
Abstract
Background:
Gender-associated epigenetic alterations are poorly investigated in male and female familial breast cancer (fBC). MicroRNAs may contribute to the different biology in men and women particularly related to RASSF1A pathways.
Methods:
Microarray technology was used to evaluate miRNA profile in 24 male and 43 female fBC. Key results were validated using RT–qPCR in an external samples set. In vitro studies were carried out to verify microRNA–target gene interaction.
Results:
Pathway enrichment analysis with the 287 differentially expressed microRNAs revealed several signalling pathways differently regulated in male and female cases. Because we previously hypothesised a peculiar involvement of RASSF1A in male fBC pathogenesis, we focussed on the MAPK and the Hippo signalling pathways that are regulated by RASSF1A. Male miR-152 and miR-497 upregulation and RASSF1A and NORE1A interacting gene downregulation were observed, confirming a possible indirect interaction between miRNAs and the two genes.
Conclusions:
For the first time, a different microRNA expression pattern in male and female fBC has been shown. Moreover, the importance of RASSF1A pathway in male fBC carcinogenesis has been confirmed, highlighting a possible role for miR-152 and miR-497 in controlling MAPK and Hippo signalling pathways, regulated by RASSF1A.
Similar content being viewed by others
Main
Breast cancer (BC) is the most frequent tumour in women in western countries. In men, it occurs at a lower incidence (∼1%), but this is increasing yearly. Furthermore, male BC has a worse prognosis. The majority of breast cancers are sporadic, but in both genders in 25% of cases a positive family history (5–7% of all BCs) is associated with BRCA1/2 germline mutations (BRCA1/2 tumours) with a consequent increased risk of the disease (Melchor and Benitez, 2013). The large majority of familial BCs (fBCs) are not explained by mutations in any known breast cancer susceptibility gene (Melchor and Benitez, 2013).
In recent years, epigenetic alterations have drawn great attention as they play a role as crucial as genetics in cancer development (Sandoval and Esteller, 2012). The concept of epigenetics refers to changes in gene activity that does not involve variations in the primary DNA sequence. However, it is still unclear whether there is a different gender-associated epigenetic pattern in familial male and female BC (fMBC and fFBC, respectively).
We previously demonstrated that in fMBC, the tumour suppressor RASSF1A (RAS association domain family protein 1 isoform A) was frequently downregulated, because it is hypermethylated in its promoter and consequently less expressed than in fFBC (Pinto et al, 2013). We thus hypothesised a particular involvement of RASSF1A in fMBC pathogenesis. RASSF1A, produced by the human RASSF1 gene on chromosome 3p21, interacts with a number of important signalling molecules regulating cell growth, survival, microtubule stabilisation, cell motility, genomic stability and apoptosis; it is therefore considered an important molecule that combines the upstream stimuli, transducing them to the specific downstream effectors (Jung et al, 2013; Volodko et al, 2014). On the other hand, we also revealed that miR17 and let7a are involved in fBC, in particular in fFBC BRCA carriers (Pinto et al, 2013).
Recently, microRNAs (miRNAs), the most widely studied class of noncoding RNAs, were used to better define and characterise various types of cancers (Esteller, 2011). MicroRNAs are short (19–24 nt), stable, endogenous noncoding RNAs that play an important role in the post-transcriptional gene silencing of crucial genes involved in all biological processes such as cell proliferation, apoptosis, development, differentiation, metabolism, immunity, ageing, cell-cycle control and stress response. They can act as either oncogenes or tumour suppressors, as they inhibit both tumour-suppressive and -oncogenic mRNAs. Very little is known yet about the role of miRNAs in fBC (Tanic et al, 2012; Murria Estal et al, 2013; Pinto et al, 2013; Tanic et al, 2013).
To the best of our knowledge, this is the first study of miRNA expression profiling by microarray technology in a cohort of fMBC and fFBC that aims (1) to understand a possible different gender-associated miRNA pattern and (2) to better investigate gender-associated different epigenetic regulation in RASSF1A pathways. The identification of target genes and pathways regulated by miRNAs in male and female fBC could provide crucial knowledge about the different biology and tumour development in the two genders. Our results demonstrated a different miRNA expression in male and female fBC with the involvement of cancer initiation, progression and cell proliferation pathways. Moreover, we revealed a possible involvement of miR-152 and miR-497 in the regulation of pathways mediated by RASSF1A, suggesting a role of the RASSF1A/NORE1A (novel Ras effector 1 or RASSF5) complex that could explain the enhanced tumourigenic potential and worse prognosis in men with fBC.
