
Abstract
Background:
Aurora kinases are key regulators of cell cycle and represent new promising therapeutic targets in several human tumours.
Methods:
Biological relevance of Aurora kinase-A and -B was assessed on osteosarcoma clinical samples and by silencing these genes with specific siRNA in three human osteosarcoma cell lines. In vitro efficacy of two Aurora kinases-targeting drugs (VX-680 and ZM447439) was evaluated on a panel of four drug-sensitive and six drug-resistant human osteosarcoma cell lines.
Results:
Human osteosarcoma cell lines proved to be highly sensitive to both drugs. A decreased drug sensitivity was observed in doxorubicin-resistant cell lines, most probably related to ABCB1/MDR1 overexpression. Both drugs variably induced hyperploidy and apoptosis in the majority of cell lines. VX-680 also reduced in vitro cell motility and soft-agar cloning efficiency. Drug association experiments showed that VX-680 positively interacts with all conventional drugs used in osteosarcoma chemotherapy, overcoming the cross-resistance observed in the single-drug treatments.
Conclusion:
Aurora kinase-A and -B represent new candidate therapeutic targets for osteosarcoma. In vitro analysis of the Aurora kinases inhibitors VX-680 and ZM447439 indicated in VX-680 a new promising drug of potential clinical usefulness in association with conventional osteosarcoma chemotherapeutic agents.
Similar content being viewed by others


Main
Osteosarcoma (OS), the most common bone tumour, mainly occurs during childhood and adolescence, and is characterised by high proliferation activity and bone turnover. It is a highly invasive and metastatic rare tumour, which can frequently develop chemoresistance. Clinical outcome of localised high-grade OS has significantly improved with the introduction of neoadjuvant chemotherapy, which is based on a combination of four different drugs: doxorubicin, methotrexate, cisplatin, and ifosfamide (Chou and Gorlick, 2006; Hattinger et al, 2010). Despite this, ∼35–40% of patients poorly respond to the chemotherapeutic treatment and relapse (Chou and Gorlick, 2006; Hattinger et al, 2010). Thus, novel effective treatment regimens are urgently needed to further improve the clinical outcome of OS.
The process of mitosis has been largely exploited for treatment of cancer, and antimitotic agents form an important group of chemotherapeutics for both adult and childhood malignancies. In the past few years the number of cell cycle-interacting drugs has significantly increased, thanks to the development of several new agents, which successfully went through the preclinical and clinical validation (Deep and Agarwal, 2008; Lens and Voest, 2010). However, before suggesting the clinical use of these anticancer drugs, it is absolutely necessary verifying whether their targets are present and biologically relevant for each specific tumour.
The Aurora kinase family members (which include Aurora kinase-A, -B, and -C) have been described as key regulators of mitosis and cell cycle, and, because of this, several drugs have been developed for inhibiting their functions (Agnese et al, 2007; Deep and Agarwal, 2008; Mountzios et al, 2008; Lens and Voest, 2010). All of them are serine/threonine protein kinases that share a high degree of sequence homology in their catalytic domains but have different subcellular distributions and functions (Ducat and Zheng, 2004; Fu et al, 2007; Lens and Voest, 2010). Aurora kinase-A (AURK-A) localises to the centrosomes during DNA duplication in the S phase and migrates to spindle poles during mitosis. Such distribution is consistent with its primary roles in centrosome regulation and mitotic spindle formation (Ducat and Zheng, 2004; Fu et al, 2007; Lens and Voest, 2010). Aurora kinase-B (AURK-B) undergoes dynamic distribution during mitosis, by localising first to the inner centromeres during prophase and metaphase and then to the spindle midzone during anaphase and through cytokinesis. Aurora kinase-B migrates as part of the chromosomal passenger complex, whose function is to ensure accurate chromosome segregation and cell body division (Ducat and Zheng, 2004; Fu et al, 2007; Lens and Voest, 2010). Aurora kinase-C (AURK-C) does not seem to have a role in mitosis in the majority of normal cells. Its expression is essentially restricted to testes, with a specific role in the regulation of chromosome segregation during spermatogenesis (Lens and Voest, 2010).
The Aurora kinases have also been indicated to act as oncogenic drivers, as their abnormal expression or activation could result in aberrant mitosis leading to the development of different cancers (Fu et al, 2007; Mountzios et al, 2008; Lens and Voest, 2010; Cheung et al, 2011). Among them, AURK-A has been the most consistently indicated factor involved in the tumorigenesis of several human cancers, being highly expressed in breast, colon, bladder, ovarian, and pancreatic cancers (Agnese et al, 2007; Fu et al, 2007; Mountzios et al, 2008; Lens and Voest, 2010). It is also interesting to note that AURK-A is located on chromosome 20q13.2, which is often amplified in many malignant diseases, including OS (Bayani et al, 2003). In several different cancer cells overexpression of AURK-B has been shown and indications have been provided for its possible involvement, together with other factors, in malignant transformation of normal cells (Mountzios et al, 2008; Lens and Voest, 2010). Interestingly, AURK-B is located on chromosome 17p13.1, which has also been found to be amplified in OS (Bayani et al, 2003).
This body of evidence has indicated the Aurora kinases as promising candidates to develop targeted drugs. To date, a growing number of inhibitors of Aurora kinases have been described (Deep and Agarwal, 2008; Mountzios et al, 2008; Katayama and Sen, 2010). Among these, VX-680 (also referred as MK-0457 or Tozasertib) and ZM447439 have been studied in several human tumours and also entered Phase I/II clinical trials (Agnese et al, 2007; Deep and Agarwal, 2008; Mountzios et al, 2008). VX-680 and ZM447439 target the ATP-binding site of Aurora kinases and inhibit all three family members; however, their efficacy appears to be mainly associated with the inhibition of AURK-A or AURK-B (Ditchfield et al, 2003; Harrington et al, 2004; Agnese et al, 2007; Deep and Agarwal, 2008; Mountzios et al, 2008; Katayama and Sen, 2010; Li et al, 2010). Clinical experience with VX-680 and ZM447439 showed that these drugs have no or very limited effects on non-cycling human normal cells (probably because their low Aurora kinases expression) and that their combination with conventional chemotherapeutic agents may improve the clinical efficacy of standard treatment regimens without increasing collateral toxicity (Agnese et al, 2007; Deep and Agarwal, 2008; Mountzios et al, 2008).
