
Abstract
Background:
Gambogic acid (GA) has been reported to have potent anticancer activity and is authorised to be tested in phase II clinical trials for treatment of non-small-cell lung cancer (NSCLC). The present study aims to investigate whether GA would be synergistic with cisplatin (CDDP) against the NSCLC.
Methods:
1-(4,5-Dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT), combination index (CI) isobologram, western blot, quantitative PCR, flow cytometry, electrophoretic mobility shift assay, xenograft tumour models and terminal deoxynucleotide transferase-mediated dUTP nick-end labelling analysis were used in this study.
Results:
The cell viability results showed that sequential CDDP-GA treatment resulted in a strong synergistic action in A549, NCI-H460, and NCI-H1299 cell lines, whereas the reverse sequence and simultaneous treatments led to a slight synergistic or additive action. Increased sub-G1 phase cells and enhanced PARP cleavage demonstrated that the sequence of CDDP-GA treatment markedly increased apoptosis in comparison with other treatments. Furthermore, the sequential combination could enhance the activation of caspase-3, -8, and 9, increase the expression of Fas and Bax, and decrease the expression of Bcl-2, survivin and X-inhibitor of apoptosis protein (X-IAP) in A549 and NCI-H460 cell lines. In addition, increased apoptosis was correlated with enhanced reactive oxygen species generation. Importantly, it was found that, followed by CDDP treatment, GA could inhibit NF-κB and mitogen-activated protein kinase (MAPK)/heme oxygenase-1 (HO-1) signalling pathways, which have been validated to reduce ROS release and confer CDDP resistance. The roles of NF-κB and MAPK pathways were further confirmed by using specific inhibitors, which significantly increased ROS release and apoptosis induced by the sequential combination of CDDP and GA. Moreover, our results indicated that the combination of CDDP and GA exerted increased antitumour effects on A549 xenograft models through inhibiting NF-κB, HO-1, and subsequently inducing apoptosis.
Conclusion:
Gambogic acid sensitises lung cancer cells to CDDP in vitro and in vivo in NSCLC through inactivation of NF-κB and MAPK/HO-1 signalling pathways, providing a rationale for the combined use of CDDP and GA in lung cancer chemotherapy.
Similar content being viewed by others


Main
Lung cancer is the leading cause of cancer deaths worldwide, which accounts for 18.2% of total cancer deaths with a high fatality (the ratio of mortality to incidence is 0.86) (Ferlay et al, 2010). Non-small-cell lung cancer (NSCLC) accounts for nearly 80% of all lung cancers (D’Addario et al, 2005). Platinum-based doublet regimens have become the major treatment option for NSCLC. However, the 1-year survival rate was increased by only 5% with platinum-based doublet regimens and the toxicity was higher when compared with non-platinum-based regimens (D’Addario et al, 2005). Therefore, there is a need for investigation and development of novel agents that can be added to and improve the platinum-based therapy.
Recently, herbal and herbal-derived drugs have been recognised as one of the attractive approaches for lung cancer therapy with little side effects (Jeong et al, 2011; Olaku and White, 2011). Furthermore, there are evidences that various herbal medicines have proven to be useful and effective in sensitising conventional agents, prolonging survival time, preventing side effects of chemotherapy, and improving quality of life in lung cancer patients (Chen et al, 2010; Jeong et al, 2011). Gambogic acid (GA) is a natural compound isolated from gamboge, a dry resin secreted from the Garcinia hanburyi tree in Southeast Asia (Wu et al, 2004). Previous investigations have demonstrated that GA could inhibit the growth of several types of human cancer cells, including lung cancer (Wu et al, 2004), leukaemia (Pandey et al, 2007), prostate cancer (Wang et al, 2012b), pancreatic cancer (Wang et al, 2012a), gastric cancer (Yu et al, 2007), and hepatocarcinoma (Guo et al, 2004) in vitro and in vivo. The possible anticancer mechanisms of GA are associated with the induction of apoptosis (Zhu et al, 2009), enhancement of reactive oxygen species (ROS) accumulation (Nie et al, 2009), inhibition of telomerase (Guo et al, 2004; Wu et al, 2004) activity, and interception of NF-κB signalling pathway (Pandey et al, 2007; Wang et al, 2012b). Currently, GA has been approved by the Chinese Food and Drug Administration for the treatment of lung cancer in phase II clinical trials (Wang et al, 2012b).
Here, we asked whether the novel anticancer agent can improve the platinum-based chemotherapy in NSCLC. Thus, in this study we investigated the effects of GA combined with cisplatin with regard to their activities against NSCLC in vitro and in vivo.
Materials and methods
Reagents
Gambogic acid (>98% purity, provided by Jiangsu Kanion Pharmaceutical Co. Ltd, Lianyungang, China) and cisplatin (Sigma-Aldrich, St Louis, MO, USA) were dissolved by DMSO to 100 mM and stored at −20 °C (Figure 1A). 1-(4,5-Dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT) was purchased from Sigma-Aldrich and was dissolved in PBS. Propidium idodide (PI) was purchased from Biosharp (St. Louis, MO, USA) and was dissolved in distilled water. The primary antibodies against PARP, caspase-3, caspase-8, caspase-9, NF-κB p65, NF-κB p50, ERK1/2, phospho-ERK1/2, p38, phospho-p38, JNK, phospho-JNK, and heme oxygenase-1 (HO-1) were purchased from Cell Signalling Technology (Danvers, MA, USA), and antibodies to β-actin, Bcl-2, Bax, survivin, X-IAP, Fas, and Bcl-XL were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA)

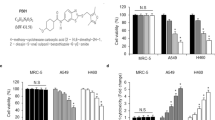
Sequence-specific potentiation of CDDP-induced growth inhibition by GA in NSCLC cells. (A) Chemical structure of GA and CDDP. (B) Timeline of in vitro experiments: (i) in studies of CDDP or GA as single agents, they were administered for 48 h followed by 48 h without drug; (ii) in studies of CDDP plus GA, they were administered concomitant for 48 h followed by 48 h without drug; (iii) in studies of GA before treatment, cells were treated with GA for 48 h followed by drug-free washout and CDDP for 48 h; (iv) in studies of CDDP before treatment, cells were treated with CDDP for 48 h followed by drug-free washout and GA for 48 h. (C) The growth curve of NSCLC cells after treated with GA, CDDP, and the combination of GA and CDDP in three sequences. (D) Analysis of the combination of GA and CDDP in NSCLC cells. A549, NCI-H460, and NCI-H1299 cells were treated for 48 h using increasing concentrations of GA and CDDP, either alone or in a fixed ratio, as described in Materials and Methods. The resultant data were analysed using Calcusyn program, and graphs from a representative experiment for each treatment schedule are shown. A CI <0.90 indicates synergism; 0.90–1.10, additive; and >1.10, antagonism. These experiments were repeated in triplicate.