Materials and methods
Samples
A training set of 67 fBC patients (24 male and 43 female cases) was enrolled through the Genetic Counseling Program at the National Cancer Research Centre ‘Giovanni Paolo II’ in Bari, Italy. The study has been approved by the Ethics Committee of Istituto Tumori ‘Giovanni Paolo II’ of Bari as satellite project of the protocol approved with n. 56/CE del 16/05/2011. Patients signed informed consent giving permission to use their pathological material. They were classified as ‘familial’ when they satisfied clinical characteristics as previously reported (Pilato et al, 2011). Male and female groups matched in terms of histology, stage, nodal involvement and proliferative activity, whereas a physiological difference in terms of ER percentage was present (fFBC: 57%; fMBC: 92%). A validation set consisting of an independent series of 23 fBCs (4 male and 19 female cases) was enrolled and classified in the same way as the training set. The normal tissue counterparts of five patients were transformed in the RNA pool used to normalise the data in the miRNA microarray analysis and as a calibrator in the real-time PCR experiments.
RNA was extracted from fresh frozen cancer specimens containing at least 70% tumour cells and from normal tissues using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Concentrations were estimated with the ND-8000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
MiRNA microarray analysis
Of each sample, 500 ng RNA was labelled using the 3 DNA Array Detection FlashTag RNA Labelling Kit (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s instructions, and analysed by the GeneChip miRNA v. 1.0 Array (Affymetrix). This contains 46 228 probes comprising 7815 probe sets and covers 71 organisms including 1100 human miRNAs derived from the Sanger miRBase and miRNA database v11 (15 April 2008, http://microrna.sanger.ac.uk). Firstly, poly (A) tailing was carried out at 37 °C for 15 min in a volume of 15 ml reaction mix that contained 1 × Reaction Buffer, 1.5 ml MgCl2 (25 mM), 1 ml ATP Mix diluted 1 : 500 and 1 ml PAP enzyme. Subsequently, FlashTag Ligation was performed at room temperature for 30 min by adding 4 ml of 5 × FlashTag Ligation Mix Biotin and 2 ml T4 DNA Ligase into 15 ml of reaction mix. Next, 2.5 ml of Stop Solution was added to stop the reaction. Each sample was hybridised on the array, washed, stained with the Affymetrix Fluidics Station 450 and scanned with the Affymetrix GeneChip Scanner 3000 7G using the Command Console software (Affymetrix).
Moreover, unsupervised average-linkage hierarchical clustering using Pearson’s correlation was performed through ‘Hierarchical clustering’ module and heat map was created with ‘Hierarchical clustering image’ module of Gene Pattern suite (Reich et al, 2006).
Microarray data set has been deposited at the ArrayExpress database under the accession number E-MTAB-2705.
Array data processing and statistical analysis
Raw data were normalised with the Robust Multiarray Average (RMA) method to remove systematic variations. Briefly, RMA corrects raw data for background using a formula that is based on a normal distribution and uses a linear model to estimate values on a log-scale. The RMA normalisation was performed using the ‘Affy’ package of the Bioconductor suite (http://www.bioconductor.org/) for R statistical language (http://cran.r-project.org/). The default settings were used. Normalised values were statistically analysed with MeV software v.4.8.1 (Dana-Farber Cancer Institute, Boston, MA, USA). Differentially expressed miRNAs were detected through t-test, with data considered statistically significant when P<0.01.
Quantitative miRNA RT–PCR analysis
Quantitative RT–PCR analysis of 11 selected miRNAs was performed on an independent series of 23 fBCs using the TaqMan microRNA Assay (Applied Biosystems, Foster City, CA, USA) on the Applied Biosystems Real-time PCR instrument 7000, in accordance with the manufacturer’s instructions.
Briefly, reverse transcriptase reactions contained: 10 ng of total RNA obtained after RNA isolation, 3 μl RT primers, 1 × RT buffer, 100 mM dNTPs, 3.33 U μl−1 MultiScribe reverse Transcriptase and 0.25 U μl−1 RNase inhibitor.
A 20 μl PCR reaction including 3 μl of RT product, 1 × TaqMan Universal PCR Master Mix and 1 × of the corresponding miRNA assay primers was incubated in 96-well plates at 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. All PCR reactions were performed in triplicate for a technical replicate, including no-template controls, and the mean of the triplicates was used.
Relative quantities of each miRNA were calculated using the ΔΔCt method after normalisation with endogenous reference RNU 48.
