy and maximises the therapeutic index. (2) The active cell-killing mechanisms, often independent of programmed death mechanisms, should decrease the emergence of acquired drug resistance. (3) Lytic death of cancer cells provides a pro-inflammatory microenvironment and the potential for induction of an anticancer vaccine response. (4) Tumour-selective expression and secretion of encoded anticancer biologics, providing a new realm of potent and cost-effective-targeted therapeutics.
Similar content being viewed by others

Main
The current upsurge of interest in oncolytic viruses reflects the culmination of incremental scientific progress gained over several years that is now being translated into the first reproducible clinical successes (Russell et al, 2012; Chiocca and Rabkin, 2014). There is also an increasing trend to demystifying oncolytic viruses and regarding them simply as a new type of drug, with appropriate consideration of their bioavailability and pharmacokinetics. Indeed, the first oncolytic virus was licensed by FDA in October 2015 to Amgen (Thousand Oaks, CA, USA) for the treatment of advanced melanoma. In this short review, we aim to highlight the main aspects of the field that have advanced in recent years with a view to explaining its current high profile, and we assess how oncolytic viruses may be best deployed in cancer treatment in the near future.
Cancer selectivity of wild-type lytic viruses
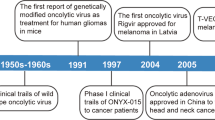
Many wild-type viruses show an intrinsic selectivity for replication within cancer cells. The first systematic studies of therapeutic potential of this approach were performed in the 1950s, involving administration of the West Nile virus Egypt 101 and wild-type adenovirus to patients with advanced disease, leading to encouraging anticancer effects (Kelly and Russell, 2007). Other wild-type viruses that have shown clinical promise include Seneca Valley virus, Newcastle disease virus and reovirus, which are still under active development in a range of tumour types. The mechanism underlying the intrinsic cancer.selectivity of several wild-type viruses is a topic of intense discussion, and is likely to reflect the activated nature of tumour cells, providing a cellular pathology that is conducive to efficient virus replication. This is considered in greater detail below, where there appears to be considerable overlap between the hallmarks of cancer and the hallmarks of virus infection (Figure 1).

The hallmarks of adenovirus infection. Cellular phenotypic adaptations associated with cancer, known as the ‘Hallmarks of cancer’ (Hanahan and Weinberg, 2011) bear many similarities to the ‘Hallmarks of virus infection’ of normal cells. This may contribute to the cancer selectivity often associated with lytic viruses, and underlies the concept of design of oncolytic viruses for cancer selectivity through phenotypic complementation.
Rational design of oncolytic viruses
Recombinant oncolytic viruses are normally designed to be inactive within non-transformed cells, but to respond to the tumour cell phenotype and achieve cancer-selective replication, cell lysis and spread of progeny virus to infect nearby cells. Advances in our understanding of both cancer cell biology and virology provide several strategies to achieve this. In perhaps the simplest approach, DNA-based oncolytic viruses that exploit cellular transcription machinery (notably, adenovirus and herpes virus) can be engineered to depend on tumour-associated transcription factors to promote virus replication, using tumour-associated promoters to regulate expression of essential viral genes. There are many example of this elegant approach using a range of cancer-selective promoters, for example, the use of human prostate-specific antigen promoter enhancer and rat probasin promoter to regulate an oncolytic adenovirus for treatment of prostate cancer (Small et al, 2006) or the telomerase promoter hTERT in controlling activity of a CD40 ligand-expressing oncolytic adenovirus (Pesonen et al, 2012).