Cell lines and cell culture
Human NSCLC cell lines A549 and NCI-H460 were obtained from the American Type Culture Collection (Manassas, VA, USA). Human NSCLC cell line NCI-H1299 was purchased from Shanghai Cell Bank (Shanghai, China). They were routinely cultured in Roswell Park Memorial Institute 1640 supplemented with 10% fetal bovine serum and maintained at 37 °C in a humidified incubator with 5% CO2.
Cell viability assay and determination of combination index
The in vitro cell viability effects of GA, CDDP alone, or combined treatments were determined by MTT assay. The cells (2 × 104 cells per ml) were seeded into 96-well culture plates. After overnight incubation, the cells were treated with various concentrations of drugs. For the combined treatment in NSCLC cells, we tested three sequences: (a) GA followed by CDDP cells were exposed to GA for 48 h, and then after washout of GA, cells were treated with CDDP for an additional 48 h; (b) CDDP followed by GA cells were exposed to CDDP for 48 h, and then after washout of CDDP, cells were treated with GA for an additional 48 h; and (c) concurrent treatment cells were exposed to both GA and ADM for 48 h. The nature of the drug interaction was analysed by using the combination index (CI) according to the method of Chou and Talalay (1984). A CI <0.90 indicates synergism; a CI between 0.90 and 1.10 indicates additive; and a CI >1.10 indicates antagonism. Data analysis was performed by the Calcusyn software (Biosoft, Oxford, UK).
Flow cytometry analysis
About 1–5 × 106 A549 and NCI-H460 cells were harvested at room temperature after pretreatment with various reagents for 24 or 48 h. The supernatant was removed and the cells were trypsinised, and then ice-cold 70% ethanol was added. Ethanol-fixed cells were resuspended in PBS containing 0.1 mg ml−1 RNase and incubated at 37 °C for 30 min. The pelleted cells were suspended in 1.0 ml of 40 μg ml−1 propidium iodide (PI) and analysed by using flow cytometer (Becton Dickinson, San Jose, CA, USA). The cell cycle distribution was estimated according to standard procedures. The percentage of cells in the different cell cycle phases (G0/G1, S, or G2/M phase) was calculated using CELLQuest (Becton Dickinson) software. The cells of sub-G1 peak were considered as apoptosis.
Western blot analysis
About 1 × 107 A549 and NCI-H460 cells were gathered after pretreatment for 24 h. The nuclear protein was prepared by a commercial kit (Thermo Scientific, Rockford, IL, USA). The tumour tissue protein was purified according to the reported method (Zheng et al, 2011). Western blot was performed as described previously (Zhang et al, 2011). In brief, an equal amount of total protein extracts from cultured cells or tissues were fractionated by 10–15% SDS–PAGE and electrically transferred onto polyvinylidene difluoride (PVDF) membranes. Mouse or rabbit primary antibodies and horseradish peroxidase (HRP)-conjugated appropriate secondary antibodies were used to detect the designated proteins. The bound secondary antibodies on the PVDF membrane were reacted with ECL detection reagents (Thermo Scientific) and exposed in ImageQuant LAS 4000mini system (GE Healthcare, Buckinghamshire, UK). Results were normalised to the internal control β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and quantified by Quantity One Software (Bio-Rad, Hercules, CA, USA).
Examination of intracellular ROS accumulation
Intracellular hydrogen peroxide levels were monitored by flow cytometry after staining with DCFH-DA (6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate; Molecular Probes, Eugene, OR, USA). Briefly, cells in a logarithmic growth phase (1 × 105 cells per ml in 35 mm polystyrene culture dishes) were treated with GA, CDDP, and CDDP plus GA for 24 h, and then labelled with 5 μmol l−1 DCFH-DA for 1 h. Next, the cells were trypsinised, washed with PBS, and then analysed by flow cytometry (Becton Dickinson). The percentage of cells displaying increased dye uptake was used to reflect an increase in ROS levels.
Electrophoretic mobility shift assays
Nuclear extracts from A549 and NCI-H460 cells were obtained with the commercially optimised NE-PER Nuclear and Cytoplasmic extraction reagents (Thermo Scientific Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. Sequence of the sense strand of the oligonucleotide probe used for electrophoretic mobility shift assay (EMSA) was as follows: triplicate repeat 5′-AGTTGAGGGGACTTTCCCAGGC-3′(NF-κB). Binding was performed at 30 °C for 20 min in a final volume of 20 μl, which contained 50 μg of nuclear extract, 5 pmol biotin-labelled specific consensus oligonucleotide, 20 μg poly(dI/dC) (Pharmacia, Freiburg, Germany), 20 μl buffer A (20 mM HEPES, pH 7.9, 20% glycerol, 100 mM KCl, 0.5 mM EDTA, 0.25% NP-40, 2 mM dithiothreitol, 0.1 mM phenylmethylsulphonyl fluoride), and 40 μl buffer B (20% Ficoll 400 (Pharmacia, Freiburg, Germany), 100 mM HEPES, pH 7.9, 300 mM KCl, 10 mM dithiothreitol, 0.1 mM phenylmethylsulphonyl fluoride). The DNA–protein complexes were resolved on a 6% non-denaturing polyacrylamide gel and transferred to a nitrocellulose membrane. Subsequently, the membrane was fixed with UV light and blocked with 5% non-fat milk. The membrane was incubated overnight at 4 °C with a streptavidin–HRP conjugate. The binding signal was detected using a Supersignal Chemiluminescent Substrate kit (Pierce). Specificity of binding was checked by incubating in the presence of an excess of ‘cold’ NF-κB oligo sequence.
Quantitative PCR analysis
Total RNA was isolated using RNeasy Mini Kit (Quiagen Inc., Valencia, CA, USA) as described in the product introduction. The RNA was reverse RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) and PCR was performed using iQ SYBR Green Supermix and the CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Primers used were GAPDH reverse primer 5′-CCCTCAACGACCACTTTGTCA-3′ and forward primer 5′-TTCCTCTTGTGCTCTTGCTGG-3′; interleukin-8 (IL-8) reverse primer 5′-TGAATTCTCAGCCCTCTTCAA-3′ and forward primer 5′-TCTGCAGCTCTGTGTGAAGG-3′; survivin reverse primer 5′-TTGCCGACAGGATGCAGAA-3′ and forward primer 5′-GCCGATCCACACGGAGTACT-3′; HO-1 reverse primer 5′-TGTTGCGCTCAATCTCCTCCT-3′ and forward primer 5′-ATGGCCTCCCTGTACCACATC-3′.