Pathway enrichment analysis
DIANA-miRPath v2.0 pathway enrichment analysis (Papadopoulos et al, 2009) was used to detect molecular pathways that are targeted by the miRNAs differentially expressed between fMBC and fFBC. This application offers a series of tools specifically focussed on miRNA-targeted pathway analysis that allows comparison of several sets of multiple miRNAs and target genes to all known KEGG pathways. Pathways showing a P-value of ⩽0.05 were considered significantly enriched. Specifically, this tool allows you: to obtain miRNA targets predicted with high accuracy based on DIANA-microT-CDS and/or experimentally verified targets from TarBase v6; to combine results with merging and meta-analysis algorithms; to perform hierarchical clustering of miRNAs and pathways based on their interaction levels; and to elaborate sophisticated visualisations such as the miRNA/pathway interaction heat map. The DIANA-miRPath v2.0 can be accessed at the following address: http://www.microrna.gr/miRPathv2.
Quantitative RT–PCR analysis of RASSF1A and NORE1A mRNA levels
The levels of RASSF1A and NORE1A mRNA were measured using the individual TaqMan RNA Assay (Applied Biosystems) on the Applied Biosystems Real-time PCR instrument 7000 in accordance with the manufacturer’s instructions.
Reverse transcriptase reactions, performed with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), contained 500 ng of total RNA obtained after RNA isolation, 1 × RT Random primers, 1 × RT buffer, 100 mM dNTPs, 1 U μl−1 MultiScribe reverse Transcriptase and 1 μl RNase inhibitor.
A 20 μl PCR reaction including 2.5 μl of RT product, 1 × TaqMan Universal PCR Master Mix and 1 × of the corresponding RNA assay primers was incubated in 96-well plates at 50 °C for 2 min and at 95 °C for 10 min followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. All PCR reactions were performed in triplicate for a technical replicate, including no-template controls, and the mean of triplicates was used.
Relative quantities of each mRNA were calculated using the ΔΔCt method after normalisation with endogenous reference RNA 18s.
Dual-luciferase reporter assay and constructs
The Firefly luciferase-UTR reporter plasmid was constructed by introducing the 3'-UTR of RASSF5/NORE1A (NM_182665.2) and RASSF1A (NM_170714) genes into pmiR-REPORT miRNA Expression Reporter Vector System (Life Technologies, Foster City, CA, USA). The 3'-UTR sequence of the two analysed genes was amplified by PCR from HEK293 cDNA. All constructs were verified by sequencing.
The reporter construct (50 ng), pSV-Renilla (2 ng, pRNL-SV40, Promega, Fitchburg, WT, USA) and hsa-miR-152 or hsa-miR-497 mimic and inhibitor or miRNA negative control (Sigma-Aldrich, St Louis, MO, USA) were transfected into HEK293 cells using Lipofectamine 2000 (Life Technologies). Two different amounts of miRNA mimic (1 and 2 pmol) were used. After 48 h, the cells were lysed in passive lysis buffer and assayed for both firefly and Renilla luciferase activity using the Dual-GLO Luciferase Assay System (Promega). Firefly luciferase activity was normalised to Renilla luciferase activity for each transfected well. Values are the mean±s.e.m. of three experimental replicates from three independent transfections. Significance was determined by a two-tailed unpaired t-test for means.
Computational and statistical analysis
The MiRWalk database (Dweep et al, 2011) was used to identify predicted and validated target genes of the deregulated miRNAs and to show miRNA/target gene interaction.
Data analysis was performed using the GraphPad Prism statistics software package (GraphPad Prism 5.01, San Diego, CA, USA). As the values were not normally distributed, nonparametric tests were used to compare miRNAs with RASSF1A and NORE1A gene levels.
The Mann–Whitney U-test was used to compare the median expression values of miRNAs and genes. Spearman’s test was used to analyse the eventual inverse correlation between miRNA and the target gene. Another test employed to confirm the association between miRNA overexpression and target gene downexpression was the two-tailed Fisher’s test considering the small size of the validation set. A P-value of ⩽0.05 was considered statistically significant.
Results
MiRNA expression profiling in fFBC and fMBC
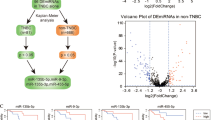
The miRNA expression analysis was performed comparing 24 male and 43 female fBC samples. We selected miRNAs annotated as ‘hsa’ in order to exclusively analyse the differential expression of human genes. The selected hsa-miRNAs (n=1100) underwent statistical analysis by t-test performed through MeV software, identifying 287 differentially expressed miRNAs. In detail, we identified 159 upregulated miRNAs in fMBC and 128 upregulated miRNAs in fFBC (Supplementary Table S1). False discovery rate (FDR) values are those that are lower than 0.05 without any P-value correction, and we decided to set a lower ‘raw’ P-value (P=0.01) in order to decrease type I errors. This is the error through which the null hypothesis of no effect is rejected when it should be accepted. An intuitive representation of this is the volcano plot (Figure 1A) that shows the global trend of significantly deregulated miRNAs (above the dotted line) with a P-value of <0.01. For males, significantly downregulated miRNAs are dotted on the left side and significantly upregulated miRNAs are on the right side: overexpressed miRNAs in males showed a greater difference in mean fluorescence with respect to females. An unsupervised hierarchical clustering analysis was performed evidencing three main sample clusters (FBC1, FBC2 and FBC3) that explain a different behaviour of male patients with respect to female patients as regards miRNA expression (Figure 1B).

