
Abstract
Background:
The role of telomerase reverse transcriptase (TERT) in gliomagenesis has been recently further strengthened by the frequent occurrence of TERT promoter mutations (TERTp-mut) in gliomas and evidence that the TERT SNP genetic rs2736100 influences glioma risk. TERTp-mut creates a binding site for Ets/TCF transcription factors, whereas the common rs2853669 polymorphism disrupts another Ets/TCF site on TERT promoter.
Methods:
We sequenced for TERTp-mut in 807 glioma DNAs and in 235 blood DNAs and analysed TERT expression by RT-PCR in 151 samples. TERTp-mut status and TERTp polymorphism rs2853669 were correlated with histology, genomic profile, TERT mRNA expression, clinical outcome and rs2736100 genotype.
Results:
TERTp-mut identified in 60.8% of gliomas (491 out of 807) was globally associated with poorer outcome (Hazard ratio (HR)=1.50). We defined, based on TERTp-mut and IDH mutation status, four prognostic groups: (1) TERTp-mut and IDH-mut associated with 1p19q codeletion, overall survival (OS)>17 years; (2) TERTp-wt and IDH-mut, associated with TP53 mutation, OS=97.5 months; (3) TERTp-wt and IDH-wt, with no specific association, OS=31.6 months; (4) TERTp-mut and IDH-wt, associated with EGFR amplification, OS=15.4 months. TERTp-mut was associated with higher TERT mRNA expression, whereas the rs2853669 variant was associated with lower TERT mRNA expression. The mutation of CIC (a repressor of ETV1-5 belonging to the Ets/TCF family) was also associated with TERT mRNA upregulation.
Conclusions:
In addition to IDH mutation status, defining the TERTp-mut status of glial tumours should afford enhanced prognostic stratification of patients with glioma. We also show that TERTp-mut, rs2853669 variant and CIC mutation influence Tert expression. This effect could be mediated by Ets/TCF transcription factors.
Similar content being viewed by others

Main
The telomerase reverse transcriptase (TERT) gene encodes a highly specialised reverse transcriptase, which adds hexamer repeats to the 3’ end of chromosomes (Aubert and Lansdorp, 2008; Cesare and Reddel, 2010). The increased telomerase activity seen in cancer leads to preservation of telomeres, allowing tumours to avoid induction of senescence (Smogorzewska and de Lange, 2004; Shay and Wright, 2011).
Somatic mutations of the TERT promoter (TERTp-mut) have recently been documented in various cancers (Griewank et al, 2013; Horn et al, 2013; Huang et al, 2013; Liu et al, 2013), but particularly in glioma (Aapola et al, 2000; Arita et al, 2013; Killela et al, 2013; Liu et al, 2013). The two most common mutations in TERT, C228T and C250T, map −124 and −146 bp, respectively, upstream of the TERT ATG site (chr5, 1,295,228 C>T and 1,295,250 C>T, respectively), creating binding sites for Ets/TCF transcription factors that are associated with a two- to four-fold increased transcriptional activity (Brennan et al, 2013; Huang et al, 2013).
There is increasing evidence that TERT variation also influences cancer susceptibility. Notably, the SNP rs2736100 is associated with glioblastoma (GBM) risk, especially for IDH1 wild-type GBM (Shete et al, 2009; Simon et al, 2010; Di Stefano et al, 2013). Recently, germline mutation of the TERT promoter at position −57 has been shown to cause familial melanoma (Horn et al, 2013).
Here, we have (1) determined the prevalence and prognostic impact of TERT promoter mutations, in 807 patients with glioma (WHO grades II, III and IV). (2) examined the relationship between TERT promoter mutation and tumour subtype and (3) assessed the contribution of germline mutations in these patients and in familial glioma and patients with glioma and melanoma.
Patients and methods
Patients and tissue samples
Collection of patient samples and clinico-pathological information was undertaken with informed consent and ethical board approval in accordance with the tenets of the Declaration of Helsinki. Patients studied fulfilled the following criteria: histologic diagnosis of primary glial tumour according to the WHO classification; complete clinical data and follow-up information available within in the neuro-oncology database (Onconeurothèque Paris). Blood DNAs from 80 patients with familial glioma requited through the Onconeurothèque service were also studied. For controls we made use of data previously generated on 1090 French individuals, which have been described previously (Shete et al, 2009).