In vivo tumour growth model
To determine the in vivo antitumour activity of GA combined with CDDP, viable A549 cells (5 × 106/100 μl PBS per mouse), as confirmed by trypan blue staining, were subcutaneously injected into the right flank of 7- to 8-week-old male SCID mice. When the average tumour volume reached 100 mm3, the mice were randomly divided into four treatment groups, including control (saline only, n=5), GA (3.0 mg kg−1 per 2 days, intravenously; n=6), CDDP (4 mg kg−1 per week, intravenously; n=6), and sequential combination (CDDP treatment one day before GA treatment, n=6). The drug doses determined from previously published work (Li et al, 2002; Yang et al, 2007), CDDP (4 mg kg−1, weekly) was generally administered at doses less than the maximum-tolerated dose in an attempt to allow any additive effects of combination treatment with platinum-based agents and GA to be more easily detected. Tumour size was measured once every 2 days with a calipre (calculated volume=shortest diameter2 × longest diameter/2). Body weight was recorded once every 2 days. After 14 days, the mice were killed and the tumours were excised and stored at −80 °C until further analysis. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Shenyang Pharmaceutical University.
Apoptosis analysis by TUNEL
Formalin-fixed tumour tissues harvested 28 days after tumour implantation were embedded in paraffin and sectioned. Terminal deoxynucleotide transferase-mediated dUTP nick-end labelling (TUNEL) system (Roche, Basel, Switzerland) was used to detect apoptosis in the tumour sections placed on slides according to the manufacturer’s protocol. Terminal deoxynucleotide transferase-mediated dUTP nick-end labelling reaction solution was substituted with TdT-free solution for a negative control. Sections were pretreated 10 min with DNase and visualised by diaminobenzidine staining. Positive nuclei were identified by brown colour.
Statistical analysis
Statistical analysis was performed using SPSS11.5 software package for Windows (SPSS, Chicago, IL, USA). Data were presented as the mean±s.e.m. Statistical significance was calculated using a Student’s t-test, with a probability level of P<0.05 considered to be statistically significant. Statistical differences among the effects of GA, CDDP, combination, or inhibitors, shown in the Figures 5A and 7A, was calculated using a one-way ANOVA with a post hoc Dunnett’s test comparing the means to the untreated control or combination treatment.
Results
GA synergised the growth inhibitory activity of CDDP on NSCLC cells at a sequence-dependent manner
The growth inhibitory effects of GA or CDDP on A549, NCI-H460, and NCI-H1299 cells were assessed by the MTT assay after 48 h exposure. A concentration-dependent inhibition of cell growth was observed with GA and CDDP, with IC50s of 3.56±0.36 and 21.88±3.21 μ M for A549 cells, 4.05±0.51 and 25.76±4.03 μ M for NCI-H460 cells, and 1.12±0.31 μ M and 25.21±4.38 μ M for NCI-H1299 cells. Because the CI method recommends a ratio of IC50 values that the drugs are equipotent, combination studies were performed at fixed 7 : 44, 2 : 13, and 1 : 25(GA : CDDP) concentration ratios in A549, NCI-H460, and NCI-H1299 cells, respectively. To determine whether the sequence of drug addition impacts on resulting synergy, cells were treated for 48 h with CDDP, followed by removal of the supernatant, and treatment for 48 h with the GA, or vice versa (see Figure 1B). The results showed that 48 h of exposure to CDDP followed by a 48-h exposure to GA led to a strong synergistic antiproliferative activity on three cell lines (Figure 1C), with the maximal CI were 0.43 for A549 cells, 0.49 for NCI-H460 cells, and 0.19 for NCI-H1299 cells (CI<0.5; Figure1D). On the contrary, the reverse sequence (GA followed by CDDP) and the concomitant treatment schedule resulted in a slightly synergistic or additive effect (Figure 1C and data not shown, CI>0.7). In addition, the antiproliferative activity of sequential treatment CDDP and GA for 48 h got close to concomitant treatment with CDDP and GA for 96 h (see Supplementary Figure 1). These results suggest that GA may synergise the growth inhibitory activities of CDDP on NSCLC cells when administered in a stepwise schedule.
GA enhanced the apoptosis-induced effect of CDDP on NSCLC cells at a sequence-dependent manner
Flow cytometry analysis after PI staining was used to detect the apoptosis induced by GA and/or CDDP. We treated A549, NCI-H460, and NCI-H1299 cells with GA and CDDP at IC50 concentrations (pharmacologically achievable concentration), or combination of both for 24 or 48 h. As shown in Figure 2A, the occurrence of apoptotic cells (sub-G1 phase cells) was increased in A549 and NCI-H460 cells after treatment with GA and CDDP for 24 h (2.7±1.6% and 16.3±3.1%, respectively, vs controls, 1.1±0.9%, for A549 cells; and 15.4±2.0% and 16.7±2.4% vs 2.1±1.3% for NCI-H460 cells), but was not changed in NCI-H1299 cells(2.3±1.0% for GA and 2.2±0.8% for CDDP, vs controls 2.2±1.2%), suggesting NCI-H1299 cells is more resistant to CDDP-induced apoptosis as compared with A549 and NCI-H460 cells. Interestingly, in the drug combinations, the sequence CDDP then GA administration produced the highest apoptotic index in three cell lines (48.7±5.6% for A549 cells; 33.5±3.8% for NCI-H460 cells; 30.3±4.5% for NCI-H1299 cells), whereas the sequence GA then CDDP and simultaneous administration of both just resulted in a moderately apoptotic index (Figure 2A). The similar results were found in the A549 and NCI-H460 cells after being treated with GA, CDDP, and combination of both for 48 h (see Supplementary Figure 2).

Sequence-specific enhancement of CDDP-induced apoptosis by GA in NSCLC cells. A549 cells were treated with GA (3.5 μ M, 24 h), CDDP (22 μ M, 24 h), or the combination (concomitant: GA 3.5 μ M and CDDP 22 μ M 24 h; GA→CDDP: GA 3.5 μ M 24 h followed by CDDP 22 μ M 24 h; CDDP→GA: CDDP 22 μ M 24 h followed by GA 3.5 μ M 24 h). NCI-H460 cells were treated with GA (4 μ M), CDDP (26 μ M), or the combination (GA 4 μ M and CDDP 26 μ M) for 24 h as above-described sequence. NCI-H1299 cells were treated with GA (1 μ M), CDDP (25 μ M), or the combination (GA 1 μ M and CDDP 25 μ M) for 24 h as above-described sequence. (A) The apoptosis and cell cycle were assessed by flow cytometry analysis. Sub-G1 is considered as apoptosis. These experiments were repeated in duplicate. Data were presented as the mean. (B) The apoptosis was assessed by western blot. Cleavage (Clv) of poly (ADP-ribose) polymerase (PARP) is considered as apoptosis. Columns, means for three replicate determinations; bars, s.d.