Representation of miRNAs deregulated in male and female cases. (A) The volcano plot shows the global trend of deregulated miRNAs. The X axis represents the difference between miRNA mean expression level in male (group B) and female (group A) cases. Significant miRNAs are located above the dotted line (green points) that indicates log10 (cutoff P-value) considered in the statistical analysis: P<0.01. (B) The heatmap shows three sample clusters (FBC1, FBC2 and FBC3). FBC2, which included fMBC named in the figure CMM, showed a different miRNA deregulation with respect to fFBC named CFM (FBC1 and FBC2). The miRNA overexpression is in red, and lower miRNA expression is in blue.
Pathway enrichment analysis
The potential impact of aberrantly expressed miRNAs in fBC on biological processes was clarified by the Diana miRPath web-based computational tool that allows a biological interpretation of miRNA profiling. Enrichment analysis was performed considering the entire number of deregulated miRNAs detected in the statistical analysis that aimed to validate miRNAs targeting genes involved in pathways retained of particular interest for fMBC. The KEGG pathway enrichment analysis revealed that almost all of the upregulated miRNAs reported above are particularly involved in focal adhesion, MAPK, regulation of actin cytoskeleton, TGF-β, axon guidance, ErbB, mTOR and in pathways involved in cancer. To understand possible gender-associated differences in RASSF1A-regulated pathways, the MAPK and Hippo pathways in which RASSF1A plays a key role have been considered. For this reason, another RASSF family member, NORE1A/RASSF5, was studied, because it acts together with RASSF1A to inhibit the proliferative stimulation induced by Ras and to promote apoptosis through activation of the MST1 kinase. A computational analysis by miRWalk database has revealed that 68 out of 159 upregulated miRNAs in fMBC had RASSF1A as a predicted or validated target, whereas 92 miRNAs had NORE1A. Nine miRNAs targeting both RASSF1A and NORE1A and other 2 miRNAs targeting only NORE1A were selected to validate the upregulation of these miRNAs in fMBC (Table 1).
Real-time miRNA validation and RASSF1A/NORE1A mRNA expression
Eleven miRNAs selected as previously explained were analysed in another validation set (n=23) of fFBC and fMBC by TaqMan microRNA assays in real-time PCR. When analysing the median miRNA expression values with the Mann–Whitney U-test, we confirmed a statistically significant overexpression of only miR-497 and miR-152 in fMBC (P=0.008 and P=0.03, respectively) with respect to fFBC. With the miRWalk database, which uses several bioinformatic target prediction algorithms (such as DIANA microT4.0, TargetScan, PITA, PicTar, miRANDA and so on), we verified that RASSF1A is a validated target of miR-152 and a predicted target of miR-497, whereas NORE1A is a predicted target of both miRNAs.
Measures of RASSF1A and NORE1A mRNA levels by TaqMan Gene Expression assays demonstrated their down-expression in male cases (Mann–Whitney test P=0.06 and P=0.02 respectively, Figure 2) with respect to fFBC. This suggests a possible epigenetic regulation of the RASSF1A/NORE1A complex by miR-152 and miR-497 in male patients.

Relative expression levels of miR-152, miR-497, RASSF1A and NORE1A in fMBC and fFBC. Relative expression levels for every sample were determined by real-time PCR and calculated by normalising with respect to the expression level of RNU48. The statistical significance of differences between fFBC and fMBC was calculated using the Mann–Whitney U-test. Bars indicate median values with interquartile range.
When analysing the whole validation set, Spearman’s test demonstrated an inverse correlation between miR-497 expression and RASSF1A (P=0.04) and NORE1A mRNA levels (Figure 3). Furthermore, an inverse correlation, even if not significant, was shown between miR-152 expression and NORE1A and RASSF1A mRNA levels (Figure 3). This inverse correlation was confirmed by the two-tailed Fisher’s test that analyses the frequencies of overexpression and down-expression of miRNAs and genes. In fact, both miR-152 and miR-497 down-expression was frequently associated with RASSF1A and NORE1A mRNA overexpression (P=0.04 only for miR-497/RASSF1A).

Correlation analysis. Relative expression levels of RASSF1A and NORE1A displayed a negative correlation with the expression levels of miR-152 and miR-497 in fBC. Statistical analysis was performed using Spearman’s coefficient. Negative correlation of miR-497 and RASSF1A was significant (r2=−0.48, P=0.04).