Molecular analysis
DNA was extracted from fresh-frozen tumours or formalin-fixed paraffin-embedded (FFPE) tumours using the QIAmp DNA minikit (Qiagen, Courtaboeuf, France) and the iPrep ChargeSwitch Forensic kit (Life Technologies, Saint Aubin, France), respectively, DNA was extracted from EDTA-venous blood samples using a standard saline method. DNAs were quantified using Nanodrop (Thermo Fisher Scientific, Villebon sur Yvette, France).
Genomic profiling was performed by CGH-array analysis or SNP array, as previously described (Idbaih et al, 2008; Gonzalez-Aguilar et al, 2012). Mutational status of IDH1, IDH2 and TP53 was determined by Sanger sequencing, as described (Sanson et al, 2009). MGMT promoter methylation status was determined by bisulphite modification and subsequent two-stage nested methylation-specific PCR (Everhard et al, 2006).
Mutation analysis of exons 1–20 of CIC was undertaken using 454 Sequencing Technology (Roche Applied Science, Meylan, France). Details of PCR primers are shown in Supplementary Table 1. All variations were then validated by Sanger sequencing using the same primers.
Genotyping of rs2736100 has been previously described (Shete et al, 2009).
The TERT promoter was amplified using GGCCGATTCGACCTCTCT (GTCCTGCCCCTTCACCTT for FFPE samples) and AGCACCTCGCGGTAGTGG primers and Sanger sequencing performed using an ABI Prism 3730 DNA Analyzer (Applied Biosystems, Villebon sur Yvette, France).
To determine TERT mRNA expression, tumours were lysed using Lysing Matrix D tube and FastPrep instrument (MP Biomedicals, Illkirch, France) and RNA extracted using the iPrep Trizol Plus RNA Kit (Life Technologies). In all, 300 ng of RNA was retrotranscribed with the Maxima First-Strand cDNA Synthesis Kit (Thermo Scientific, Villebon sur Yvette, France). The cDNA obtained was used as a template for the determination of TERT mRNA expression by qPCR using a QuantiFast assay (Qiagen). The ΔΔCp method was applied to normalise to the expression of TERT mRNA, using both the expression of β actin and a non-tumour brain tissue sample.
Statistical analysis
The χ2 test was used to compare the distribution of categorical variables and unpaired t-test or Mann–Whitney test associations with continuous variables.
Overall survival (OS) was defined as the time between the diagnosis and death or last follow-up. Patients who were still alive at last follow-up were considered as a censored event in the analysis. Progression-free survival (PFS) was defined as the time between the diagnosis and recurrence or last follow-up. Patients who were recurrence free at last follow-up were considered as a censored event in analysis. To identify clinical and/or genomic factors associated with OS or PFS, survival curves were calculated by the Kaplan–Meier method and differences between curves assessed using the log-rank test. Variables with a significant P-value were then used to develop a multivariate Cox model. In all analyses a P-value of ⩽0.05 (two-sided) was considered to be statistically significant.
Results
Somatic and constitutional TERTp-mut status
Tumours from 807 patients (451 male; median age at diagnosis 51.0 years, range, 17.3–89.1; 206 grade II, 206 grade III and 395 grade IV) were screened for TERTp-mut. Complete patient characteristics are shown in Supplementary Table 2.
Tumours from 491 of the 807 patients (60.8%) were TERTp-mut–355 C228T (72.3%) and 136 C250T (27.7%). One GBM and two grade II oligodendrogliomas carried both C250T and C228T. These three cases were considered as TERT C228T mutant in all subsequent analyses. To confirm the mutations were somatic, we screened germline DNA of 91 of the cases. No mutation was detectable in germline DNA. We also investigated for the presence of TERTp-mut, in 80 familial glioma patients and 64 glioma patients with a second cancer −14 with melanoma (Supplementary Table 2). In none of the cases was a −149, −124 or −57 mutation identified.
rs2853669 genotypes were available for 385 of the tumours. The distribution of genotypes showed no significant departure from HWE (39 CC, 161 CT, 185 TT P=0.650). There was no difference in the distribution of genotypes between the TERTp mut and the TERTp wt cases (TT 45.8 vs 53.5%, CT 44.3 vs 36.0% and CC: 10.0 vs 10.5%, respectively).
We then investigated a purported association between somatic TERTp-mut and rs2736100 genotype in 518 glioma patients, finding no association in the whole group (allele A frequency 371 out of 616=60% vs 249 out of 420=59%, P=0.9) or when stratifying by IDH status and tumour class (Supplementary Table 3).