Very scarce data have been reported concerning the efficacy of Aurora kinase inhibitors in OS cells (Ditchfield et al, 2003; Gizatullin et al, 2006) and no information is available about the impact of AURK-A and AURK-B in OS pathogenesis and clinical outcome. In the present study, we have assessed the biologic relevance of both kinases in OS clinical samples and have evaluated the in vitro efficacy of VX-680 and ZM447439 on a panel of drug-sensitive and drug-resistant human OS cell lines, either as single agents or in combination with the conventional chemotherapeutic drugs used in OS chemotherapy.
Materials and Methods
Drugs
Cisplatin (CDDP), doxorubicin (DX), and methotrexate (MTX) were purchased, respectively, from Teva Italia (Milan, Italy), Wyeth Lederle (Latina, Italy), and Sandoz (Varese, Italy). CBA Research, Inc. (Lexington, KY, USA) provided CBT-1. Stock solutions of CDDP (500 μg ml−1) and MTX (25 mg ml−1) were stored at 4 °C. Stock solution aliquots of DX (2 mg ml−1) and CBT-1 (0.01 M) were stored at −20 °C. VX-680 and ZM447439 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and were dissolved in dimethylsulphoxide (DMSO) at 20 and 1 mM concentrations, respectively. Stock solution aliquots were stored at −20 °C. For all drugs, working concentrations were prepared by diluting stock solutions in culture medium just before use.
Cell cultures
The cell line panel used for this study included U-2OS and Saos-2 human OS cell lines, which were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA), and the IOR/OS9 and IOR/OS18 human OS cell lines that were established from clinical specimens obtained from OS patients at the Experimental Oncology Laboratory of the Orthopaedic Rizzoli Institute (Benini et al, 1999; Ottaviano et al, 2010). The human OS cell lines resistant to DX (U-2OS/DX580 and Saos-2/DX580), MTX (U-2OS/MTX300; Saos-2/MTX300), and CDDP (U-2OS/CDDP4 μg and Saos-2/CDDP6 μg) were established by exposing the drug-sensitive U-2OS and Saos-2 cell lines to stepwise increasing concentrations of each drug, as previously described (Serra et al, 1993, 2004; Pasello et al, 2008). DNA fingerprint analysis of 14 polymorphic short-term tandem repeat (STR) sequences was performed for all cell lines as previously described (Ottaviano et al, 2010). For U-2OS and Saos-2 cell lines, STR profile completely agreed with that provided by the ATCC (Ottaviano et al, 2010). STR profile of drug-resistant variants was identical to those of their corresponding parental cell lines. All cell lines were cultured in Iscove's modified Dulbecco's medium (IMDM), supplemented with penicillin (100 U ml−1)/streptomycin (100 μg ml−1) (Invitrogen Ltd, Paisley, UK) and 10% heat-inactivated fetal bovine serum (FBS; Biowhittaker Europe, Cambrex-Verviers, Belgium). Normal human osteoblast cultures (purchased from Lonza Walkersville Inc., Walkersville, MD, USA) were cultured in OGM-osteoblast medium (Lonza Walkersville Inc) for 2–3 weeks before being processed for RNA extraction. All cell cultures were maintained at 37 °C in a humidified 5% CO2 atmosphere.
Clinical samples
The series of clinical samples included 21 conventional OS (primary, high-grade tumours located in the extremities of patients younger than 40 years). All samples were obtained from surgical biopsies obtained at diagnosis before any treatment. All patients were treated with neoadjuvant chemotherapy protocols based on DX, MTX, CDDP, and ifosfamide and continuously followed up. Median follow-up was 91 months (range 63–218 months). Written informed consent for using their biologic material for research purposes was obtained from each patient entering the study, which was approved by the Istituto Ortopedico Rizzoli Ethical Committee.
RNA isolation
For cell lines, RNA was extracted from cell pellets by using the TRIzol reagent (Invitrogen) according to standard procedures. After isolation, RNA concentration and quality were evaluated by spectrophotometry using the NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, USA) and by electrophoresis on a 1.5% agarose gel. For clinical samples, total RNA was extracted from snap-frozen specimens by using the TRIzol method. Before extraction, all samples were histologically examined for tissue quality in order to isolate RNA only from representative specimens. Fragmentation of cRNA, hybridisation to hg-u133 plus 2.0 microarrays, and scanning were performed as described by (Molenaar et al, 2012). After normalisation of expression data with the MAS5.0 algorithm, gene expression profiles were analysed by using the freely available R2 web application (http://r2.amc.nl).
Aurora kinases and ABCB1 silencing by siRNA
Cells were seeded in 6-well or in 96-well plates in drug-free IMDM 10% FBS without antibiotics. After 24 h, the medium was replaced with FBS- and antibiotic-free IMDM supplemented with Lipofectamine 2000 (Invitrogen) and 25 nM Dharmacon ON-TARGET plus SMARTpool siRNA (Thermo Fisher Scientific, Waltham, MA, USA) specific for AURK-A (L-003545-01, Human AURKA), AURK-B (L-003326-00, Human AURKB), or scrambled SMARTpool siRNA (D-001810-02-20). The 10 nM IDT Screening DsiRNA Duplex pool (Integrated DNA Technologies, Coralville, IA, USA) was used to silence ABCB1 (HSC.RNAI.N000927.12.1_10 nm;.12.2_10 nm;.12.3_10 nm) and scrambled oligonuleotide was IDT Negative Control (DS NC1—trifecta; Integrated DNA Technologies).
Controls were cultured in the same media without siRNA. After 5 h, transfection medium was replaced with IMDM 10% FBS without antibiotics and cells were maintained in culture for 24, 48, and 72 h for U-2OS cell line and for 48, 72, and 96 h for Saos-2 and IOR/OS18 cell lines. After evaluation of cell morphology, cells were harvested and counted with the Trypan blue dye exclusion method (to assess the extent of growth inhibition) and processed for cell cycle analysis, RNA and protein extraction. In ABCB1-silencing experiments, the medium of Saos-2/DX580 and U-2OS/DX580 was replaced with IMDM 10% FBS without antibiotics added or not with VX-680 or ZM447439 at different dosages. After 96 h, the harvested cells were processed for RNA extraction and protein evaluation by immunoflorescence. Growth inhibition was assesed by means of the 3-(4,5-dimethylthiazol-2-yl)-2,5-dephenyltetrazolium bromide (MTT) assay.