Functional analysis to test RASSF1A/NORE1A regulation by miR-152 and miR-497
To experimentally test whether miR-152 and miR-497 directly target the 3'-UTR of RASSF1A and NORE1A in vitro, HEK293 cells were co-transfected with RASSF1A and NORE1A reporter construct along with a synthetic mimic of miR-152 and miR-497. As shown in Figure 4A, the overexpression of miR-497 did not have any reduction on the luciferase activity of the vector containing the 3'-UTR of NORE1A, whereas there was a reduction, but not significant, on the luciferase activity of the vector containing the 3'-UTR of RASSF1A when compared with the control. Similar results were obtained when we considered the expression of miR-152 with respect to RASSF1A and NORE1A.

Dual-luciferase reporter analysis. HEK293 cells were co-transfected with the indicated reporter constructs and with a miR-152 and miR-497 mimic (A) and inhibitor (B). The luciferase activity was normalised with respect to the level of Renilla luciferase (pRNL) and the relative luciferase activity (RLA) was calibrated to 1 that refers to the RLA of cells transfected with the hsa-miR-negative control.
Consistently, the use of specific miR-497 and miR-152 inhibitors did not have any consequence on the NORE1A and RASSF1A levels (Figure 4B). The simultaneous use of the specific miR-497 and miR-152 inhibitors led to an increase in the RASSF1A and NORE1A levels even if they did not reach a statistically significant overexpression level (Figure 4B).
Discussion
Clinical experience has demonstrated that men with BC have a worse prognosis than women, probably because of an advanced stage and an older age at diagnosis and a higher incidence of lymph node metastasis (Ottini et al, 2010; Callari et al, 2011; Ruddy and Winer, 2013; Sousa et al, 2013). Male and female BCs clinically differ in the frequency of their histological types and in the expression of hormone receptors and HER2. From a biological point of view, the lower incidence rate in men has greatly limited acquisition of knowledge about male BC compared with female. A positive family history of BC is an important high risk factor for both genders (Melchor and Benitez, 2013). However, it is still unclear whether there are distinct gender-associated epigenetic alterations that may contribute to the different biology of the two tumours. In the past 10 years, miRNAs were used to better define and characterise various types of cancers (Esteller, 2011), but very little is known about the association of miRNAs to fBC.
In this study we analysed a miRNA expression profile by microarray technology in a cohort of fMBC and fFBC to enhance knowledge about a possible distinct gender-associated miRNA pattern. We profiled 24 fMBCs and 43 fFBCs, identifying 287 differentially expressed miRNAs between the two groups. In detail, we identified 159 upregulated miRNAs in fMBC and 128 upregulated miRNAs in fFBC. To understand the potential impact of aberrantly expressed miRNAs on biological processes in fBC, a pathway enrichment analysis was performed with the Diana miRPath web-based computational tool that allowed a biological interpretation of miRNA profiling. This analysis revealed several signalling pathways regulated differently by miRNAs in male and female fBCs. This emphasises the presence of many differences in the biology of the two genders, such as cancer initiation and progression pathways, and could explain the worse prognosis of fMBC. Only few reports describe miRNAs in fBC without distinction between genders. We previously reported the involvement of miR-17 and let-7a in fFBC, in particular in BRCA carriers, in which they were significantly overexpressed (Pinto et al, 2013). Tanic, with a miRNA expression profiling, individuated 17 downregulated miRNAs and the upregulation of miR-21 and miR-300 in fBC compared with normal control tissues (Tanic et al, 2012), demonstrating that the altered expression of these miRNAs could be related to cell proliferation and migration pathways, in particular to the MAPK signalling pathway. Moreover, the same authors used miRNAs to subclassify BRCAX tumours in four different subgroups characterised by a specific miRNA signature (Tanic et al, 2013). Hereditary BCs have been distinguished from nonhereditary BCs by Murria Estal et al (2013), who demonstrated that BRCA1, BRCA2 and sporadic BCs were associated with specific clusters of hyperexpressed miRNAs, whereas in BRCAX all these miRNAs were hypoexpressed.
In our previous paper, we showed a different gender-associated regulation of the oncosuppressor RASSF1A in fBC (Pinto et al, 2013). We demonstrated a higher down-expression of RASSF1A mRNA level in fMBC compared with fFBC because of its promoter hypermethylation underlining a greater inactivation of this oncosuppressor in fMBC than in fFBC. Interestingly, of the 159 upregulated miRNAs in fMBC, the miRWalk database revealed that 68 miRNAs had RASSF1A as a predicted or validated target. The KEGG enrichment pathway analysis of our microarray data revealed that almost all of the deregulated miRNAs were particularly involved in cell proliferation and in cancer initiation and progression. To better understand the possible biological gender-associated differences in RASSF1A-regulated pathways because of epigenetic alterations, we focussed our attention on the MAPK and Hippo signalling pathways.