Case–control comparison of showed a stronger association with rs2736100 with IDH-wt gliomas but not with TERTp-mut gliomas (Supplementary Table 4). Collectively, these data imply there is no association between TERTp-mut and rs2736100 genotype. In addition, we did not find any significant association between TERT promoter mutation and the other gliomas susceptibility SNPs rs11979158, rs2252586, rs4295627, rs4977756, rs498872 and rs6010620 (data not shown).
TERTp-mut is associated with GBM and EGFR amplification, and with oligodendroglioma, 1p19q codeletion and CIC mutation
TERTp-mut was associated with an older age at diagnosis in all gliomas (median age 56.1 years for TERT mutated patients vs 40.0 years; t-test P<0.0001) and when stratified by grade (median age at diagnosis 40.4 years vs 36.1 for grade II, P=0.008; 53.3 vs 37.8 for grade III, P<0.0001 and 59.6 vs 53.6 years for grade IV, P<0.0001).
TERTp-mut was more frequent in GBM than in grade II or III tumours (299 out of 395=75.8% vs 189 out of 412=45.9%; χ2 test P<10−17), more frequent in oligodendrogliomas than in astrocytomas/oligoastrocytomas for grade III (52 out of 81=64.2% vs 46 out of 125=30.7%: χ2 test P=0.0001) and for grade II (70 out of 119=58.8% vs 21 out of 87=24.1%; χ2 test P<10−6). Additionally, there was no difference in the ratio of C228T/C250T mutations among the different grades (Table 1).
TERTp-mut was identifiable in 87.9% (94 out of 107 of gliomas with 1p19q codeletion (90 oligodendrogliomas, 17 oligoastrocytomas; 26 (24.3%) on C250T and 68 (63.6%) on C228T) as compared with 58.8% of non-codeleted gliomas (341 out of 580, χ2 test P<0.0001). EGFR amplification was present in 183 tumours (142 GBM) and was mutually exclusive with 1p19q codeletion: 163 (89.1%) having TERTp-mut (121, C228T) as compared with 51.8% (323 out of 624) of EGFR non-amplified tumours (χ2 test P<0.0001). The association of TERT promoter mutations with other molecular alterations commonly seen in glioma is detailed in Supplementary Table 5 and Supplementary Figure 1. We investigated whether there was a relationship between CIC inactivating mutations and TERTp-mut in grades II and III. CIC mutation was associated with TERTp-mut in 85% of the cases (28 out of 33), compared with 61% (25 out of 41) in CIC-wt tumours (χ2 test P<0.04).
TERTp-mut is associated with increased TERT mRNA expression
We investigated the transcriptional consequences of TERTp-mut in 153 tumours for which mRNA was available. We found a three-fold increase in mRNA expression between TERTp-mut and non-mutated groups (mean±s.e.m. 1.03±0.37 vs 3.44±0.88 AU; Mann–Whitney test P<0.0001. Figure 1A).

Expression of TERT mRNA in gliomas. The Mann–Whitney test was used to compare the expression of the different groups. (A) Expression of TERT mRNA according to TERT promoter mutation status. TERTp mutation (C228T n=88 or C250T n=30) is associated with higher TERT mRNA expression compared with TERTp-wt group (n=35) (P⩽ 0.0001 in both cases). (B) Expression of TERT mRNA according to rs2853669 status. Variant allele carriers (n=70) present a lower TERT expression than TT homozygotes (n=66) (P=0.0053). (C) Expression of TERT mRNA according to TERTp and rs2853669 status. TERT mRNA expression is lower for the variant allele carriers (n=62) compared with TT (n=56) in TERTp-mut subgroup (P=0.0079). For TERTp-wt group, only seven CC+CT samples and eight TT samples were available. (D) Expression of TERT mRNA according to CIC mutation status. TERT mRNA expression is increased in CIC mutant tumours (n=18) compared with CIC wild type (n=11; P=0.043). (E) Impact of rs2853669 and CIC mutational status TERT expression. In the CIC-wt cohort, TERT expression was lower in CC+CT subgroup, as compared with TT subgroup (P=0.0159). For the variant allele carriers (CC+CT), expression of TERT was increased in the CIC mutant group (n=8), as compared with CIC wt (n=5) (P=0.0016). *P<0.05; **P<0.01; ***P<0.0001.
Since the presence of the rs2853669 -C allele disrupts an Ets2 binding site (Rachakonda et al, 2013), we investigated the effect of rs2853669 genotype on TERT mRNA expression. Tumours harbouring the variant allele (CC+CT) showed a two-fold reduction in TERT expression, as compared with TT homozygotes (respective means±s.e.m. 2.97±1.01 and 6.57±2.04 AU; Mann–Whitney test P=0.005). This relationship was also seen in the TERTp mutant cohort, however we did not evidence any significant association in TERTp wt tumours (Figure 1B and C).