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
For single gene expression analyses, 500 ng of total RNA were reverse transcribed using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol. cDNAs were aliquoted and stored at −20 °C until use. To quantify the fold-change of each kinase gene expression between the silenced samples and controls, the TaqMan Gene Expression Assays AURKA (Assay ID: Hs01582072_m1) and AURKB (Assay ID: Hs00177782_m1) were used on the ABI PRISM 7900 SDS instrument (Applied Biosystems). The reference gene chosen was GAPDH (Assay ID: Hs99999905_m1). To detect the ABCB1-mRNA IDT PrimeTime Std qPCR Assay 6-FAM/ZEN/IBFQ (Assay ID: Hs.PT.56. 39182776, Integrated DNA Technologies) was used. All amplification reactions for each sample were performed in triplicate using the TaqMan Universal PCR Master Mix (Applied Biosystems) in a total volume of 25 μl and the standard programme of 40 cycles (95 °C for 5 s, 60 °C for 20 s, 72 °C for 30 s) after an initial incubation step at 95 °C for 10 s. Each gene and its variant were analysed in at least two independent experiments. The delta (Δ) CT values of GAPDH were used to normalise all other genes tested from the same cDNA aliquot. The fold-differences in gene expression of silenced samples compared with non-treated cells (controls) were calculated as 2-ΔΔCT, using controls as calibrators, where ΔCT=CT of target genes–CT of reference gene, and ΔΔCT=ΔCT of variant–ΔCT of calibrator.
Protein analysis by western blot
Cells were scraped, washed twice in cooled PBS, and then lysed in RIPA buffer. The cell suspensions were shaken in ice for 30 min. The lysates were centrifuged at 13 000 r.p.m. for 15 min at 4 °C. Equal amounts of cell lysates were resolved by SDS–PAGE and then transferred to a PVDF membrane (Immobilon P-Transfer membrane, Millipore, Billerica, MA, USA). The membranes were incubated in blocking solution consisting of 5% powered milk in TBST at room temperature for 1 h and then with the anti-Aurora-A kinase mouse monoclonal antibody (AbD serotec, Oxford, UK) or the anti-Aurora-B (N-term) rabbit polyclonal antibody (Epitomics, CA, USA). Purified mouse monoclonal antibodies specific for human Caspase 2 (Cell Signaling Technology, Danvers, MA, USA), Caspase 3 (Cell Signaling Technology), and poly ADP-ribose polymerase-1 (PARP-1; BD Biosciences, Franklin Lakes, NJ, USA) were used to assess apoptosis markers. To verify the protein loading of each sample, the same membranes were immunostained with an anti-beta-actin monoclonal antibody (Chemicon International, Temecula, CA, USA). Protein bands were visualised by using an enhanced chemiluminescence detection system (Liteablot Plus, Euroclone, Milan, Italy) and autoradiography. For each band, the amount of protein was determined by densitometric analysis and normalised to that of beta-actin.
Protein analysis by immunofluorescence
For immunofluorescence staining, cells were harvested, washed once in PBS, twice with a Hepes 0.01 M solution (Sigma-Aldrich Co., St. Louis, MO, USA) in HBSS (Sigma-Aldrich Co.), and then fixed with PFA (4% in PBS) for 5 min. After a wash in Hepes 0.01 M, cells were permeabilised with a Saponin 0.1% solution (Sigma-Aldrich Co.) in Hepes 0.01 M for 5 min and incubated with the primary antibody anti-P-glycoprotein mouse mAb MRK16 (Kamiya, Seattle, WA, USA) diluted 1 : 100 in Saponin 0.1% for 40 min. Cell were washed once with Saponin 0.1% and then treated with the secondary antibody anti-mouse FITC antibody (1 : 100 in Saponin 0.1%, Sigma-Aldrich Co.) for 40 min followed by washing twice with Saponin 0.1% and once with Hepes 0.01 M. For the negative control, the primary antibody was replaced by Saponin 0.1%. Samples were analysed by flow cytometry (FACSCalibur, Becton Dickinson, San Jose, CA, USA).
In vitro drug sensitivities of human OS cell lines
Drug sensitivity of each cell line was calculated from the drug dose–response curves obtained by using a standard MTT assay kit (Roche Diagnostics GmbH, Mannheim, Germany) and expressed as IC50 (drug concentration resulting in 50% inhibition of cell growth after 96 h of in vitro treatment). In DX-resistant variants, in vitro efficacies of VX-680 and ZM447439 were also assessed when ABCB1 was inhibited with CBT-1 or siRNA. Cells were treated for 96 h with different dosages of each Aurora kinase inhibitor ±0.5–1 μ M CBT-1, 10 nM ABCB1 siRNA, or scrambled oligonucleotide. The extent of growth inhibition in the presence of CBT-1, siRNA ABCB1, or scrambled was compared with that obtained with VX-680 or ZM447439 alone.
Evaluation of drug–drug interactions
To evaluate the in vitro interactions between VX-680 or ZM447439 and conventional chemotherapeutic drugs, human OS cell lines were incubated with different regimens of two-drugs combinations. Cell lines were treated with combinations of increasing and decreasing drug dosages defined on the basis of the ratio of the specific IC50 values obtained in each cell line. Drug interaction effects were evaluated after 96 h of combined treatment. To define the type of interaction in terms of synergism, antagonism, or additivity, the combination index (CI) of each two-drugs combination was calculated with the equation of Chou–Talalay by using the CalcuSyn software (Biosoft, Stapleford, UK), as previously described (Pasello et al, 2008). By following the range of CI values indicated in the CalcuSyn software manual, we classified the drug–drug interaction as synergistic when CI was lower than 0.90, as additive when 0.90⩽CI⩽1.10, or as antagonistic when CI was higher than 1.10.