An alternative rational design strategy for cancer.selectivity involves genetic removal of essential virus functions that can be trans-complemented by the acquired features of the tumour cell phenotype. In this approach, the genetically attenuated virus will be inactive within non-transformed cells and activated only when it infects tumour cells. This concept can be illustrated by considering similarities between the ‘Hallmarks of cancer’, elaborated so effectively by Hanahan and Weinberg (2011) to highlight the changes that occur to cells upon malignant transformation and what we might term the ‘hallmarks of virus infection’ – referring to the changes that occur following virus infection of a normal cell. Many of the ‘hallmarks’ are shared between the malignant phenotype and the phenotype of normal cells infected with virus, even though the reason for their existence is so different. The similarities between viral infection and malignant transformation provided the rationale, even 40 years ago, for the use of virus genes (e.g., SV40 Large T) to provide a model for the study of malignant transformation. The precise hallmarks of virus infection vary slightly between different virus types, and Figure 1 overviews some of the cellular hallmarks of adenovirus infection.
Interestingly, although none of the ‘multicellular’ cancer hallmarks are thought to be shared by adenovirus-infected cells (notably, metastasis and angiogenesis), virtually all of the hallmarks that relate to the individual cancer cell phenotype are mirrored in virus-infected cells. These include deregulation of metabolism, away from oxidative phosphorylation and towards a more glycolytic phenotype that produces nutrients for anabolism and synthesis of new cells and viruses. Similarly, adenovirus-infected cells activate cell cycle-associated enzymes and resist activation of p53-dependent apoptosis. All of these ‘hallmarks of virus infection’ have direct correlates in the tumour cell phenotype. By engineering viruses genetically to lack the ability to mediate any or all of these cellular changes, which are part of the normal viral infectious cycle, it is possible to create a virus that relies on phenotypic complementation by tumour cells and consequently shows cancer-selective replication.
The first agent to exploit this ‘phenotype-complementation’ principle was the adenovirus Onyx-015, which lacked the virus protein E1B55KDa. This deletion was intended to remove the ability of Onyx-015 to inactivate cellular p53, hence successful completion of the virus life cycle would be dependent on the presence of inactivated p53 in tumour cells. Onyx-015 was developed successfully in a series of phase I and II clinical trials in the United States (Kirn, 2001) and eventually formed the basis of the oncolytic adenovirus H101, which received a product licence (‘Oncorine’) in China. Subsequently, it emerged that the cancer selectivity resulting from deletion of E1B55KDa was more related to nuclear export of late virus mRNA, which occurred more efficiently in many cancer cells than in non-transformed cells (O’Shea et al, 2005), nevertheless the principle of phenotypic complementation in tumour cells was maintained.
Exploiting cell cycle deregulation
Some oncolytic viruses have been engineered to depend on a deregulated cell cycle in tumour cells. These include adenoviruses containing mutant E1A protein that fails to bind retinoblastoma protein and liberate E2F. A related approach is employed by the conditionally replicating vaccinia virus Jx594 (also known as Pexa-Vec) that lacks its own thymidine kinase (TK) gene and is hence dependent on a high-thymidine nucleotide pool for successful replication (Liu et al, 2008). This agent, engineered also to express granulocyte-macrophage colony-stimulating factor (GM-CSF), has been assessed in a range of early-phase clinical trials, where it showed good activity particularly following direct intratumoural injection (Heo et al, 2013). Recently, the more virulent vaccinia strain Western Reserve, has been engineered for tumour selectivity by deleting both the TK and vaccinia growth factor genes (Zeh et al, 2015).
Exploiting deregulated interferon defences
Many tumour cells have acquired deficiencies in interferon antiviral defences, most likely either to help evade immune surveillance or as a means to cope with accumulating genetic damage (Stojdl et al, 2000). Cells with active ras mutations are reported to have particular deficiencies in RNA-dependent protein kinase (Farassati et al, 2001), and evidence is emerging that other acquired mutations can inactivate a range of interferon-defence pathways. Notably a recent publication (Li et al, 2015) shows that tumour-associated PTEN deficiencies may underpin inactivation of several interferon defence pathways simultaneously, by preventing downstream activation and nuclear import of IRF3 (interferon regulatory factor 3). Loss of interferon defences is thought to form the basis for the cancer selectivity of wild-type reovirus (developed as Reolysin by Oncolytics Biotech Inc. (Calgary, AB, Canada)), which is unable to replicate successfully in cells with functional interferon systems. More complex viruses, with intrinsic interferon-defence strategies, have also been engineered for increased cancer selectivity by making them more vulnerable in interferon-competent cells. These include adenoviruses deleted in VA genes and vaccinia deleted in B18R (Kirn et al, 2007); however, the interferon-vulnerable agent currently attracting most attention is the oncolytic herpes virus cancer vaccine, talimogene laherparepvec (T-Vec), which lacks the viral protein ICP34.5 to remove neurovirulence and make it susceptible to antiviral defences in non-transformed cells. Talimogene laherparepvec also contains several other engineered changes, for example, its ability to escape immune detection in normal cells is compromised, and it expresses GM-CSF locally within infected tumour cells for secretion into the tumour microenvironment (Senzer et al, 2009).