Several signalling pathways may be regulated by RASSF1A because of the presence of various domains in RASSF1A. The RA domain, located at the C-terminal (RASSF1- RASSF6) or N-terminal (RASSF7RASSF10), is responsible for the binding with Ras and other small GTPase molecules. The SARAH domain mediates interaction between RASSF1/RASSF6 and the mammalian sterile-20-like kinases (MSTs) (Jung et al, 2013; Volodko et al, 2014).
NORE1A, another RASSF family member, is considered a tumour suppressor as it acts with RASSF1A to inhibit the proliferative stimulation induced by Ras, promoting apoptosis through activation of the MST1 kinase. The miRWalk database revealed that 92 out of 159 upregulated miRNAs in fMBC had NORE1A as the predicted or validated target (Avruch et al, 2006; Bee et al, 2010; Volodko et al, 2014).
These two tumour suppressors are considered key regulators of the MAPK and Hippo signalling pathways, and loss of RASSF1A/NORE1A function may enhance the proliferative processes reducing apoptosis (Avruch et al, 2012; Fausti et al, 2012; Liu et al, 2012; Fernandes et al, 2013). Although promoter methylation is apparently the major mechanism of NORE1A and RASSF1A silencing (Lee et al, 2010; Dumitrescu, 2012; Jiang et al, 2012; Alvarez et al, 2013; Pinto et al, 2013), other mechanisms, such as miRNA interfering, exist. Real-time PCR experiments validated miR-152 and miR-497 overexpression in fMBC. As these two miRNAs have RASSF1A and NORE1A as predicted or validated targets, we analysed their expression levels compared with the mRNA levels of the two oncosuppressor targets, demonstrating the inverse correlation between miRNA and target gene expression. However, in vitro analysis has demonstrated that hsa-miR-152 and hsa-miR-497 do not regulate NORE1A and RASSF1A expression by the direct binding to the 3′-UTR region of the genes, but probably the relation of miRNA/target gene is mediated by other target genes.
Currently, little is known about the role of miR-497 and miR-152 in BC, and in particular in fBC. MiR-497 has been identified as downregulated in women affected by BC (Li et al, 2011; Shen et al, 2012; Luo et al, 2013; Wang et al, 2013; Tahiri et al, 2014). In the study of Lehmann et al (2010), in which 319 miRNAs were profiled in MBC and compared with controls, it was reported to be one of the two most prominently downregulated miRNA. Moreover, miR-497 has already been shown to be a regulator of the MAPK/ERK pathway, as it is one of the regulators of MEK1 in addition to RAF1 and ERK1 (Zheng et al, 2012).
MiR-152 is a member of the miR-148/miR-152 family, a family whose role is controversial (Chen et al, 2013). Members of this family seem to be upregulated in nontumoural diseases and downregulated in tumours such as gastric, prostate, ovarian, breast cancer and so on (Zhou et al, 2012; Chen et al, 2013). In the miRWalk database, RASSF1A was a validated target of miR-152 even if the reported reference considered the action of miR-152 precursor on RASSF1A level mediated by DNMT-1 (Braconi et al, 2010). Moreover, recent papers have demonstrated an emerging role for this miRNA in angiogenesis regulation (Xu et al, 2013; Zheng et al, 2013).
Our study seems to overturn the function of the oncosuppressor that is normally attributed to miR-152 and miR-497, defining them as oncogenic miRNAs in fMBC.
In conclusion, we demonstrated for the first time epigenetic differences between male and female fBCs in terms of miRNA expression, highlighting a possible role for miR-152 and miR-497 in the control of RASSF1A-regulated pathways (Figure 5). The loss of RASSF1A and NORE1A mRNA could explain the enhanced tumourigenic potential and higher aggressiveness of breast cancer in men compared with women because of a reduction in apoptosis and the increase of proliferation process. For this reason, distinct, gender-associated management of the disease and therapeutic approaches may be required. However, given the fact that a single mRNA can be targeted by a large number of miRNAs and that a single miRNA can target several genes, further epigenetic studies are needed to better understand gender-associated differences in RASSF1A-regulated pathways. Moreover, epigenetics includes several other alterations in addition to miRNA interference such as DNA methylation, histone modifications and chromatin modifications that may be useful to investigate and better characterise fBC and in particular gender-associated epigenetic patterns.

Schematic representation of MAPK and Hippo signalling pathway regulation. A schematic representation of the possible regulation of MAPK and Hippo signalling pathways due to RASSF1A/NORE1A inactivation by miR-152 and miR-497 in fFBC (F) and fMBC (M).