Cell cycle analysis
Assessment of VX-680 and ZM447439 effects on cell cycle was performed by seeding drug-sensitive and drug-resistant cell lines in IMDM 10% FBS. After 24 h, the medium was changed with IMDM 10% FBS without (control) or with VX-680 or ZM447439 at dosages corresponding to the IC50 or 2-fold-IC50 concentrations of each cell line. Additional controls were also included in these experiments by culturing cells in IMDM 10% FBS supplemented with DMSO concentrations corresponding to those of drug-treated samples. After 48 and 72 h of drug exposure, cells were incubated with 10 mM bromodeoxyuridine (Sigma-Aldrich Co.) for 1 h in a humidified 5% CO2 atmosphere at 37 °C, harvested, and fixed in 70% ethanol for 30 min. After DNA denaturation with 2 N HCl, cells were processed for indirect immunofluorescence with the B44 anti-bromodeoxyuridine mouse monoclonal antibody (Becton Dickinson) diluted 1 : 8, followed by an anti-mouse FITC antibody (Sigma-Aldrich Co.) diluted 1 : 200. For the simultaneous determination of DNA content, cell suspensions were stained with 20 mg ml−1 propidium iodide (Sigma-Aldrich Co.). All samples were analysed by flow cytometry (FACSCalibur, Becton Dickinson).
Soft-agar assay
Anchorage-independent growth was determined in 0.33% agarose (SeaPlaque, FMC BioProducts, Rockland, ME, USA) with a 0.5% agarose underlay. The following number of cells were seeded in 60 mm dishes: 10 000 for U-2OS/DX580, Saos-2/MTX300, and Saos-2/CDDP6 μg; 25 000 for IOR/OS18; 33 000 for U-2OS, Saos-2, Saos-2/DX580, and U-2OS/CDDP4 μg; 50 000 for U-2OS/MTX300; and 100 000 for IOR/OS9. Cells were plated in a semisolid medium (IMDM+10% FBS containing 0.33% agarose) containing the IC50 concentration of VX-680 or ZM447439, or with IMDM 10% FBS supplemented with DMSO concentrations corresponding to those of treated cells (non-treated DMSO controls). Dishes were incubated at 37 °C in a humidified atmosphere containing 5% CO2, and colonies with more than 50 cells were counted after 15 days.
Motility assay
Motility assays were performed by seeding 300 000 cells per well in 6-well plates. After 24 h, ∼0.6 mm-wide scrapes were made using a 1-ml micropipette tip. Culture media was changed to remove cellular debris and cells were incubated with VX-680 or ZM447439 dosages corresponding to the IC50 concentration of each cell line. Untreated controls were cultured in IMDM 10% FBS without or with DMSO concentrations corresponding to those of drug-treated samples. Scrapes were photographed at the beginning of treatments and at different time points with the Olympus IX51 inverted microscope (X40 magnification; Olympus Optical, Tokyo, Japan). Scrape widths were measured with the ImageJ software (National Institutes of Health, Bethesda, MD, USA). Motility capability was estimated by substracting the width of scrape at each time point from that of the initial scrape, obtaining an estimation of the scrape-healing capability.
Statistics
Differences among means were analysed with the Student’s t-test. Kaplan–Meier and log-rank methods were used to draw and evaluate the significance of survival curves. Differences in motility capability were tested by two-way ANOVA for repeated measures (analyses were performed with the GraphPad Prism v5 software; GraphPad Software Inc., La Jolla, CA, USA).
Results
Analysis of kinase expression level on human OS clinical samples and cell lines
By mining the data obtained from gene expression profiles with the R2 bioinformatic tool (http://r2.amc.nl), AURK-A and AURK-B proved to be significantly overexpressed in human OS clinical cases in comparison with human normal muscles and other normal tissues (Figure 1A). By using as cut-off the median expression level of each kinase to distinguish the low-expressing- (expression level lower than the cut-off value) from high-expressing patients (expression level equal or higher than the cut-off value), a trend towards a worse outcome in terms of event-free (AURK-A) and overall survival (AURK-A and AURK-B) probability for high expressors was found (Figures 1B and C). These findings, together with the observation that three out of the four outlier patients (identified by the dots located above the grey area of the box plots in Figure 1A) with the highest expression level of AURK-A and/or AURK-B relapsed, suggest that both kinases are involved in OS pathogenesis and impact on clinical outcome.

Gene expression profiling results obtained by using the hg-u133 plus 2.0 Affymetrix microarrays and analysed with the R2 web application (http://r2.amc.nl). (A) Box plot graphical presentation of the AURK-A and AURK-B expression levels in 21 human osteosarcoma clinical samples in comparison with human normal muscles and other normal tissues. Numbers inside parenthesis indicate the number of analysed samples. Asterisks indicate statistically significant differences by ANOVA test (P<0.01). (B) Event-free and (C) overall survival probability of the 21 high-grade OS patients stratified according to the median expression level of AURK-A or AURK-B. The group of high expressors (HIGH) included patients showing kinase expression levels equal or higher than the cut-off value.
Knockdown of AURK-A and AURK-B
In order to verify whether AURK-A and AURK-B are functionally and biologically relevant for OS cell growth, RNA interference approaches were used to knock down each kinase in three human OS cell lines (U-2OS, Saos-2, and IOR/OS18) showing different expression of each kinase (Supplementary Table S1). Small interfering RNA transfection experiments proved to significantly silence both kinases in all three cell lines. A significant knockdown of both AURK-A and AURK-B mRNA and proteins was obtained in U-2OS and Saos-2 cell lines, whereas in IOR/OS18 cell line the knockdown was remarkably more relevant at the protein than at the mRNA level (Table 1). The silencing specificity was confirmed by the fact that transfection with scrambled siRNA (SCR) did not decrease the expression of AURK-A and AURK-B compared with non-treated control (NT-CTR) cells (Table 1). The effects of AURK-A and AURK-B silencing were also estimated through the evaluation of cell morphology and growth inhibition. Cell lines did not show any evident cell distress or significant morphological changes as consequence of AURK-A or AURK-B silencing (data not shown). However, kinases silencing produced an evident growth inhibition in all three cell lines but with different efficiencies (Figure 2). Silencing of AURK-A and AURK-B was associated with a marked growth inhibition in U-2OS and Saos-2 cell lines, which was already evident 48 h after treatment with siRNA and was also maintained for longer times. The IOR/OS18 cell line showed a similar marked growth inhibition after AURK-B silencing, which was, however, less relevant after AURK-A knockdown (probably because of its inherent higher levels of AURK-A compared with U-2OS and Saos-2, as shown in Supplementary Table S1).