To further confirm the above data, PARP cleavage, another marker for apoptosis, was determined by western blot. As shown in Figure 2B, PARP cleavage was slightly or moderately induced by GA or CDDP as single agents, and then simultaneous administration or the sequence of GA followed by CDDP in three NSCLC cell lines, whereas the sequence of CDDP followed by GA resulted in an obvious cleavage of PARP. Taken together, these results are well conformed to the MTT data, suggesting that the synergism of CDDP followed by GA might be executed via apoptosis-inducing action in NSCLC cells. The A549 and NCI-H460 cell lines were selected for the next experiments, due to their sensitivity to CDDP- plus GA-induced apoptosis and the representative to different NSCLC subtypes (A549 from adenocarcinoma and NCI-H460 from large-cell carcinoma).
The sequential combination of CDDP and GA induced an enhancement of apoptosis in caspases-dependent pathway
As apoptosis is usually associated with the activation of the caspase cascade, we further investigated the involvement of caspases in combined treatment-induced apoptosis. First, we determined caspase-3, which is known to be a key effector caspase in various forms of apoptosis that degrades several cellular proteins (Crawford and Wells, 2011). After 24 h treatment, the sequential combination of CDDP and GA could cause an obvious cleavage of procaspase-3 when compared with GA or CDDP monotreatment in A549 and NCI-H460 cells (Figure 3). Furthermore, the activities of initiator caspase (caspase-8 and -9) were also investigated. As shown in Figure 3, the sequential combination led to an enhanced cleavage of procaspase-8 when compared with GA or CDDP monotreatment in A549 and NCI-H460 cells. Similarly, the cleavages of caspase-9 were obviously increased after treatment with CDDP and GA. Taken together, these results indicate that the sequential combination of CDDP and GA induces cell death by stimulating caspase-dependent apoptotic pathways.

The sequential combination of CDDP and GA induced an enhancement of apoptosis in caspase-dependent pathway. A549 and NCI-H460 cells were treated with dimethyl sulphoxide (DMSO) control, CDDP, GA, or sequential combination (CDDP→GA) as mentioned above. Then, protein extracts were immunoblotted with the specified antibodies against caspase-8, caspase-9, and caspase-3. Activation levels of caspases (cleavage bands) were determined. Mean±s.d. for three replicate determinations.
The effects of the sequential combination of CDDP and GA on apoptosis-related proteins
To further study the role of apoptosis induced by CDDP plus GA, we evaluated the expression of Fas, Bax, Bcl-2, Bcl-XL, survivin and X-IAP proteins by western blot performed on whole-cell lysates from control and treated A549 and NCI-H460 cells and the results were graphically presented in Figures 4A–C. In A549 cells, the sequential combination treatment significantly increased Fas and Bax proteins levels by ∼4.1- and ∼7.8-fold compared with ∼1.05- and ∼2.8-fold increase by GA or ∼2.0- and ∼4.2-fold CDDP alone, respectively (Figure 4A). The combination significantly decreased the expression of Bcl-2 to ∼0.08-fold compared with a ∼0.91-fold decrease by GA or ∼0.32-fold CDDP alone, but had no affect on the expression of Bcl-XL (Figure 4B). In addition, the combination significantly decreased survivin and X-IAP expression to ∼0.20- and ∼0.11-fold compared with control, respectively (Figure 4C). The similar data was found in NCI-H460 cells (see Figures 4A–C). These results demonstrate that the Fas, Bcl-2 family, and IAPs family proteins may also be involved in the synergism.

The effects of GA, CDDP, and the sequential combination on apoptosis-related proteins of A549 and NCI-H460 cells. A549 and NCI-H460 cells were treated with DMSO control, CDDP, GA, or sequential combination (CDDP→GA) as mentioned above. Then, protein extracts were immunoblotted with the specified antibodies against Fas, Bax (A), Bcl-XL, Bcl-2 (B), survivin, and XIAP (C). Protein expression levels (relative to β-actin) were determined. Mean±s.d. for three replicates were determined.
The sequential combination of CDDP and GA induced an enhancement of ROS accumulation through suppression of MAPK/HO-1 signalling pathway
There is growing evidence that ROS is important for the induction of apoptosis in cancer cells (Raj et al, 2011). To determine whether the ability of GA to enhance the apoptosis-inducing effect of CDDP was mediated by ROS, we used flow cytometry to measure the intracellular ROS in A549 and NCI-H460 cells. The cells were treated with GA, CDDP, or the combination of both for 24 h, and DCF fluorescence was recorded as a measure of intracellular ROS levels. As shown in Figure 5A, the levels of intracellular ROS were significantly (P<0.05) higher in cells that had been treated with GA, CDDP, and the sequential combination of CDDP and GA than that in control cells. Furthermore, treatment with the combination of CDDP and GA led to a further increase in the accumulation of ROS compared with treatment with GA or CDDP alone (P<0.05). To confirm that ROS generation is responsible for enhancing CDDP- plus GA-induced apoptosis, the cells were pretreated with NAC (10 mM), an inhibitor of ROS production (Raj et al, 2011; Zhang et al, 2011), for 4 h, and then incubated with the sequential combination of CDDP and GA. Our data indicate that pretreatment with NAC not only reduced the level of intracellular ROS (Figure 5A) but also decreased the amount of apoptosis caused by the combined treatment of CDDP and GA in A549 and NCI-H460 cells (Figure 5B). Taken together, these results demonstrate that ROS mediates the ability of GA to enhance CDDP-induced apoptosis.

The effects of GA, CDDP, and the sequential combination on ROS accumulation and the expression of HO-1, ERK1/2, phosphorylated (p)-ERK1/2, p38, phosphorylated-p38, JNK, and phosphorylated-JNK. A549 and NCI-H460 cells were treated with dimethyl sulphoxide (DMSO) control, CDDP, GA, or sequential combination (CDDP→GA) as mentioned above. (A) The ROS release was assessed by flow cytometry analysis. The cells were labelled with 5 μmol l−1 DCFH-DA for 1 h before the treatment with drugs. (B) The apoptosis was measured by flow cytometry analysis. Sub-G1 is considered as apoptosis. (C) The expression of HO-1 was detected by western blot. (D) The mRNA expression of HO-1 was detected by quantitative polymerase chain reaction (PCR). (E) The expression of ERK1/2, phosphorylated-ERK1/2, p38, phosphorylated-p38, JNK, and phosphorylated-JNK was detected by western blot. Protein expression levels (relative to β-actin) were determined. Columns, means for three replicate determinations; bars, s.d. P<0.05 (*the sequential combination compared with the untreated controls, GA, or CDDP single treatments; #GA or CDDP single treatments compared with the untreated controls).