Identifying oncolytic viruses by bioselection
The complexity of the virus–cell interface and our current incomplete understanding of virus biology makes it impossible to predict the optimum design of oncolytic viruses. One interesting alternative approach is to step away from rational design in favour of a bioselection approach. Avoiding rational design means that the bioselected viruses may be complemented simultaneously by several of the ‘hallmarks of cancer’, including some hallmarks that may be currently unknown (Bauzon and Hermiston, 2012). To achieve this, a large library of chimeric adenoviruses was generated, by growing several adenovirus types together under conditions designed to promote recombination. This library was incubated with different cancer cells, and the viruses capable of replicating and lysing cancer cells most quickly were collected from the cell supernatant. In this way, the library was repeatedly enriched until a small number of highly efficient viruses were obtained (Kuhn et al, 2008). These most successful cancer-killing viruses were then screened on normal cells to identify preferred candidates for development. One particular agent, ColoAd1 (now known as ‘Enadenotucirev’ or ‘EnAd’) was bioselected for its ability to kill colorectal cancer cells. Enadenotucirev is chimeric between adenovirus types Ad11p and Ad3, and was fast tracked for development because of its impressive combination of potency and cancer cell selectivity. The virus is currently undergoing several early-phase clinical trials. Enadenotucirev is derived predominantly from Ad11p, although it has an E2B region that is chimeric between Ad3 and Ad11p, is missing several E3 genes and has a small deletion in E4 (Kuhn et al, 2008). Although it is highly selective for carcinoma cells and fails to replicate in any normal cells, the precise mechanism(s) of EnAd’s selectivity is still the subject of intense investigation. This exemplifies the power of bioselection, providing clinical candidates with the desired biological properties for translational development and simultaneously yielding new insights into virus biology.
Challenges of delivering oncolytic viruses
Systemic delivery is a major goal in the field of oncolytic viruses, allowing the agent to access disseminated tumour deposits. However, despite the compelling simplicity of a self-amplifying molecularly targeted cytotoxic agent, their translational development has been difficult, with many early-clinical trials not living up to expectation. At least in part this reflects a lack of attention to the principles of clinical pharmacology in developing these nanoparticulate therapeutics. For example, oncolytic viruses have sometimes been administered intravenously (i.v.) to access disseminated cancer deposits; however, the attention has often focused predominantly on the mechanism of virus interaction with cancer cells rather than on features of the delivery process, including circulation kinetics, susceptibility to first pass hepatic clearance, and the difficulty for a virus to reach and infect extravascular tumour cells (Fisher, 2006). Consequently, the pharmacodynamic observations of many early-i.v. trials of virotherapy have been limited to an acute inflammation presenting as transient flu-like symptoms associated with reversible elevation of cytokines, as the virus particles are rapidly sequestered and eliminated by the innate immune defences (Small et al, 2006).
Instability of therapeutic virus particles in the often hostile environment of the human blood stream has received relatively little attention. Even today many workers characterise the ability of viral therapeutics to function in dilutions of human serum, whereas if given i.v. in clinical studies, the agents will be required to survive in neat human blood. Similarly, little attention has been paid to the inactivating effects of complement, or the consequences of the ability of human erythrocytes to function as ‘virus traps’ by sequestering virus particles and targeting them for rapid elimination (Carlisle et al, 2009). All of these components combine to restrict i.v. delivery of therapeutic viruses, culminating in a number of unsuccessful clinical studies.