Growth inhibition (GI) induced by AURK-A (left) or AURK-B (right) silencing in the three human OS cell lines U-2OS, Saos-2, IOR/OS18. CTR, control, non-treated cells; SCR, cells transfected with scrambled siRNA.
In vitro sensitivity to Aurora kinases-targeting drugs
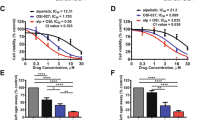
The in vitro sensitivity to the Aurora kinases-targeting drugs VX-680 and ZM447439 was estimated on the basis of drug–dosage response curves after 96 h of treatment, from which the IC50 value (drug concentration that induced 50% growth inhibition compared with untreated controls) for each cell line was determined (Table 2). All drug-sensitive cell lines proved to be highly sensitive to both drugs, showing IC50 values in a submicromolar/low micromolar range (from 0.2 to 5.5 μ M). In the group of drug-sensitive cell lines, VX-680 proved to be slightly more effective than ZM447439, showing a mean IC50 value of 0.7±0.3 compared with 3.0±1.8 μ M. Aurora kinases-targeting drugs efficacy was also assessed on six different drug-resistant variants. No evidence of cross-resistance was found with MTX, showing MTX-resistant variants IC50 values similar to those of drug-sensitive cell lines. All DX-resistant variants showed a decrease of in vitro sensitivity to both VX-680 and ZM447439, indicating the possible presence of cross-resistance mechanisms. As the most relevant mechanism of drug resistance developed by these cell lines is the overexpression of ABCB1 (Serra et al, 1993; Hattinger et al, 2009), additional experiments were performed in order to verify whether this membrane transporter may also mediate resistance against these two drugs. The in vitro efficacy of VX-680 and ZM447439 was assessed either after knocking down ABCB1 gene expression by specific siRNA (Supplementary Table S2) or by inhibiting the ABCB1 activity with CBT-1 in both DX-resistant cell lines. As shown in Figure 3, both ABCB1-mRNA silencing and CBT-1 increased the efficacy of both anti-kinase drugs, indicating that ABCB1 overexpression negatively interferes with VX-680 and ZM447439 activity. VX-680 also showed a decreased activity in CDDP-resistant variants; however, it is not clear at this moment which mechanism may be responsible for this partial cross-resistance. The reduced activity of VX-680 and ZM447439 in DX- and CDDP-resistant variants was not due to an increase of their intracellular content of AURK-A or AURK-B, which was similar to those of parental cell lines (data not shown).

Reversal of cross-resistance against Aurora kinase-targeting drugs in U-2OS/DX580 and Saos-2/DX580 cell lines. Columns represent the fold increase in VX-680 and ZM447439 sensitivity of cells transfected with scrambled oligonucleotide (SCR), with ABCB1 silencing siRNAs or in the presence of two different doses of the ABCB1 inhibitor CBT-1 (CBT-1 0.5 μ M and CBT-1 1 μ M). The fold increase in drug sensitivity was calculated by dividing the VX-680 and ZM447439 IC50 values of controls (cell treated with VX-680 or ZM447439 only) by the IC50 values of silenced- or CBT-1-treated cells.
Cell cycle perturbations and apoptosis induced by Aurora kinases-targeting drugs
Cell cycle perturbations induced by Aurora kinases-targeting drugs were assessed by flow cytometry after treatment of both drug-sensitive and drug-resistant cell lines with their corresponding IC50 dose of VX-680 or ZM447439. Incubation of drug-sensitive cell lines with VX-680 induced a clear hyperploidisation (most probably due to endoreduplication), which emerged after 48 h of treatment (Figure 4A). This effect was maintained even after 72 h of drug exposure (data not shown). In the Saos-2 cell line, the drug-induced hyperploidisation was still present but less evident compared with the other three cell lines (Figure 4A). The drug-induced hyperploidisation did not allow a reliable estimation of cell distribution among the different cell cycle phases, as the peak of hyperploid cells overlapped that of the G2/M phase of the original cell population. A similar phenomenon occured with ZM447439, which produced the same evidence in all drug-sensitive cell lines with the only exception of IOR/OS9 (Figure 4A).

Cell cycle perturbations induced by treatment with the IC50 doses of VX-680 and ZM447439 in drug-sensitive ( A ) and drug-resistant ( B ) human OS cell lines. Graphs were derived from cell cycle analyses performed after 48 h of drug treatment. The intensity of propidium iodide fluorescence (representative for the DNA content) is plotted on x axis. The intensity of the incorporated BrdU fluorescence (representative for the DNA synthesis) is plotted on y axis. The different cell populations were identified according to the scheme shown in the plot on the top left of panel A. CTR, control non-treated cells cultured in drug-free and DMSO-free medium; DMSO, control non-treated cells cultured in the presence of DMSO concentrations corresponding to those of drug-treated samples; VX-680 or ZM447439, cells treated with IC50 dosage of each Aurora kinase inhibitor drug.
In drug-resistant variants of U-2OS and Saos-2 cell lines, similar but less homogeneous results were obtained (Figure 4B). After 48 h of treatment, VX-680 induced hyperploidisation in all Saos-2 drug-resistant variants and in U-2OS/DX580, whereas ZM447439 induced hyperploidisation only in Saos-2/MTX300, Saos-2/CDDP6μg, and U-2OS/CDDP4μg. As described for drug-sensitive cell lines, the same picture without any significant change was observed after 72 h of drug exposure (data not shown). To verify whether treatment with Aurora kinases-targeting drugs also induced apoptosis, cleaveage, or degradation of Caspase 2, Caspase 3, and PARP-1 was assessed by western blot. As shown in Figure 5 and in Supplementary Figures S1A and B, both drugs proved to variably induce apoptosis in all cell lines, with a non-homogeneous involvement of the apoptotic markers here considered. VX-680 induced apoptosis mainly through cleavage or degradation of Caspase 2 (with the only exception of Saos-2/DX580 and Saos-2/MTX300) and/or PARP-1 (U-2OS, Saos-2, IOR/OS18, U-2OS/CDDP4 μg, and Saos-2/MTX300), whereas Caspase 3 was cleaved only in Saos-2, Saos-2/DX580, and Saos-2/MTX300 cell lines. Apoptosis induction after treatment with ZM447439 was revealed in a lower number of cell lines compared with VX-680. In particular, ZM447439 mainly induced apoptosis through cleavage or degradation of Caspase 2 (U-2OS, Saos-2, IOR/OS18, U-2OS/MTX300, and U-2OS/CDDP4 μg) and/or PARP-1 (U-2OS, Saos-2, IOR/OS18, and Saos-2/MTX300), whereas Caspase 3 was cleaved only in the Saos-2 cell line.