As it is reported that HO-1 inhibits the production of ROS and contributes to CDDP resistance (Abraham et al, 2007; Kim et al, 2008), we assessed whether HO-1 is involved in the enhanced accumulation of ROS induced by the combined treatment of CDDP and GA in A549 and NCI-H460 cells. As shown in Figures 5C and D, treatment of two cell lines with CDDP alone led to the increasing expression of HO-1 in mRNA and protein levels, whereas the treatment with GA alone and the sequential combination of CDDP and GA resulted in the decreasing expression of HO-1. These data indicate that the inhibition of HO-1 is involved in the enhanced accumulation of ROS caused by the combined treatment of CDDP and GA. Furthermore, we also detected the proteins of mitogen-activated protein kinase (MAPK) pathway, which is demonstrated to the upstream signalling pathway of HO-1 (Kim et al, 2008), in A549 and NCI-H460 cells. Our results indicated that CDDP alone prompted the phosphorylation of ERK, p38, and JNK, but sequential combination treatment inhibited the CDDP-induced phosphorylation of ERK, p38, and JNK (See Figure 5E). This pattern is similar to HO-1, suggesting that GA might inhibit CDDP-induced upregulation of HO-1 through MAPK pathway.
GA sensitised NSCLC cells to CDDP through inhibition of NF-κB pathway
It is well known that Bcl-2, survivin and X-IAP are the target genes of NF-κB signalling. The above-mentioned data prompt us to speculate whether NF-κB is responsible for the synergistic effect. Therefore, we examined the expression and subcellular location of NF-κB proteins, including p65 and p50, using western blot method. As shown in Figure 6A in A549 and NCI-H460 cells, CDDP could increase the nuclear expression of p65 and p50. However, addition of GA after CDDP treatment could reverse this increase of p65 and p50 in the nucleus. These data suggest that GA might sensitise A549 and NCI-H460 cells to CDDP through inhibition of CDDP-induced NF-κB activation.

The effects of GA, CDDP, and their combination on nuclear factor- κ B (NF- κ B) activation. A549 and NCI-H460 cells were treated with dimethyl sulphoxide (DMSO) control, CDDP, GA, or sequential combination as mentioned above. After 24/48 h of these treatments, cells were harvested, and nuclear, cytosolic, and whole-cell extracts were prepared as described in Materials and Methods. (A) The extracts were analysed for p65 and p50 levels by western blot analysis using specific antibodies as described in Material and Methods. Membrane was stripped and reprobed for β-actin and histone 3 as cytoplasmic and nuclear controls, respectively. (B) EMSA was performed in nuclear extract for NF-κB of A549 and NCI-H460 cells as detailed in Material and Methods. Competition (Compe) reaction was performed using unlabelled consensus NF-κB oligos. The cells treated with TNF-α (10 ng ml−1, 1 h) were applied as positive control. (C)The mRNAs were analysed for IL-8 and survivin levels by quantitative PCR using specific primers as described in Materials and Methods. GAPDH was used as control. These experiments were repeated in duplicate or triplicate.
Moreover, we also detected the effect of CDDP and/or GA on the NF-κB-binding ability in A549 and NCI-H460 cells. Our results showed that CDDP-induced NF-κB activation in A549 and NCI-H460 cells, whereas concomitant treatment or GA followed by CDDP treatment did not show any considerable change in NF-κB DNA-binding activity after 24 h of treatment (Figure 6B). However, in sequence CDDP followed by GA treatment, GA completely inhibited CDDP-induced NF-κB activation. Specificity of the band was confirmed by competition with unlabelled wild-type oligos (see Figure 6B). Furthermore, we also measured the expression of the classical target genes of NF-κB, including IL-8 and servivin, in mRNA level. The data indicated that CDDP alone treatment induced the increased expression of IL-8 and servivin, but the combination treatment could suppress the increase of IL-8 and servivin (see Figure 6C). Taken together, these results demonstrate that GA could inhibit CDDP-induced activation of NF-κB pathway in NSCLC cells.
Inhibition of MAPK/ERK, MAPK/JNK, and NF-κB pathways contributed to the sequential combination of CDDP- and GA-induced ROS release and apoptosis
To further explore the role of MAPK and NF-κB pathways in the combination of CDDP- and GA-induced ROS release and apoptosis, we applied the specific inhibitors for MAPK and NF-κB pathways and analysed the ROS release and PARP cleavage. As shown in Figure 7A, single treatment with SB203580 (a specific inhibitor of MAPK/p38), SP600125 (a specific inhibitor of MAPK/JNK), PD98095 (a specific inhibitor of MAPK/ERK) and PDTC (a specific inhibitor of NF-κB) induced a slight or moderate accumulation of ROS in A549 and NCI-H460 cells. Interestingly, in contrast to SB203580, pretreatment with SP600125, PD98095, or PDTC for 1 h resulted in a significant increase in the accumulation of ROS induced by CDDP combined with GA (see Figure 7A), indicating that the MAPK/JNK, MAPK/ERK, and NF-κB pathways, but not MAPK/p38 pathway, has a role in regulating the sequential combination-induced ROS release of A549 and NCI-H460 cells. Importantly, similar pattern was found in PARP cleavage experiment. Pretreatment with SP600125, PD98095 or PDTC led to a marked enhancement of PARP cleavage caused by CDDP combined with GA (shown in Figure 7B). Taken together, our results confirm that inhibition of MAPK/ERK, MAPK/JNK, and NF-κB pathways contributes to the combination of CDDP- and GA-induced ROS release and apoptosis in NSCLC cells.

The effects of the specific inhibitors of MAPK and NF- κ B pathways on the combination of CDDP and GA-induced ROS release and apoptosis. A549 and NCI-H460 cells were treated with dimethyl sulphoxide (DMSO) control or single specific inhibitors (SB203580, 12.5 μ M; SP600125, 12.5 μ M; PD98095, 12.5 μ M; PDTC, 10 μ M) or triple combination (pretreatment with specific inhibitor for 1 h followed by CDDP and GA sequence treatment). (A) The ROS release was assessed by flow cytometry analysis. (B) The apoptosis was assessed by western blot. Cleavage (Clv) of poly (ADP-ribose) polymerase (PARP) is considered as apoptosis. Columns, means for three replicate determinations; bars, s.d. P<0.05 (*the sequential combination compared with triple treatments).