Recently, however, the physical properties of virus particles that are needed to allow systemic delivery have received greater consideration. Some viruses appear to infect cells bearing Fc receptors (such as circulating monocytes) more efficiently in the presence of anti-virus antibodies (Lyons et al, 2006; Ilett et al, 2014), and there are encouraging signs emerging that some free viruses can target disseminated tumours following i.v. injection. For example, in our research group EnAd was prioritised for clinical development by i.v. delivery on the basis of its stability to whole human blood (Di et al, 2014) and the normally low level of pre-existing neutralising antibodies to type 11 adenovirus (Vogels et al, 2003). This agent is now under development by PsiOxus Therapeutics Ltd (Milton Park, Oxfordshire, UK) in a range of early-phase clinical trials. The dosage and administration protocol were carefully optimised to achieve maximum circulation of infectious virus particles and i.v. kinetics showed evidence not only of good circulation but also time-dependent rises in some patients suggested significant virus replication (Calvo et al, 2014). Primary and metastatic colorectal tumours resected at various times after i.v. delivery of virus showed extensive infection of tumour cells by the virus, evidenced using anti-hexon immunohistochemistry (Gil Martin et al, 2014). This demonstrates not only that the EnAd can access many tumour cells from an i.v. delivery, but that it infects them and replicates successfully, expressing encoded proteins in situ.
Mechanisms of oncolytic virus-mediated cytotoxicity and immunogenicity
Oncolytic viruses express highly evolved multifunctional proteins that attempt to take control of infected cancer cells and commandeer them as virus factories. Such viruses are therefore ideally placed to kill cancer cells actively, without reliance on cellular death pathways (Guo et al, 2014). When under control of many types of oncolytic virus, the cell’s resources are diverted away from cellular activities into a pseudo-activated state that supports production of multiple copies of the virus genome and capsids, leading to intracellular packaging of complete virus particles. Typically, the virus then coordinates cell death at the optimal time in the virus life cycle, leading to release of infectious virus particles.
For most oncolytic viruses it seems evident that successful release of cell-associated virus particles requires membrane lysis and release of cell contents. This is distinct from the neat death process mediated by conventional apoptosis, which if carried out successfully would presumably degrade all the intracellular virus particles alongside the cell’s own genome. Accordingly, although some oncolytic viruses, particularly simple RNA viruses, do appear to activate cellular apoptosis pathways, others clearly mediate a more active death process, with mechanisms often described as necrosis, oncosis or inflammatory apoptosis (Baird et al, 2008; Whilding et al, 2013). The ability of many viruses to mediate pathways of death that are at least partly independent of apoptosis provides the enticing possibility that virotherapy may be able to evade apoptosis-related pathways of acquired drug resistance.
Although oncolytic viruses may work effectively as single-agent treatments, they appear to show non-overlapping toxicities with many standard therapeutics and may well form important components of combination therapies. In addition, the abilities of some DNA viruses to inhibit cellular DNA repair pathways may allow them to function effectively as cancer-selective radiosensitisers. Accordingly, combination of oncolytic viruses with many other agents is currently being widely explored.
Whereas apoptosis is a non-inflammatory mechanism of cell death, it is widely accepted that lytic pathways of cell death are highly immunogenic (Guo et al, 2014). Oncolytic viruses may therefore create a pro-inflammatory environment of dying cancer cells that may be exploited in creating anticancer immune responses (Lichty et al, 2014). In recent years, the concept has attracted considerable scientific attention, with several oncolytic viruses being considered also as ‘oncolytic vaccines’.