Since Ets/TCF transcription factors, including ETV1-4 transcription factors are controlled by CIC (Dissanayake et al, 2011), we also investigated a specific relationship with CIC mutation. We found TERT mRNA expression was two-fold higher in CIC mutant tumours, compared with CIC wild-type gliomas (Mann–Whitney test P=0.043) for the whole group (Figure 1D), and for the carriers of the variant allele (Figure 1E). The variant allele C was also associated with a decrease in TERT mRNA expression in the CIC wt group.
Prognostic impact of TERTp-mut is dependent on tumour grade
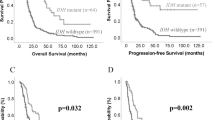
For patients with grade III and IV gliomas TERTp-mut was associated with a significantly shorter PFS and OS (Figure 2; Supplementary Table 6). For example in grade III gliomas, median OS of TERT promoter normal patients was twice longer (62.6 vs 29.4 months) than OS of TERT promoter mutated (log-rank test P=0.013). This was in sharp contrast with low-grade gliomas, where OS was better for patients with TERTp-mut (>16 years vs 97.5 months, P=0.013). There was no difference in outcome between C228T and C250T TERTp-mut in any of the analyses.

Prognostic impact of TERT promoter mutation status on overall survival and PFS, according to grade. Survivals were compared using the log-rank test (Mantel Cox). In grade II gliomas (n=206), TERTp mutation is associated with better survival (median >16 years vs 97.5 months; P=0.013). There is also a trend for better PFS (median 41.3 vs 33.3 months; P=0.068) (A) whereas in grade III (B; n=206) and grade IV gliomas (C; n=395), TERTp mutation is associated with poorer survival (median 29.4 vs 62.6 months P=0.013 and 13.8 vs 18.4 months P<0.0001) and PFS (median 15.1 vs 22.4 months P=0.006 and 8.3 vs 10.4 months P<0.0001).
In a multivariate Cox model analysis incorporating IDH mutation, age at diagnosis, 1p19q codeletion, MGMT promoter methylation, Karnofsky performance status, WHO grade and extension of surgery (Table 2) TERTp-mut was seen to be an independent negative prognostic factor for OS (Hazard ratio (HR)=1.50; 95% CI: 1.07–2.09, P=0.018).
TERTp-mut is associated with specific prognostic and molecular subgroups
Given TERTp-mut is associated with both 1p19q codeletion and EGFR amplification, which are mutually exclusive alterations with opposite prognostic effects and TERTp-mut had a different effect in low- and high-grade gliomas, prompted us to refine our survival analysis (Figure 3). Gliomas can be stratified into four distinct prognostic groups according to IDH and TERTp-mut status: (1) TERTp-mut and IDH-mut, highly associated with 1p19q codeletion (83.9%, 94 out of 11), OS> 17 years; (2) TERTp-wt and IDH-mut, associated with TP53 mutation (67.7%, 67 out of 99, OS=97.5 months); (3) TERTp-wt and IDH-wt, with no specific association (all negative), OS=31.6 months; (4) TERTp-mut and IDH-wt, highly associated with EGFR amplification (44.1%, 161 out of 365, OS=15.0 months) (Figure 4).

Association of TERT promoter mutations with the major genetic alterations in gliomas ( n =806). Each tumour is represented by a column. A yellow box indicates the presence of the genetic alteration, the absence in blue, and the cases not assessed are indicated in grey. The stratification has been done using four groups: IDH mut-TERTp mut, IDH mut-TERTp wt, IDH wt-TERTp wt and IDH wt TERTp mut. TERTp mutation is associated with two mutually exclusive alterations: 1p19q codeletion and EGFR amplification.

Prognostic stratification of gliomas according to IDH and TERT promoter mutation status ( n =804). (A) Overall survival. (B) Progression-free survival. We identified four prognostic subgroups, (1) TERTp-mut and IDH-mut (OS> 17 years, PFS 46.9 months), (2) TERTp-wt and IDH-mut (OS=97.5 months, PFS 28.6 months), (3) TERTp-wt and IDH-wt, (OS=31.6 months, PFS14.1 months) and (4) TERTp-mut and IDH-wt (OS=15.0 months, PFS 8.5 months).