The sequential combination of CDDP and GA inhibited tumour growth in vivo, induced apoptosis, inactivated NF-κB, and downregulated HO-1 in tumour tissues
To further investigate whether GA synergises CDDP against tumour growth in vivo, A549 tumours were implanted in SCID mice. Our data showed that CDDP combined with GA or used individually cause inhibitory effects on the growth of A549 tumours (Figure 8A). The tumour volumes were all decreased when compared with those of control group, whereas the degree of tumour reduction differed; the volume of combination group was significantly smaller than that of GA group or CDDP group (P<0.05). In the control group, the mean tumour volume was 1613 mm3 at the end of the experiment. When mice were treated with CDDP combined with GA, the tumour inhibition rate was 69.3%, whereas those of mice treated with CDDP and GA alone were 57.2% and 29.0%, respectively. These results demonstrate that the antitumour effect of GA combined with CDDP is superior to that of the drugs used individually. Furthermore, we found that the body weights of GA group showed no significant loss as compared with mice in the control group, whereas CDDP treatment could significantly decrease the body weights after treatment for 9 days. This result is consistent with the previous reports (Yamori et al, 1997; Villena-Heinsen et al, 1998). The combination-treated mice showed a slight but not significant body weight loss as compared with that in the control group (see Figure 8B). Thus, the combination of CDDP and GA produced much more potent tumour growth inhibitory effects, without increasing the toxicities to the animals.

The antitumour effect of CDDP and GA on A549 human xenograft models. (A) The mice transplanted with A549 human xenografts were randomly divided into four groups and given injection of CDDP (4 mg kg−1 per week, intravenously), GA (2 mg kg−1 per 2 days, intravenously), and the sequential combination or vehicle for a period of 3 weeks. The tumour volumes are expressed as mean±s.d. (n=5–6 per group). (B) The average body weight of each group is expressed as mean±s.d. (n=5–6 per group). (C) Representative photographs of TUNEL staining of tumour tissues in different groups. (D) Expression of p50, p65, survivin, and HO-1 extracted from the tumour tissues of drug-administered mice of four groups were detected. Extracted tumour proteins (50 μg per lane, mixed from two tumours in the same group) were subjected to SDS–PAGE and blotted with antibody. P<0.05 (*GA, CDDP single administration or the combination administration compared with the control; #the combination administration compared with CDDP single administration).
To confirm the underlying mechanisms of the synergistic effect in vitro, we next assessed the effect of monotreatment or combined treatment of CDDP and GA on the apoptosis and the expression levels of NF-κB-related proteins and HO-1 in tumour tissues from drug-administered mice. As shown in Figure 8C, the combination therapy could induce an enhanced apoptosis in the tumour tissues from the mice. Moreover, the nuclear protein expression of p65 and p50 was remarkably decreased with increasing p65 and p50 expression in the cytoplasm in tumour tissues of combination-treated mice (shown in Figure 8D), consistent with the results in vitro. The inhibition of NF-κB was further confirmed by the decreasing expression of survivin, its target gene, in tumour tissues of combination-treated mice. In addition, compared with CDDP-treated mice, the expression of HO-1 was decreased in tumour tissues of combination-treated mice (see Figure 8D and Supplementary Figure 3). These results provide further verification to the involvement of NF-κB and HO-1 in the synergistic tumour growth inhibitory effect of GA and CDDP in vivo.
Discussion
In this study, our results illustrated for the first time that GA is capable of sensitising NSCLC cells to CDDP-mediated apoptosis in a sequence-dependent manner, which is evidenced by the activation of caspase-3, caspase-8, caspase-9, and PARP. Furthermore, GA acts synergistically with CDDP to reduce the tumour burden in the A549 xenograft model. Importantly, we revealed the underlying mechanisms of apoptosis induction and CDDP sensitisation by GA, which involves the inhibition of NF-κB and MAPK/HO-1 signalling pathways accompanied with ROS accumulation.
Cisplatin is one of the most widely used drugs for treating NSCLC (Rinaldi et al, 2006). It works by binding to DNA leading to different types of DNA lesions and subsequently inducing apoptosis (Galluzzi et al, 2012). The efficacy of CDDP in NSCLC treatment is limited owing to resistance, either intrinsic or acquired following CDDP chemotherapy (Koberle et al, 2010; Galluzzi et al, 2012). The mechanism of CDDP resistance has been studied in several types of NSCLC cell lines and appears to be multifactorial, including alteration of drug transport, enhancement of drug detoxification system, change of DNA damage tolerance mechanisms, and disruption of apoptotic cell death pathways (Koberle et al, 2010). Among them, apoptosis inhibition is considered as the most important factor of CDDP resistance (Koberle et al, 2010; Galluzzi et al, 2012). Gambogic acid was reported to induce apoptosis of cancer cells through both death receptor apoptotic pathway and mitochondrial apoptotic pathway (Li et al, 2012; Zhao et al, 2013). Thus, it is possible that GA could sensitise CDDP in NSCLC by promoting apoptosis. Our results showed that CDDP and GA were observed in vitro to act synergistically to induce apoptosis in human NSCLC cells. Western blot analyses showed that CDDP combined with GA synergistically increased the execution of caspase-dependent apoptosis. Importantly, we also found that, compared with monotreatment, enhanced apoptosis was triggered by the combination therapy in the tissues from the drug-administered mice. The above data demonstrate that GA might become an ideal agent for overcoming CDDP resistance.
It is becoming increasingly evident that several antiapoptotic pathways including those regulated by NF-κB are critical in mediating chemotherapeutic resistance in NSCLC cells (Li et al, 2005; Baby et al, 2007; Chen et al, 2011). Nuclear factor-κB is a transcription factor that regulates the expression of numerous genes that are critical for survival. It is activated by diverse stimuli that include proinflammatory cytokines, cellular stress, and growth factors, as well as chemotherapeutic agents (Chen et al, 2011). These stimuli can prompt the active NF-κB (p65–p50 subunits) translocate into the nucleus where it binds with NF-κB-specific DNA-binding sites to activate transcriptionally the expression of several survival genes. The activation of NF-κB leads to increased expression of the IAPs, namely X-IAP and survivin (Chen et al, 2011), and the Bcl-2 family of proteins, including Bcl-2 and Bcl-xL (Chen et al, 2011), which counteract the action of proapoptotic proteins including Bax. Constitutive activation of NF-κB has been detected in NSCLC and has been implicated in imparting resistance to CDDP (Baby et al, 2007; Chen et al, 2011). Therefore, inhibition of NF-κB signalling may serve as a critical target for enhancing the efficacy of CDDP in NSCLC. Recently, GA has been identified as a potential anticancer agent that inhibits NF-κB signalling (Pandey et al, 2007; Nie et al, 2009; Wang et al, 2012b). Our data indicated that the sequential treatment with GA almost completely inhibited the NF-κB activation induced by CDDP. Furthermore, three of the NF-κB apoptosis-related targets, X-IAP, survivin and Bcl-2, were downregulated by GA and the combination between CDDP and GA. Therefore, the inhibition of NF-κB by GA could be responsible for the synergistic apoptosis-inducing effect of GA and CDDP in NSCLC cells and tumour xenograft.