Western blot analyses for Caspase 2, Caspase 3, and PARP-1 in the four drug-sensitive human osteosarcoma cell lines after treatment with VX-680 and ZM447439. Lines with U-2OS+CDDP5 μg and Saos-2+CDDP10 μg were used as positive controls to detect Caspase 3 and PARP-1 cleavage induced by the treatment for 48 h with CDDP 5 μg ml−1 or CDDP 10 μg ml−1, respectively. CTR, control non-treated cells cultured in drug-free and DMSO-free medium; DMSO, control non-treated cells cultured in the presence of DMSO concentrations corresponding to those of drug-treated samples; VX-680 or ZM447439, cells treated with IC50 or two-fold IC50 (VX-680 × 2 or ZM447439 × 2) dosage of each Aurora kinase inhibitor drug.
Effect of VX-680 and ZM447439 on soft-agar colony formation and cell motility
The in vitro activity of Aurora kinase inhibitors was also assessed in terms of inhibition of soft-agar colony formation in the presence of VX-680 or ZM447439 IC50 concentrations. Both drugs strongly and significantly inhibited the colony formation ability in all cell lines (Supplementary Table S3). The impact of VX-680 and ZM447439 on OS cells motility was assessed by using a wound-healing migration assay (Supplementary Figures S2A and B). VX-680 decreased migration capability in several OS cell lines, reaching a statistically significant inhibition in IOR/OS9, U-2OS/CDDP4 μg, Saos-2/MTX300, and Saos-2/CDDP6 μg. A clear trend towards an inhibition of migration capability was also observed for U-2OS/DX580, Saos-2, and Saos-2/DX580, whereas IOR/OS18, U-2OS, and U-2OS/MTX300 did not show any response to VX-680 treatment (Supplementary Figure S2A). Unlike VX-680, ZM447439 did not show any significant inhibition of migration capability, with the only exception of Saos-2/CDDP6 μg (Supplementary Figure S2B).
Interaction of Aurora kinase inhibitors with conventional chemotherapeutics
Drug–drug interaction analyses considered the combination of VX-680 and ZM447439 with the conventional drugs that are most commonly used in OS chemotherapy in order to verify whether this association may improve the efficacy of conventional drugs and/or overcome the cross-resistance effects observed in single-drug treatment experiments. As shown in Table 3, the combination of VX-680 with DX, MTX, or CDDP proved to be additive or synergistic in most cell lines, showing an antagonistic interaction with only MTX in U-2OS and with DX in Saos-2 drug-sensitive cell lines. It is worthwhile to note that the association of VX-680 with all three conventional chemotherapeutic agents produced an invariably positive interaction in all drug-resistant variants, including those that appeared to be partially cross-resistant to this kinase inhibitor. On the contrary, association of ZM447439 with conventional chemoterapeutics mostly produced antagonistic interactions.
Discussion
Protein kinases have been proved to be involved in many key tumour cell processes including signalling, survival, proliferation, genomic instability, angiogenesis, migration, and metastatic dissemination (Lens and Voest, 2010; Barouch-Bentov and Sauer, 2011). For these reasons, pharmacological modulation of kinase functions is an attractive therapeutic option, also because protein kinases can be targeted by several small molecules and drugs. ‘Druggable’ kinases have therefore become one of the largest drug target families, with several approved inhibitors, many of which have already entered clinical trials and many more are under preclinical development (Cohen, 2002; Mountzios et al, 2008; Katayama and Sen, 2010; Lens and Voest, 2010; Barouch-Bentov and Sauer, 2011).
Although OS has not been extensively studied as other types of cancer, some findings have suggested that kinases may be involved in the pathogenesis and biological aggressiveness of this tumour (Messerschmitt et al, 2008; Yamaguchi et al, 2009). On the other hand, the value of tyrosine kinase inhibitors as adjuvant to standard OS treatment has yet to be further extensively explored. In this study, we have analysed the impact on OS cell biology of two of the most relevant cell cycle-related, druggable kinases: AURK-A and AURK-B. In particular, we assessed their biologic relevance for OS cell growth and malignancy, as well as the in vitro efficacy of the two Aurora kinase inhibitors VX-680 and ZM447439.
The Aurora family of protein kinases has emerged as a crucial factor of mitosis and cell proliferation in several different human tumours and has been found to be frequently overexpressed in human cancers, thus becoming an interesting candidate therapeutic target (Agnese et al, 2007; Mountzios et al, 2008; Saeki et al, 2009; Lens and Voest, 2010). However, like for other kinases, the biological impact of Aurora kinases needs to be evaluated and carefully preclinically validated inside each tumour type before being indicated as a possible target for new therapeutic approaches.
Our analysis of AURK-A and AURK-B gene expression in a series of 21 human high-grade OS clinical samples showed that both kinases were expressed at higher levels in tumour cells compared with several human normal tissues and that the high expression of these molecules was associated with a trend towards a worse clinical outcome. These findings indicated that AURK-A and AURK-B are probably involved in the OS pathogenesis and may also have an impact on the clinical outcome.
On the basis of this evidence, additional studies based on siRNA silencing were performed to determine the dependency of OS cells on AURK-A and AURK-B expression. Silencing experiments were carried out on three different human OS cell lines with AURK-A and AURK-B expression levels comparable to those observed in OS clinical samples. The three cell lines used for silencing experiments were selected on the basis of their relative kinase expression, in order to study cells with different expression levels of AURK-A or AURK-B. Knockdown of both kinases produced a relevant cell growth inhibition, indicating that the expression of AURK-A and AURK-B is important for proliferation of human OS cells.