TERTp-mut confers a poor prognosis except if associated with 1p19q codeletion
We considered the prognostic impact of the above classification in grades II, III and IV (Figure 5; Supplementary Table 7). In grades II and III, TERTp-mut was predictive of a longer survival in the IDH mutated group, but shorter survival in the IDH wt group. This finding can be explained by the fact that 94 out of 114 of TERTp-mut-IDH-mut are 1p19q codeleted. Indeed in the GBM group that do not include any 1p19q codeletion, TERTp-mut is associated with a particularly poor prognosis in IDH-wt tumours (OS=13.8 vs16.5 months, P=0.006) but surprisingly also in IDH-mut (OS=13.8 vs 29.1 months, P=0.022) (Figure 6). In contrast TERTp-mut was associated with a poorer outcome in IDH-wt gliomas irrespective of grade (OS: 76.2 vs 94.8 months in grade II P=ns; 18.0 vs 36.5 months in grade III P=0.007; 13.7 vs 17.5 months in grade IV P=0.006).

Prognostic stratification of gliomas according to IDH and TERT promoter mutation status (four subgroups) in grade II ( n =205), grade III ( n =206) and grade IV ( n =394). Median survivals are indicated in months if not otherwise stated. TERTp mutation is associated with a poorer outcome in IDH-wt gliomas whatever the grade. TERTp mutation is associated with better outcome in IDH mut grades II and III (OS: >16 years vs 107.8 months in grade II P=0.004; >16 years vs 102.4 in grade III P=0.0005). In contrast, the survival of grade IV with TERTp mutation and IDH-mut is extremely poor compared with TERTp-wt (13.8 vs 29.1 months; P=0.022).

Prognostic stratification of gliomas according to grade in the four molecular subgroups ( IDH mut TERTp mut n =122; IDH mut TERTp wt n =180; IDH wt TERTp wt n =114, IDH wt TERTp mut n =340). There is a dramatic difference of survival between grade IV (OS=13.8 months) and grades II+III (OS> 16 years) in the IDH-mut-TERT-mut group (P<0.0001). In contrast, the group 2 (IDH-mut-TERT-wt) is much more homogeneous through grades II–IV.
Discussion
Given that 60% of tumours being TERTp-mut, TERT is the most frequently mutated gene in gliomas thus far identified (Arita et al, 2013; Killela et al, 2013; Liu et al, 2013). We found TERTp-mut glioma patients were older, consistent with previous reports of other malignancies (Griewank et al, 2013; Killela et al, 2013). Unlike melanomas, in which germline TERTp mutations have been reported to cause familial melanoma (Horn et al, 2013), we found no evidence that TERTp-mut contributes substantially to predisposition to gliomas or the glioma/melanoma syndrome.
Our data showed that TERTp-mut is generally associated with poorer outcome in high-grade gliomas, consistently with previous data, on glioma (Killela et al, 2013, 2014), and other tumours (Rachakonda et al, 2013). In contrast, however we observed a trend for better outcome in low-grade gliomas. Stratifying tumours by both IDH1/2 and TERTp-mut status provides insight into this apparent paradox, identified four molecular subtypes of gliomas with distinct prognosis, In IDH mutated tumours, TERTp-mut is largely confined to 1p19q codeleted oligodendroglial tumours that have the best outcome (Kaloshi et al, 2007; van den Bent et al, 2013). Mutation of CIC, recently identified (Bettegowda et al, 2011; Yip et al, 2012) is also primarily a feature of this group (Figure 3). In contrast, if TERTp-mut is associated with IDH1/2 wild-type tumours, then it is mainly seen in the context of GBM (276 out of 340) with almost half of them (124 out of 276) having an EGFR amplification which is associated with poor outcome. In our study, this subgroup also included 54 grade III gliomas that had a particularly poor OS of 20.1 months. Taken together, our data show that the prognostic impact of TERTp-mut is highly contextual and depends on the histologic and genomic background of the tumour.
From a mechanistic point of view, TERTp mutation leads to the creation of a putative binding site for Ets/TCF transcription factors (Huang et al, 2013), leading to a two- to four-fold higher expression of telomerase (Arita et al, 2013; Huang et al, 2013; Nault et al, 2013; Rachakonda et al, 2013). The activity of telomerase reverse transcriptase is closely correlated with TERT mRNA level. The expression of TERT is regulated by many transcription factors binding motives located in its promoter and by epigenetic and chromatin remodelling mechanisms (Kyo et al, 2008; Zhu et al, 2010). Among the complex regulation of telomerase expression, rs2853669 has been shown to modulate both TERT expression and impact on prognosis in bladder cancer (Rachakonda et al, 2013). Indeed, the presence of the variant allele disrupts a pre-existing Ets2 binding site and results in the decrease of TERT expression in our series. However, unlike bladder cancer, rs2853669 variant does not modify the prognostic impact of TERTp mutation in our glioma series (data not shown). Our data also suggest a link between CIC mutation and TERT expression in the context of glial tumours. Indeed, the presence of the variant allele of rs2853669 did not result in a reduction of TERT expression in the CIC mutant subgroup.