Change history
09 December 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alvarez C, Tapia T, Cornejo V, Fernandez W, Munoz A, Camus M, Alvarez M, Devoto L, Carvallo P (2013) Silencing of tumor suppressor genes RASSF1A, SLIT2, and WIF1 by promoter hypermethylation in hereditary breast cancer. Mol Carcinog 52 (6): 475–487.
Avruch J, Praskova M, Ortiz-Vega S, Liu M, Zhang XF (2006) Nore1 and RASSF1 regulation of cell proliferation and of the MST1/2 kinases. Methods Enzymol 407: 290–310.
Avruch J, Zhou D, Fitamant J, Bardeesy N, Mou F, Barrufet LR (2012) Protein kinases of the Hippo pathway: regulation and substrates. Semin Cell Dev Biol 23 (7): 770–784.
Bee C, Moshnikova A, Mellor CD, Molloy JE, Koryakina Y, Stieglitz B, Khokhlatchev A, Herrmann C (2010) Growth and tumor suppressor NORE1A is a regulatory node between Ras signaling and microtubule nucleation. J Biol Chem 285 (21): 16258–16266.
Braconi C, Huang N, Patel T (2010) MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 51 (3): 881–890.
Callari M, Cappelletti V, De Cecco L, Musella V, Miodini P, Veneroni S, Gariboldi M, Pierotti MA, Daidone MG (2011) Gene expression analysis reveals a different transcriptomic landscape in female and male breast cancer. Breast Cancer Res Treat 127 (3): 601–610.
Chen Y, Song YX, Wang ZN (2013) The microRNA-148/152 family: multi-faceted players. Mol Cancer 12: 43.
Dweep H, Sticht C, Pandey P, Gretz N (2011) miRWalk—database: prediction of possible miRNA binding sites by "walking" the genes of three genomes. J Biomed Inform 44 (5): 839–847.
Dumitrescu RG (2012) Epigenetic markers of early tumor development. Methods Mol Biol 863: 3–14.
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12 (12): 861–874.
Fausti F, Di Agostino S, Sacconi A, Strano S, Blandino G (2012) Hippo and rassf1a pathways: a growing affair. Mol Biol Int 2012: 307628.
Fernandes MS, Carneiro F, Oliveira C, Seruca R (2013) Colorectal cancer and RASSF family—a special emphasis on RASSF1A. Int J Cancer 132 (2): 251–258.
Jiang Y, Cui L, Chen WD, Shen SH, Ding LD (2012) The prognostic role of RASSF1A promoter methylation in breast cancer: a meta-analysis of published data. PLoS One 7 (5): e36780.
Jung HY, Jung JS, Whang YM, Kim YH (2013) RASSF1A suppresses cell migration through inactivation of HDAC6 and increase of acetylated alpha-tubulin. Cancer Res Treat 45 (2): 134–144.
Lee CK, Lee JH, Lee MG, Jeong SI, Ha TK, Kang MJ, Ryu BK, Hwangbo Y, Shim JJ, Jang JY, Lee KY, Kim HJ, Chi SG (2010) Epigenetic inactivation of the NORE1 gene correlates with malignant progression of colorectal tumors. BMC Cancer 10: 577.
Lehmann U, Streichert T, Otto B, Albat C, Hasemeier B, Christgen H, Schipper E, Hille U, Kreipe HH, Langer F (2010) Identification of differentially expressed microRNAs in human male breast cancer. BMC Cancer 10: 109.
Li D, Zhao Y, Liu C, Chen X, Qi Y, Jiang Y, Zou C, Zhang X, Liu S, Wang X, Zhao D, Sun Q, Zeng Z, Dress A, Lin MC, Kung HF, Rui H, Liu LZ, Mao F, Jiang BH, Lai L (2011) Analysis of MiR-195 and MiR-497 expression, regulation and role in breast cancer. Clin Cancer Res 17 (7): 1722–1730.
Liu AM, Wong KF, Jiang X, Qiao Y, Luk JM (2012) Regulators of mammalian Hippo pathway in cancer. Biochim Biophys Acta 1826 (2): 357–364.
Luo Q, Li X, Gao Y, Long Y, Chen L, Huang Y, Fang L (2013) MiRNA-497 regulates cell growth and invasion by targeting cyclin E1 in breast cancer. Cancer Cell Int 13 (1): 95.
Melchor L, Benitez J (2013) The complex genetic landscape of familial breast cancer. Hum Genet 132 (8): 845–863.
Murria Estal R, Palanca Suela S, de Juan Jimenez I, Egoavil Rojas C, Garcia-Casado Z, Juan Fita MJ, Sanchez Heras AB, Segura Huerta A, Chirivella Gonzalez I, Sanchez-Izquierdo D, Llop Garcia M, Barragan Gonzalez E, Bolufer Gilabert P (2013) MicroRNA signatures in hereditary breast cancer. Breast Cancer Res Treat 142 (1): 19–30.