Findings obtained on clinical samples and derived from silencing experiments provided thus the rationale for extending the study to test the Aurora kinase inhibitors efficacy. Among the several inhibitors that have been described so far, we decided to consider VX-680 and ZM447439 as they have already been preclinically and, successfully, clinically validated in Phase I/II studies and proved to be active in different human tumour cells (Ditchfield et al, 2003; Harrington et al, 2004; Agnese et al, 2007; Mountzios et al, 2008; Katayama and Sen, 2010; Lens and Voest, 2010; Li et al, 2010). The in vitro activity of both drugs was assessed on a panel of four drug-sensitive (three of which were the same that were used for the silencing experiments) and six drug-resistant human OS cell lines. VX-680 and ZM447439 proved to be highly active in all drug-sensitive and MTX-resistant cell lines, which presented IC50 values in the submicromolar/low micromolar range. Both drugs presented an evident decrease of activity in DX-resistant variants and, at a lower extent, in CDDP-resistant variants, suggesting the possible existence of cross-resistance mechanisms.
As the most relevant mechanism of resistance developed by U-2OS- and Saos-2 DX-resistant variants is the overexpression of the ABCB1 (MDR1) membrane transporter (Serra et al, 1993; Hattinger et al, 2009), we explored whether this membrane efflux pump may be responsible for the observed reduced efficacy of VX-680 and ZM447439. The reversion of cross-resistance obtained by silencing the ABCB1 gene in DX-resistant variants or by treating these cells with the ABCB1 inhibitor CBT-1 (Robey et al, 2008; Kelly et al, 2012) confirmed the involvement of this transporter in the decrease of VX-680 and ZM447439 activity. These data are in agreement with evidence reported in other experimental models, in which it has been demonstrated that VX-680 can be recognised as substrate by ABCB1 (Cheung et al, 2011). The same evidence emerged from the combined treatment with ZM447439 and CBT-1, providing the new indication that this drug may also be a substrate of ABCB1.
At this moment, we do not have any explanation for the decreased cell growth inhibition activity of VX-680 that we observed in CDDP-resistant variants. Ongoing studies should clarify whether this drug can be detoxified by glutathione enzymes, which are strictly connected with the CDDP resistance of these cell lines (Pasello et al, 2008).
Drug combination experiments, in which Aurora kinase inhibitors were associated with conventional chemotherapeutics in both parental, drug-sensitive U-2OS and Saos-2 cell lines and their drug-resistant variants, showed that the association of VX-680 with either DX, MTX, or CDDP produced almost invariably additive or synergistic interactions, even overcoming the DX- and CDDP cross-resistance described above. The positive interaction of VX-680 with conventional drugs may therefore support a further preclinical and clinical evaluation of the activity of this drug, which proved to be able to potentiate the efficacy of conventional cytotoxic agents on OS cells. It is worthwhile noting that the synergistic effect of the combined treatment of VX-680 with CDDP that we observed in CDDP-resistant variants may suggest the involvement of Aurora kinases in CDDP resistance of OS cells, as described for other tumours (Shahzad et al, 2009). Unlike VX-680, drug association experiments with ZM447439 produced mainly negative interactions for presently unknown reasons.
The preclinical evaluation of VX-680 and ZM447439 activity on OS cells was implemented with the analysis of their effects on cell cycle, apoptosis, cellular motility, and growth in anchorage-independent conditions.
Treatment with VX-680 and ZM447439 produced hyperploidisation in the majority of OS cell lines, which was most probably also responsible for the observed slower cell proliferation. These results can be explained by the effects that Aurora kinases inhibition may produce on cell cycle progression and, consequently, on cellular DNA ploidy (Katayama and Sen, 2010; Nair et al, 2012). In different experimental models, it has been reported that inhibition of AURK-A and/or AURK-B can produce different cell cycle and ploidy alterations, which can be also related to the status of P53: cell lines with wild-type or active P53 usually undergo apoptosis or growth arrest in a pseudo-G1 phase, whereas cell lines with mutated or inactive P53 tend to progress along cell cycle phases and become polyploid (Ditchfield et al, 2003; Agnese et al, 2007; Nair et al, 2012). However, in our study, we did not find this strict association with P53 status. In fact, although our panel included two cell lines (U-2OS and IOR/OS9) carrying wild-type P53 and two cell lines (Saos-2 and IOR/OS18) with partially deleted, non-functional P53 (Ottaviano et al, 2010), we did not observe significant differences between them in terms of tendency to become hyper(poly)ploid or to undergo apoptosis after treatment with VX-680 or ZM447439. This evidence indicated that, in our experimental models, the hyperploidy induced by these two drugs is independent from the P53 status, as described for other human tumour cells (Nair et al, 2012). On the other hand, in our cell lines, the rapid hyperploidisation induced by VX-680 and ZM447439 treatment appeared to be associated with an impaired capacity to proliferate, which may drive cells toward apoptosis, as indicated by our results and as described in other systems (Ganem and Pellman, 2007; Katayama and Sen, 2010; Nair et al, 2012). In our cell lines, we obtained evidence that VX-680 and ZM447439 trigger cells to apoptosis by activating Caspase 2 and inducing mitotic catastrophe, as described for other agents that, like these Aurora kinase inhibitors, disrupt the mitotic spindle (Hassa, 2009).
As in other experimental models inhibition of AURK-A and AURK-B have been shown to decrease the anchorage-independent growth in soft-agar and the cellular migration and motility (Guan et al, 2007; Arlot-Bonnemains et al, 2008; Wan et al, 2009; Ulisse et al, 2010; Baldini et al, 2011), we evaluated whether also in OS cells VX-680 and ZM447439 may influence these features, which are related to the in vivo tumorigenic and metastatic potential. Both drugs significantly reduced the anchorage-independent growth in soft-agar in all OS cell lines. VX-680 also decreased migration capability in the majority of cell lines, whereas ZM447439 decreased migration capability of the Saos-2/CDDP6 μg cell line only. These findings further support the potential therapeutic value of these two Aurora kinases inhibitors, and in particular of VX-680, which appeared to decrease the malignant phenotype of human OS cells.