Among the 40% gliomas lacking TERTp mutation, ∼50% harbour an IDH mutation (mostly astrocytomas (43 out of 180) and oligoastrocytomas (91 out of 180), which are frequently TP53 mutated. In this group, mutations in the ATRX gene (alpha thalassaemia/mental retardation syndrome X-linked), or in its partner death domain-associated protein (DAXX), which are involved in alternative lengthening telomere (ALT) phenotype, have been frequently documented (Jiao et al, 2012; Kannan et al, 2012; Liu et al, 2012; Killela et al, 2013) and are mutually exclusive with telomerase reactivation. The IDH-wt and TERTp-wt group includes mostly GBM tumours (58%, 66 out of 114). The ‘triple negative’ low-grade gliomas, characterised by a poorer outcome also conform to these categories (Metellus et al, 2010). Telomere maintenance mechanism has not been investigated yet in this subgroup.
A more detailed analysis shows the four group classification, recently reported (Killela et al, 2014) is an oversimplification (see Supplementary Figure 2 and Figure 5); for example, the TERTp-mut-IDH-mut is indicative of better outcome for grades II and III with an OS>17 years, but is associated with a poorer outcome in GBM (OS=13.8 months), whereas in GBM the best group prognostic is those patients with TERTp-wt-IDH-mut group (OS=29.1 vs 13.8 months for the TERTp-mut-IDH mut group). This discrepancy is unlikely to be solely explained by the relationship between TERTp mutation and 1p19q codeletion, present in 89% (94 out of 106) of our TERTp-mut-IDH-mut grades II and III, but none of our grade IV tumours. In fact, the survival of patients with TERTp-mut-IDH-mut grades II and III, without 1p19q codeletion was not significantly different from those with 1p19q codeleted tumours (median OS=10 years) (Supplementary Figure 3).
In conclusion, our data confirm the high frequency of TERTp-mut in glioma and show that these mutations clusterise into specific entities, with distinct clinical significances. TERTp mutations are mostly associated with poor outcome, except for 1p19q codeleted grade II and grade III, and for EGFR amplified grade III and grade IV (Supplementary Figure 2A and B, respectively). A telomere maintenance mechanism (either TERTp mutation or ATRX/DAXX mutations) is involved in >80% of gliomas and appears therefore as a unique feature in these tumors, offering the prospect of new therapeutic approaches.
Change history
11 November 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aapola U, Kawasaki K, Scott HS, Ollila J, Vihinen M, Heino M, Shintani A, Minoshima S, Krohn K, Antonarakis SE, Shimizu N, Kudoh J, Peterson P (2000) Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 65 (3): 293–298.
Arita H, Narita Y, Fukushima S, Tateishi K, Matsushita Y, Yoshida A, Miyakita Y, Ohno M, Collins VP, Kawahara N, Shibui S, Ichimura K (2013) Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol 126 (2): 267–276.
Aubert G, Lansdorp PM (2008) Telomeres and aging. Physiol Rev 88 (2): 557–579.
Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, Velculescu VE, Oba-Shinjo SM, Marie SK, Vogelstein B, Bigner D, Yan H, Papadopoulos N, Kinzler KW (2011) Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 333 (6048): 1453–1455.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, Zou L, Vegesna R, Shukla SA, Ciriello G, Yung WK, Zhang W, Sougnez C, Mikkelsen T, Aldape K, Bigner DD, Van Meir EG, Prados M, Sloan A, Black KL, Eschbacher J, Finocchiaro G, Friedman W, Andrews DW, Guha A, Iacocca M, O'Neill BP, Foltz G, Myers J, Weisenberger DJ, Penny R, Kucherlapati R, Perou CM, Hayes DN, Gibbs R, Marra M, Mills GB, Lander E, Spellman P, Wilson R, Sander C, Weinstein J, Meyerson M, Gabriel S, Laird PW, Haussler D, Getz G, Chin L (2013) The somatic genomic landscape of glioblastoma. Cell 155 (2): 462–477.
Cesare AJ, Reddel RR (2010) Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11 (5): 319–330.