The first oncolytic vaccines are currently being developed for local delivery direct into individual tumour nodules, and they show good therapeutic activity. A recent study of T-Vec demonstrated statistically significant improved durable responses in patients with unresectable melanoma and a trend towards improved overall survival. As might be predicted, this oncolytic vaccine is particularly effective when combined with immune checkpoint inhibitors (Puzanov et al, 2014). However, our increasing understanding of the nature of local immune suppression within tumour deposits raises the enticing question of whether this sort of cancer vaccine would work most effectively if it were administered i.v, allowing the virus to infect disseminated tumour deposits and mediate local immune stimulation to reverse local suppressive effects. This would be particularly exciting in the case of a virus that was capable of stimulating an anticancer immune response and simultaneously expressing encoded checkpoint inhibitors in tumour masses throughout the body (see below).
‘Armed’ oncolytic viruses
Oncolytic virotherapy provides the possibility of direct cytotoxicity, due to viral lysis, coupled with potential stimulation of the immune system following creation of the pro-inflammatory cell lysis environment. However, a major additional feature of this technological approach is the possibility to encode anticancer biologics and express them selectively within cancer cells. If the agents are designed for secretion, or if they are functional when released by virus-mediated cell lysis, this provides an opportunity to modulate the tumour microenvironment and/or to mediate effects on other cells in the tumour microenvironment. Indeed, encoding therapeutic biologics within oncolytic viruses combines the very desirable properties of cancer-targeted expression with the ability to self-amplify in situ, achieving potentially very high and exquisitely selective expression of agents within tumour deposits, with minimal systemic effects.
There is an exciting array of possible therapeutic ‘arming’ approaches, including cytokines, chemokines, enzymes, imaging agents and antibodies (Bauzon and Hermiston, 2014; Figure 2). At present, the possibility to arm oncolytic viruses to encode immune modulators is attracting most attention, with several oncolytic viruses designed to express and secrete GM-CSF in an attempt to improve local presentation of tumour antigens (Breitbach et al, 2015). However, the effects of GM-CSF are now known to vary depending on the local concentration, with some evidence of immune suppression at high concentrations (Clive et al, 2010); hence, GM-CSF may be not be the ideal arming agent. Accordingly, there is an exciting opportunity to express alternative immune modulatory agents such as checkpoint inhibitors, cytokines or coordinated combinations of biologics to modulate the tumour immune environment, maximising therapeutic outputs while simultaneously avoiding the systemic toxicities engendered when these agents are given i.v. as free proteins. Worldwide there are now many studies actively exploring these therapeutic strategies, as the concept of localised expression of potent biologics within tumours is very attractive and potentially game changing for clinical management of cancer.

Armed oncolytic viruses. Oncolytic viruses can be ‘armed’ to express therapeutic proteins within infected tumour cells, and secrete them into the tumour microenvironment. This can provide high-level tumour-selective expression of biologics, maximising local activity and minimising systemic toxicities. If the virus is suitable for systemic delivery, this provides the intriguing concept of localised therapy in disseminated tumours following intravenous delivery. AntiCTLA4=checkpoint inhibitor antibody; BiTE=Bi-specific T-cell engager, for modulating activity and tropism of T cells; NIS=sodium iodide symporter (for SPECT imaging of virus activity); NTR=nitroreductase TNF=tumour necrosis factor.
References
Baird SK, Aerts JL, Eddaoudi A, Lockley M, Lemoine NR, McNeish IA (2008) Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene 27 (22): 3081–3090.
Bauzon M, Hermiston T (2014) Armed therapeutic viruses - a disruptive therapy on the horizon of cancer immunotherapy. Front Immunol 5: 74.
Bauzon M, Hermiston TW (2012) Oncolytic viruses: the power of directed evolution. Adv Virol 2012: 586389.
Breitbach CJ, Parato K, Burke J, Hwang TH, Bell JC, Kirn DH (2015) Pexa-Vec double agent engineered vaccinia: oncolytic and active immunotherapeutic. Curr Opin Virol 13: 49–54.