Heme oxygenase-1 is considered as an another critical protein in mediating chemotherapeutic resistance in NSCLC cells (Raval and Lee, 2010). Heme oxygenase-1 catalyses the first and rate-limiting step in the degradation of heme to biliverdin, carbon monoxide (CO), and ferrous iron. Biliverdin is further converted to bilirubin by biliverdin reductase. Biliverdin and bilirubin are the most potent endogenous ROS scavengers (Abraham et al, 2007). By reducing ROS production, HO-1 plays critical roles in antioxidant defence and antiapoptotic (Raval and Lee, 2010). Recent study demonstrates that HO-1 protein is upregulated in NSCLC and its overexpression contributes to resistance to CDDP (Kim et al, 2008; Degese et al, 2012; Jeon et al, 2012). Our results indicate that treatment with CDDP alone led to the increase of HO-1 in vitro and in vivo, whereas the combination of CDDP and GA resulted in the reduction of HO-1. Consistent with the downregulation of HO-1 expression in NSCLC cells, the combination of CDDP and GA also resulted in the accumulation of ROS that is responsible for enhancing CDDP/GA-induced apoptosis. These data suggest that GA sensitises CDDP to NSCLC cells through inhibiting CDDP-induced HO-1 upregulation and then afterwards prompting the accumulation of ROS.
Mitogen-activated protein kinase, including ERK, p38, and JNK, are components of signalling cascades that respond to extracellular stimuli by targeting transcription factors, resulting in the regulation of gene expression. It is well known that many oxidative and chemical stressors could lead to the activation of MAPK signalling pathways and subsequent activation of the Nrf2 transcriptional factor that binds to the antioxidant response element in the HO-1 promoter (Gong and Cederbaum, 2004; Kocanova et al, 2007), causing an increase in its transcription. In our experiment, we demonstrated that treatment with CDDP alone resulted in an increase in the phosphorylation of MAPK proteins, which also modulate the Nrf2-HO-1 pathway, whereas sequential combination with CDDP and GA led to a decreased expression of HO-1 and increased generation of ROS simultaneously. These results demonstrated that GA might inhibit CDDP-induced upregulation of HO-1 through MAPK/ERK and MAPK/JNK pathways. In addition, Liu et al (2004) reported that the elevated HO-1 was associated with increased activity of the nuclear NF-κB and the inhibition of NF-κB led to the block of its induction. In the present study, we demonstrated that the expression of HO-1 was associated with the alteration in the NF-κB activity in NSCLC cells, suggesting that NF-κB signalling pathway is also involved in the regulation of HO-1 by the combination of CDDP and GA.
The identification of optimal dosing regimens and schedules is necessary for evaluating cancer therapeutics in clinical study, especially when therapies are combined (Hirai et al, 2010). Preclinical results are valuable to guide the design of clinical study protocols. In this study, the sequential treatment with CDDP followed by GA caused a synergistic cell growth inhibition. On the other hand, the reverse sequence and the simultaneous drug treatment resulted in antagonistic or additive efficacy. One possible reason for this could be that only the treatment with CDDP followed by GA caused an obviously inhibition of NF-κB and subsequently induction of apoptosis, but not in GA followed by CDDP sequence treatment or simultaneous treatment (Figure 6B).
In summary, we verified for the first time that GA sensitises NSCLC cells to CDDP in vitro and in vivo. More importantly, our findings revealed that GA could inhibit the activation of NF-κB and MAPK/HO-1 pathways induced by CDDP, and subsequently enhance ROS generation, thus potentiating the execution of apoptosis triggered by CDDP. Taken together, these data suggest that the combination of CDDP and GA is an attractive novel potential therapy for the treatment of NSCLC. Further studies are warranted to demonstrate the efficacy of the CDDP–GA combination in the clinical setting.
Change history
21 January 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abraham NG, Asija A, Drummond G, Peterson S (2007) Heme oxygenase -1 gene therapy: recent advances and therapeutic applications. Curr Gene Ther 7 (2): 89–108.
Baby J, Pickering BF, Vashisht Gopal YN, Van Dyke MW (2007) Constitutive and inducible nuclear factor-kappaB in immortalized normal human bronchial epithelial and non-small cell lung cancer cell lines. Cancer Lett 255 (1): 85–94.
Chen S, Flower A, Ritchie A, Liu J, Molassiotis A, Yu H, Lewith G (2010) Oral Chinese herbal medicine (CHM) as an adjuvant treatment during chemotherapy for non-small cell lung cancer: a systematic review. Lung Cancer 68 (2): 137–145.
Chen W, Li Z, Bai L, Lin Y (2011) NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front Biosci 16: 1172–1185.
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55.
Crawford ED, Wells JA (2011) Caspase substrates and cellular remodeling. Annu Rev Biochem 80: 1055–1087.
D'Addario G, Pintilie M, Leighl NB, Feld R, Cerny T, Shepherd FA (2005) Platinum-based versus non-platinum-based chemotherapy in advanced non-small-cell lung cancer: a meta-analysis of the published literature. J Clin Oncol 23 (13): 2926–2936.
Degese MS, Mendizabal JE, Gandini NA, Gutkind JS, Molinolo A, Hewitt SM, Curino AC, Coso OA, Facchinetti MM (2012) Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer 77 (1): 168–175.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127 (12): 2893–2917.
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G (2012) Molecular mechanisms of cisplatin resistance. Oncogene 31 (15): 1869–1883.
Gong P, Hu B, Cederbaum AI (2004) Diallyl sulfide induces heme oxygenase-1 through MAPK pathway. Archiv Biochem Biophys 432: 252–260.
Guo QL, You QD, Wu ZQ, Yuan ST, Zhao L (2004) General gambogic acids inhibited growth of human hepatoma SMMC-7721 cells in vitro and in nude mice. Acta Pharmacol Sin 25 (6): 769–774.
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9 (7): 1956–1967.
Jeon WK, Hong HY, Seo WC, Lim KH, Lee HY, Kim WJ, Song SY, Kim BC (2012) Smad7 sensitizes A549 lung cancer cells to cisplatin-induced apoptosis through heme oxygenase-1 inhibition. Biochem Biophys Res Commun 420 (2): 288–292.
Jeong SJ, Koh W, Kim B, Kim SH (2011) Are there new therapeutic options for treating lung cancer based on herbal medicines and their metabolites? J Ethnopharmacol 138 (3): 652–661.
Kim HR, Kim S, Kim EJ, Park JH, Yang SH, Jeong ET, Park C, Youn MJ, So HS, Park R (2008) Suppression of Nrf2-driven heme oxygenase-1 enhances the chemosensitivity of lung cancer A549 cells toward cisplatin. Lung Cancer 60 (1): 47–56.