Di Stefano AL, Enciso-Mora V, Marie Y, Desestret V, Labussiere M, Boisselier B, Mokhtari K, Idbaih A, Hoang-Xuan K, Delattre JY, Houlston RS, Sanson M (2013) Association between glioma susceptibility loci and tumour pathology defines specific molecular etiologies. Neuro Oncol 15 (5): 542–547.
Dissanayake K, Toth R, Blakey J, Olsson O, Campbell DG, Prescott AR, MacKintosh C (2011) ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem J 433 (3): 515–525.
Everhard S, Kaloshi G, Criniere E, Benouaich-Amiel A, Lejeune J, Marie Y, Sanson M, Kujas M, Mokhtari K, Hoang-Xuan K, Delattre JY, Thillet J (2006) MGMT methylation: a marker of response to temozolomide in low-grade gliomas. Ann Neurol 60 (6): 740–743.
Gonzalez-Aguilar A, Idbaih A, Boisselier B, Habbita N, Rossetto M, Laurenge A, Bruno A, Jouvet A, Polivka M, Adam C, Figarella-Branger D, Miquel C, Vital A, Ghesquieres H, Gressin R, Delwail V, Taillandier L, Chinot O, Soubeyran P, Gyan E, Choquet S, Houillier C, Soussain C, Tanguy ML, Marie Y, Mokhtari K, Hoang-Xuan K (2012) Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res 18 (19): 5203–5211.
Griewank KG, Murali R, Schilling B, Scholz S, Sucker A, Song M, Susskind D, Grabellus F, Zimmer L, Hillen U, Steuhl KP, Schadendorf D, Westekemper H, Zeschnigk M (2013) TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. Br J Cancer 109 (2): 497–501.
Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R (2013) TERT promoter mutations in familial and sporadic melanoma. Science (New York, NY) 339 (6122): 959–961.
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339 (6122): 957–959.
Idbaih A, Marie Y, Lucchesi C, Pierron G, Manie E, Raynal V, Mosseri V, Hoang-Xuan K, Kujas M, Brito I, Mokhtari K, Sanson M, Barillot E, Aurias A, Delattre JY, Delattre O (2008) BAC array CGH distinguishes mutually exclusive alterations that define clinicogenetic subtypes of gliomas. Int J Cancer 122 (8): 1778–1786.
Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, Friedman H, Friedman AH, Riggins GJ, Holdhoff M, Burger P, McLendon R, Bigner DD, Vogelstein B, Meeker AK, Kinzler KW, Papadopoulos N, Diaz LA, Yan H (2012) Frequent ATRX, CIC, and FUBP1 mutations refine the classification of malignant gliomas. Oncotarget 3 (7): 709–722.
Kaloshi G, Benouaich-Amiel A, Diakite F, Taillibert S, Lejeune J, Laigle-Donadey F, Renard MA, Iraqi W, Idbaih A, Paris S, Capelle L, Duffau H, Cornu P, Simon JM, Mokhtari K, Polivka M, Omuro A, Carpentier A, Sanson M, Delattre JY, Hoang-Xuan K (2007) Temozolomide for low-grade gliomas: predictive impact of 1p/19q loss on response and outcome. Neurology 68 (21): 1831–1836.
Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT (2012) Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 3 (10): 1194–1203.
Killela PJ, Pirozzi CJ, Healy P, Reitman ZJ, Lipp E, Rasheed BA, Yang R, Diplas BH, Wang Z, Greer PK, Zhu H, Wang CY, Carpenter AB, Friedman H, Friedman AH, Keir ST, He J, He Y, McLendon RE, Herndon JE 2nd, Yan H, Bigner DD (2014) Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget 5 (6): 1515–1525.
Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, Hruban RH, Jallo GI, Mandahl N, Meeker AK, Mertens F, Netto GJ, Rasheed BA, Riggins GJ, Rosenquist TA, Schiffman M, Shih IeM, Theodorescu D, Torbenson MS, Velculescu VE, Wang TL, Wentzensen N, Wood LD, Zhang M, McLendon RE, Bigner DD, Kinzler KW, Vogelstein B, Papadopoulos N, Yan H (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA 110 (15): 6021–6026.
Kyo S, Takakura M, Fujiwara T, Inoue M (2008) Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci 99 (8): 1528–1538.
Liu X, Wu G, Shan Y, Hartmann C, von Deimling A, Xing M (2013) Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle (Georgetown, Tex) 12 (10): 1637–1638.
Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, Fleming A, Hadjadj D, Schwartzentruber J, Majewski J, Dong Z, Siegel P, Albrecht S, Croul S, Jones DT, Kool M, Tonjes M, Reifenberger G, Faury D, Zadeh G, Pfister S, Jabado N (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124 (5): 615–625.
Metellus P, Coulibaly B, Colin C, de Paula AM, Vasiljevic A, Taieb D, Barlier A, Boisselier B, Mokhtari K, Wang XW, Loundou A, Chapon F, Pineau S, Ouafik L, Chinot O, Figarella-Branger D (2010) Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol 120 (6): 719–729.
Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, Zucman-Rossi J (2013) High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 4: 2218.
Rachakonda PS, Hosen I, de Verdier PJ, Fallah M, Heidenreich B, Ryk C, Wiklund NP, Steineck G, Schadendorf D, Hemminki K, Kumar R (2013) TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci USA 110 (43): 17426–17431.
Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY (2009) Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27 (25): 4150–4154.
Shay JW, Wright WE (2011) Role of telomeres and telomerase in cancer. Semin Cancer Biol 21 (6): 349–353.
Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY, Hoang-Xuan K, El Hallani S, Idbaih A, Zelenika D, Andersson U, Henriksson R, Bergenheim AT, Feychting M, Lonn S, Ahlbom A, Schramm J, Linnebank M, Hemminki K, Kumar R, Hepworth SJ, Price A, Armstrong G, Liu Y, Gu X, Yu R, Lau C, Schoemaker M, Muir K, Swerdlow A, Lathrop M, Bondy M, Houlston RS (2009) Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 41 (8): 899–904.
Simon M, Hosking FJ, Marie Y, Gousias K, Boisselier B, Carpentier C, Schramm J, Mokhtari K, Hoang-Xuan K, Idbaih A, Delattre JY, Lathrop M, Robertson LB, Houlston RS, Sanson M (2010) Genetic risk profiles identify different molecular etiologies for glioma. Clin Cancer Res 16 (21): 5252–5259.
Smogorzewska A, de Lange T (2004) Regulation of telomerase by telomeric proteins. Annu Rev Biochem 73: 177–208.
van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Enting RH, French PJ, Dinjens WN, Vecht CJ, Allgeier A, Lacombe D, Gorlia T, Hoang-Xuan K (2013) Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31 (3): 344–350.
Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, Birol I, Chesnelong C, Chiu R, Chuah E, Corbett R, Docking R, Firme M, Hirst M, Jackman S, Karsan A, Li H, Louis DN, Maslova A, Moore R, Moradian A, Mungall KL, Perizzolo M, Qian J, Roldan G, Smith EE, Tamura-Wells J, Thiessen N, Varhol R, Weiss S, Wu W, Young S, Zhao Y, Mungall AJ, Jones SJ, Morin GB, Chan JA, Cairncross JG, Marra MA (2012) Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol 226 (1): 7–16.
Zhu J, Zhao Y, Wang S (2010) Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell 1 (1): 22–32.
Acknowledgements
This study is supported by grants from the Institut National du cancer (INCa), the Association pour la Recherche contre le Cancer (ARC), the Ligue Nationale contre le Cancer (LNCC), and Association pour la Recherche sur les Tumeurs cérébrales (ARTC). ALDS is supported by an investigator fellowship from Collegio Ghislieri, Pavia, Italy. VG and SM are supported by a grant from the Association pour la Recherche contre le Cancer (ARC). SM is supported by a grant from the Ligue Nationale contre le Cancer (LNCC).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Labussière, M., Di Stefano, A., Gleize, V. et al. TERT promoter mutations in gliomas, genetic associations and clinico-pathological correlations. Br J Cancer 111, 2024–2032 (2014). https://doi.org/10.1038/bjc.2014.538
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.538
Keywords
This article is cited by
-
Genomic profiles of IDH-mutant gliomas: MYCN-amplified IDH-mutant astrocytoma had the worst prognosis
Scientific Reports (2023)
-
Predicting TERT promoter mutation status using 1H-MR spectroscopy and stretched-exponential model of diffusion-weighted imaging in IDH-wildtype diffuse astrocytic glioma without intense enhancement
Neuroradiology (2023)
-
The road-map for establishment of a prognostic molecular marker panel in glioma using liquid biopsy: current status and future directions
Clinical and Translational Oncology (2022)
-
Synergistic activation of mutant TERT promoter by Sp1 and GABPA in BRAFV600E-driven human cancers
npj Precision Oncology (2021)
-
Germline ALK variations are associated with a poor prognosis in glioma and IDH-wildtype glioblastoma
Journal of Neuro-Oncology (2021)