Calvo E, Gil-Martin M, Machiels J-P, Rottey S, Cubillo A, Salazar R, Mardjuadi F, Geboes K, Ellis C, Beadle J, Blanc C (2014) A first-in-class, first-in-human phase I study of enadenotucirev, an oncolytic Ad11/Ad3 chimeric group B adenovirus, administered intravenously in patients with metastatic epithelial tumors. J Clin Oncol 32 (5s): abstract 3103.
Carlisle RC, Di Y, Cerny AM, Sonnen AF, Sim RB, Green NK, Subr V, Ulbrich K, Gilbert RJ, Fisher KD, Finberg RW, Seymour LW (2009) Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood 113 (9): 1909–1918.
Chiocca EA, Rabkin SD (2014) Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res 2 (4): 295–300.
Clive KS, Tyler JA, Clifton GT, Holmes JP, Mittendorf EA, Ponniah S, Peoples GE (2010) Use of GM-CSF as an adjuvant with cancer vaccines: beneficial or detrimental? Expert Rev Vaccines 9 (5): 519–525.
Di Y, Seymour L, Fisher K (2014) Activity of a group B oncolytic adenovirus (ColoAd1) in whole human blood. Gene Ther 21 (4): 440–443.
Farassati F, Yang AD, Lee PW (2001) Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol 3 (8): 745–750.
Fisher K (2006) Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther 8 (4): 301–313.
Gil Martin M, Cubillo A, Machiels J, Rottey S, Mardjuadi F, Geboes K, Salazar R, Beadle J, Ellis C, Fisher K, Blanc C, Calvo E (2014) A phase 1 study of enadenotucirev, an oncolytic Ad11/Ad3 chimeric group B adenovirus, administered intravenously - analysis of dose expansion and repeat cycle cohorots in patients with metastatic colorectal cancer (mCRC). Ann Oncol 25 (suppl 4): lv361–lv372.
Guo ZS, Liu Z, Bartlett DL (2014) Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol 4: 74.
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5): 646–674.
Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, Cho M, Lim HY, Chung HC, Kim CW, Burke J, Lencioni R, Hickman T, Moon A, Lee YS, Kim MK, Daneshmand M, Dubois K, Longpre L, Ngo M, Rooney C, Bell JC, Rhee BG, Patt R, Hwang TH, Kirn DH (2013) Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 19 (3): 329–336.
Ilett E, Kottke T, Donnelly O, Thompson J, Willmon C, Diaz R, Zaidi S, Coffey M, Selby P, Harrington K, Pandha H, Melcher A, Vile R (2014) Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol Ther 22 (10): 1851–1863.
Kelly E, Russell SJ (2007) History of oncolytic viruses: genesis to genetic engineering. Mol Ther 15 (4): 651–659.
Kirn D (2001) Oncolytic virotherapy for cancer with the adenovirus dl1520 (Onyx-015): results of phase I and II trials. Expert Opin Biol Ther 1 (3): 525–538.
Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH (2007) Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med 4 (12): e353.
Kuhn I, Harden P, Bauzon M, Chartier C, Nye J, Thorne S, Reid T, Ni S, Lieber A, Fisher K, Seymour L, Rubanyi GM, Harkins RN, Hermiston TW (2008) Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One 3 (6): e2409.
Li S, Zhu M, Pan R, Fang T, Cao YY, Chen S, Zhao X, Lei CQ, Guo L, Li CM, Jokitalo E, Yin Y, Shu HB, Guo D (2015) Pivotal role for PTEN in the induction of type I interferon and antiviral response in vivo. Nature Immunol e-pub ahead of print 21 December 2015 doi:10.1038/ni.3311.
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC (2014) Going viral with cancer immunotherapy. Nat Rev Cancer 14 (8): 559–567.
Liu TC, Hwang T, Park BH, Bell J, Kirn DH (2008) The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther 16 (9): 1637–1642.
Lyons M, Onion D, Green NK, Aslan K, Rajaratnam R, Bazan-Peregrino M, Phipps S, Hale S, Mautner V, Seymour LW, Fisher KD (2006) Adenovirus type 5 interactions with human blood cells may compromise systemic delivery. Mol Ther 14 (1): 118–128.