Koberle B, Tomicic MT, Usanova S, Kaina B (2010) Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta 1806 (2): 172–182.
Kocanova SBE, Matroule JY, Piette J, Golab J, de Witte P (2007) Induction of heme-oxygenase 1 requires the p38MAPK and PI3K pathways and suppresses apoptotic cell death following hypericin-mediated photodynamic therapy. Apoptosis 12: 731–741.
Li C, Qi Q, Lu N, Dai Q, Li F, Wang X, You Q, Guo Q (2012) Gambogic acid promotes apoptosis and resistance to metastatic potential in MDA-MB-231 human breast carcinoma cells. Biochem Cell Biol 90 (6): 718–730.
Li D, Williams JI, Pietras RJ (2002) Squalamine and cisplatin block angiogenesis and growth of human ovarian cancer cells with or without HER-2gene overexpression. Oncogene 21: 2805–2814.
Li Y, Ahmed F, Ali S, Philip PA, Kucuk O, Sarkar FH (2005) Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res 65 (15): 6934–6942.
Liu ZM, Chen GG, Ng EK, Leung WK, Sung JJ, Chung SC (2004) Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene 23 (2): 503–513.
Nie F, Zhang X, Qi Q, Yang L, Yang Y, Liu W, Lu N, Wu Z, You Q, Guo Q (2009) Reactive oxygen species accumulation contributes to gambogic acid-induced apoptosis in human hepatoma SMMC-7721 cells. Toxicology 260 (1–3): 60–67.
Olaku O, White JD (2011) Herbal therapy use by cancer patients: a literature review on case reports. Eur J Cancer 47 (4): 508–514.
Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB (2007) Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood 110 (10): 3517–3525.
Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, Lee SW (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475 (7355): 231–234.
Raval CM, Lee PJ (2010) Heme oxygenase-1 in lung disease. Curr Drug Targets 11 (12): 1532–1540.
Rinaldi M, Cauchi C, Gridelli C (2006) First line chemotherapy in advanced or metastatic NSCLC. Ann Oncol 17 (Suppl 5): v64–v67.
Villena-Heinsen C, Friedrich M, Ertan AK, Farnhammer C, Schmidt W (1998) Human ovarian cancer xenografts in nude mice: chemotherapy trials with paclitaxel, cisplatin, vinorelbine and titanocene dichloride. Anticancer Drugs 9 (6): 557–563.
Wang C, Zhang H, Chen Y, Shi F, Chen B (2012a) Gambogic acid-loaded magnetic Fe(3)O(4) nanoparticles inhibit Panc-1 pancreatic cancer cell proliferation and migration by inactivating transcription factor ETS1. Int J Nanomed 7: 781–787.
Wang X, Deng R, Lu Y, Xu Q, Yan M, Ye D, Chen W (2012b) Gambogic acid as a non-competitive inhibitor of ATP-binding cassette transporter B1 reverses the multidrug resistance of human epithelial cancers by promoting ATP-binding cassette transporter B1 protein degradation. Basic Clin Pharmacol Toxicol 112 (1): 25–33.
Wu ZQ, Guo QL, You QD, Zhao L, Gu HY (2004) Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol Pharm Bull 27 (11): 1769–1774.
Yamori T, Sato S, Chikazawa H, Kadota T (1997) Anti-tumor efficacy of paclitaxel against human lung cancer xenografts. Jpn J Cancer Res 88 (12): 1205–1210.
Yu J, Guo QL, You QD, Zhao L, Gu HY, Yang Y, Zhang HW, Tan Z, Wang X (2007) Gambogic acid-induced G2/M phase cell-cycle arrest via disturbing CDK7-mediated phosphorylation of CDC2/p34 in human gastric carcinoma BGC-823 cells. Carcinogenesis 28 (3): 632–638.
Yang Y, Yang L, You QD, Nie FF, Gu HY, Zhao L (2007) Differential apoptotic induction of gambogic acid, a novel anticancer natural product, on hepatoma cells and normal hepatocytes. Cancer Lett 256: 259–266.
Zhang H, Yang JY, Zhou F, Wang LH, Zhang W, Sha S, Wu CF (2011) Seed oil of Brucea javanica induces apoptotic death of acute myeloid leukemia cells via both the death receptors and the mitochondrial-related pathways. Evid Based Complement Alternat Med 2011: 965016.
Zhao W, You CC, Zhuang JP, Zu JN, Chi ZY, Xu GP, Yan JL (2013) Viability inhibition effect of gambogic acid combined with cisplatin on osteosarcoma cells via mitochondria-independent apoptotic pathway. Mol Cell Biochem 382 (1-2): 243–252.
Zheng L, Yang W, Zhang C, Ding WJ, Zhu H, Lin NM, Wu HH, He QJ, Yang B (2011) GDC-0941 sensitizes breast cancer to ABT-737 in vitro and in vivo through promoting the degradation of Mcl-1. Cancer Lett 309 (1): 27–36.
Zhu X, Zhang H, Lin Y, Chen P, Min J, Wang Z, Xiao W, Chen B (2009) Mechanisms of gambogic acid-induced apoptosis in non-small cell lung cancer cells in relation to transferrin receptors. J Chemother 21 (6): 666–672.
Acknowledgements
We gratefully acknowledge the financial support from Scientific Research Fund of Liaoning Provincial Education Department (L2011179), Jiangsu Planned Projects for Postdoctoral Research Funds (0902115C), and China Postdoctoral Science Foundation funded project (20100481213, 2012T50474).
Author information
Authors and Affiliations
Corresponding authors
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wang, LH., Li, Y., Yang, SN. et al. Gambogic acid synergistically potentiates cisplatin-induced apoptosis in non-small-cell lung cancer through suppressing NF-κB and MAPK/HO-1 signalling. Br J Cancer 110, 341–352 (2014). https://doi.org/10.1038/bjc.2013.752
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.752
Keywords
This article is cited by
-
Synergic effect of anticancer peptide CIGB-552 and Cisplatin in lung cancer models
Molecular Biology Reports (2022)
-
Gambogic acid triggers vacuolization-associated cell death in cancer cells via disruption of thiol proteostasis
Cell Death & Disease (2019)
-
Gambogenic acid inhibits fibroblast growth factor receptor signaling pathway in erlotinib-resistant non-small-cell lung cancer and suppresses patient-derived xenograft growth
Cell Death & Disease (2018)
-
Targeting EHMT2 reverses EGFR-TKI resistance in NSCLC by epigenetically regulating the PTEN/AKT signaling pathway
Cell Death & Disease (2018)
-
Melatonin increases the effect of 5-fluorouracil-based chemotherapy in human colorectal adenocarcinoma cells in vitro
Molecular and Cellular Biochemistry (2018)