Ottini L, Palli D, Rizzo S, Federico M, Bazan V, Russo A (2010) Male breast cancer. Crit Rev Oncol Hematol 73 (2): 141–155.
Papadopoulos GL, Alexiou P, Maragkakis M, Reczko M, Hatzigeorgiou AG (2009) DIANA-mirPath: integrating human and mouse microRNAs in pathways. Bioinformatics 25 (15): 1991–1993.
Pilato B, Martinucci M, Danza K, Pinto R, Petriella D, Lacalamita R, Bruno M, Lambo R, D'Amico C, Paradiso A, Tommasi S (2011) Mutations and polymorphic BRCA variants transmission in breast cancer familial members. Breast Cancer Res Treat 125 (3): 651–657.
Pinto R, Pilato B, Ottini L, Lambo R, Simone G, Paradiso A, Tommasi S (2013) Different methylation and microRNA expression pattern in male and female familial breast cancer. J Cell Physiol 228 (6): 1264–1269.
Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP (2006) GenePattern 2.0. Nat Genet 38 (5): 500–501.
Ruddy KJ, Winer EP (2013) Male breast cancer: risk factors, biology, diagnosis, treatment, and survivorship. Ann Oncol 24 (6): 1434–1443.
Sandoval J, Esteller M (2012) Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 22 (1): 50–55.
Shen L, Li J, Xu L, Ma J, Li H, Xiao X, Zhao J, Fang L (2012) miR-497 induces apoptosis of breast cancer cells by targeting Bcl-w. Exp Ther Med 3 (3): 475–480.
Sousa B, Moser E, Cardoso F (2013) An update on male breast cancer and future directions for research and treatment. Eur J Pharmacol 717 (1-3): 71–83.
Tahiri A, Leivonen SK, Luders T, Steinfeld I, Ragle Aure M, Geisler J, Makela R, Nord S, Riis ML, Yakhini Z, Kleivi Sahlberg K, Borresen-Dale AL, Perala M, Bukholm IR, Kristensen VN (2014) Deregulation of cancer-related miRNAs is a common event in both benign and malignant human breast tumors. Carcinogenesis 35 (1): 76–85.
Tanic M, Andres E, Rodriguez-Pinilla SM, Marquez-Rodas I, Cebollero-Presmanes M, Fernandez V, Osorio A, Benitez J, Martinez-Delgado B (2013) MicroRNA-based molecular classification of non-BRCA1/2 hereditary breast tumours. Br J Cancer 109 (10): 2724–2734.
Tanic M, Yanowsky K, Rodriguez-Antona C, Andres R, Marquez-Rodas I, Osorio A, Benitez J, Martinez-Delgado B (2012) Deregulated miRNAs in hereditary breast cancer revealed a role for miR-30c in regulating KRAS oncogene. PLoS One 7 (6): e38847.
Volodko N, Gordon M, Salla M, Ghazaleh HA, Baksh S (2014) RASSF tumor suppressor gene family: Biological functions and regulation. FEBS Lett 588 (16): 2671–2684.
Wang S, Li H, Wang J, Wang D (2013) Expression of microRNA-497 and its prognostic significance in human breast cancer. Diagn Pathol 8: 172.
Xu Q, Jiang Y, Yin Y, Li Q, He J, Jing Y, Qi YT, Li W, Lu B, Peiper SS, Jiang BH, Liu LZ (2013) A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J Mol Cell Biol 5 (1): 3–13.
Zheng D, Radziszewska A, Woo P (2012) MicroRNA 497 modulates interleukin 1 signalling via the MAPK/ERK pathway. FEBS Lett 586 (23): 4165–4172.
Zheng X, Chopp M, Lu Y, Buller B, Jiang F (2013) MiR-15b and miR-152 reduce glioma cell invasion and angiogenesis via NRP-2 and MMP-3. Cancer Lett 329 (2): 146–154.
Zhou X, Zhao F, Wang ZN, Song YX, Chang H, Chiang Y, Xu HM (2012) Altered expression of miR-152 and miR-148a in ovarian cancer is related to cell proliferation. Oncol Rep 27 (2): 447–454.
Acknowledgements
We thank Caroline Oakley for manuscript revision. This work was supported by PON 01.01297 ‘VIRTUALAB’. No additional external funding was received for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Pinto, R., De Summa, S., Danza, K. et al. MicroRNA expression profiling in male and female familial breast cancer. Br J Cancer 111, 2361–2368 (2014). https://doi.org/10.1038/bjc.2014.535
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.535
Keywords
This article is cited by
-
Obesity and male breast cancer: provocative parallels?
BMC Medicine (2015)