O’Shea CC, Soria C, Bagus B, McCormick F (2005) Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell 8 (1): 61–74.
Pesonen S, Diaconu I, Kangasniemi L, Ranki T, Kanerva A, Pesonen SK, Gerdemann U, Leen AM, Kairemo K, Oksanen M, Haavisto E, Holm SL, Karioja-Kallio A, Kauppinen S, Partanen KP, Laasonen L, Joensuu T, Alanko T, Cerullo V, Hemminki A (2012) Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res 72 (7): 1621–1631.
Puzanov I, Milhern M, Andtbacka R, Minor D, Hamid O, Li A, Chastain M, Gorski K, Anderson A, Vanderwalde A, Chou J, Kaufman H (2014) Primary analysis of a phase 1b multicenter trial to evaluate safety and efficacy of talimogene laherparepvec (T-VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB-IV melanoma. J Clin Oncol 32 (5s): abstract 9029.
Russell SJ, Peng KW, Bell JC (2012) Oncolytic virotherapy. Nat Biotechnol 30 (7): 658–670.
Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K, Goldsweig H, Marshall T, Love C, Coffin R, Nemunaitis JJ (2009) Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 27 (34): 5763–5771.
Small EJ, Carducci MA, Burke JM, Rodriguez R, Fong L, van Ummersen L, Yu DC, Aimi J, Ando D, Working P, Kirn D, Wilding G (2006) A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther 14 (1): 107–117.
Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC (2000) Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med 6 (7): 821–825.
Vogels R, Zuijdgeest D, van Rijnsoever R, Hartkoorn E, Damen I, de Bethune MP, Kostense S, Penders G, Helmus N, Koudstaal W, Cecchini M, Wetterwald A, Sprangers M, Lemckert A, Ophorst O, Koel B, van Meerendonk M, Quax P, Panitti L, Grimbergen J, Bout A, Goudsmit J, Havenga M (2003) Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity. J Virol 77 (15): 8263–8271.
Whilding LM, Archibald KM, Kulbe H, Balkwill FR, Oberg D, McNeish IA (2013) Vaccinia virus induces programmed necrosis in ovarian cancer cells. Mol Ther 21 (11): 2074–2086.
Zeh HJ, Downs-Canner S, McCart JA, Guo ZS, Rao UN, Ramalingam L, Thorne SH, Jones HL, Kalinski P, Wieckowski E, O'Malley ME, Daneshmand M, Hu K, Bell JC, Hwang TH, Moon A, Breitbach CJ, Kirn DH, Bartlett DL (2015) First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther 23 (1): 202–214.
Acknowledgements
We are grateful to Cancer Research UK for supporting our research (grant number C552/A177020).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
LS and KF both hold equity in PsiOxus Therapeutics Ltd. LS is Chair of Psioxus’ Scientific Advisory Committee and KF serves as its Chief Scientific Officer.
PowerPoint slides
Rights and permissions
This work is licensed under the Creative Commons Attribution-Non-Commercial-Share Alike 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Seymour, L., Fisher, K. Oncolytic viruses: finally delivering. Br J Cancer 114, 357–361 (2016). https://doi.org/10.1038/bjc.2015.481
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.481
Keywords
This article is cited by
-
Extracellular Vesicles: a Trojan Horse Delivery Method for Systemic Administration of Oncolytic Viruses
Regenerative Engineering and Translational Medicine (2023)
-
CD19-targeted BiTE expression by an oncolytic vaccinia virus significantly augments therapeutic efficacy against B-cell lymphoma
Blood Cancer Journal (2022)
-
Development of Duramycin-Based Molecular Probes for Cell Death Imaging
Molecular Imaging and Biology (2022)
-
Evaluation of MMR live attenuated vaccine oncolytic potential using Ehrlich ascites carcinoma in a murine model
Medical Oncology (2022)
-
Journey from Serendipity to Biologics
Pharmaceutical Chemistry Journal (2022)