Conclusions
In conclusion, targeted therapies against specific kinases have been indicated to have an acceptable toxicity profile and to produce fewer short-term and long-term collateral toxicity compared with conventional chemotherapy (Cohen, 2002; Agnese et al, 2007; Mountzios et al, 2008; Katayama and Sen, 2010). Therefore, targeting these molecules offers the hope for not only an increased treatment efficacy and cure probability but also for an improved quality of life, an aspect that has a very high relevance for tumours that, like OS, mainly affect young people with long life expectancies. However, in this study we provided evidence indicating that the use of these new agents in multimodal treatment of OS must be considered in association with conventional chemotherapy and based on a preliminary knowledge of the presence of possible mechanisms of resistance (most of all the ABCB1 expression level) in each patient, which may limit the efficacy of this approach. The fact that the association of VX-680 with standard chemotherapeutics proved to enhance the activity of each drug and to overcome the partial cross-resistance observed in the single-drug administration’s further supports the potential clinical value of Aurora kinase inhibition approaches. Moreover, as ABCB1 overexpression has been demonstrated to negatively impact on the clinical outcome of 40–45% high-grade OS patients (Hattinger et al, 2010) and as, in this study, this same mechanism has been shown to partially decrease the efficacy of Aurora kinase-targeting drugs, the findings here on the possibility of reverting the ABCB1-associated mutlidrug resistance with CBT-1 may open up additional new therapeutic perspectives also for ABCB1-overexpressing OS patients.
Change history
12 November 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Agnese V, Bazan V, Fiorentino FP, Fanale D, Badalamenti G, Colucci G, Adamo V, Santini D, Russo A (2007) The role of aurora-A inhibitors in cancer therapy. Ann Oncol 18 (Suppl 6): vi47–vi52.
Arlot-Bonnemains Y, Baldini E, Martin B, Delcros JG, Toller M, Curcio F, Ambesi-Impiombato FS, D'Armiento M, Ulisse S (2008) Effects of the aurora kinase inhibitor VX-680 on anaplastic thyroid cancer-derived cell lines. Endocr Relat Cancer 15 (2): 559–568.
Baldini E, Arlot-Bonnemains Y, Sorrenti S, Mian C, Pelizzo MR, De Antoni E, Palermo S, Morrone S, Barollo S, Nesca A, Moretti CG, D'Armiento M, Ulisse S (2011) Aurora kinases are expressed in medullary thyroid carcinoma (MTC) and their inhibition suppresses in vitro growth and tumorigenicity of the MTC derived cell line TT. BMC Cancer 11: 411–423.
Barouch-Bentov R, Sauer K (2011) Mechanisms of drug resistance in kinases. Expert Opin Investig Drugs 20 (2): 153–208.
Bayani J, Zielenska M, Pandita A, Al-Romaih K, Karaskova J, Harrison K, Bridge JA, Sorensen P, Thorner P, Squire JA (2003) Spectral karyotyping identifies recurrent complex rearrangements of chromosomes 8 17 and 20 in osteosarcomas. Genes Chromosomes Cancer 36 (1): 7–16.
Benini S, Baldini N, Manara MC, Chano T, Serra M, Rizzi S, Lollini PL, Picci P, Scotlandi K (1999) Redundancy of autocrine loops in human osteosarcoma cells. Int J Cancer 80 (4): 581–588.
Cheung CH, Lin WH, Hsu JT, Hour TC, Yeh TK, Ko S, Lien TW, Coumar MS, Liu JF, Lai WY, Shiao HY, Lee TR, Hsieh HP, Chang JY (2011) BPR1K653 a novel aurora kinase inhibitor exhibits potent anti-proliferative activity in MDR1 (P-gp170)-mediated multidrug-resistant cancer cells. PLoS One 6 (8): e23485.
Chou AJ, Gorlick R (2006) Chemotherapy resistance in osteosarcoma: current challenges and future directions. Expert Rev Anticancer Ther 6 (7): 1075–1085.
Cohen P (2002) Protein kinases—the major drug targets of the twentyfirst century? Nat Rev Drug Discov 1 (4): 309–315.
Deep G, Agarwal R (2008) New combination therapies with cell cycle agents. Curr Opin Investig Drugs 9 (6): 591–604.
Ditchfield C, Johnson VL, Tighe A, Ellston R, Haworth C, Johnson T, Mortlock A, Keen N, Taylor SS (2003) Aurora B couples chromosome alignment with anaphase by targeting BubR1 Mad2 and Cenp-E to kinetochores. J Cell Biol 161 (2): 267–280.
Ducat D, Zheng Y (2004) Aurora kinases in spindle assembly and chromosome segregation. Exp Cell Res 301 (1): 60–67.
Fu J, Bian M, Jiang Q, Zhang C (2007) Roles of aurora kinases in mitosis and tumorigenesis. Mol Cancer Res 5 (1): 1–10.
Ganem NJ, Pellman D (2007) Limiting the proliferation of polyploid cells. Cell 131 (3): 437–440.
Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI (2006) The Aurora Kinase Inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res 66 (15): 7668–7677.
Guan Z, Wang XR, Zhu XF, Huang XF, Xu J, Wang LH, Wan XB, Long ZJ, Liu JN, Feng GK, Huang W, Zeng YX, Chen FJ, Liu Q (2007) Aurora-A a negative prognostic marker increases migration and decreases radiosensitivity in cancer cells. Cancer Res 67 (21): 10436–10444.
Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C, Hercend T, Diu-Hercend A, Su M, Golec JM, Miller KM (2004) VX-680 a potent and selective small-molecule inhibitor of the aurora kinases suppresses tumor growth in vivo. Nat Med 10 (3): 262–267.
Hassa PO (2009) The molecular "Jekyll and Hyde" duality of PARP1 in cell death and cell survival. Front Biosci 14: 72–111.
Hattinger CM, Pasello M, Ferrari S, Picci P, Serra M (2010) Emerging drugs for high-grade osteosarcoma. Expert Opin Emerg Drugs 15 (4): 615–634.
Hattinger CM, Stoico G, Michelacci F, Pasello M, Scionti I, Remondini D, Castellani GC, Fanelli M, Scotlandi K, Picci P, Serra M (2009) Mechanisms of gene amplification and evidence of coamplification in drug-resistant human osteosarcoma cell lines. Genes Chromosomes Cancer 48 (4): 289–309.
Katayama H, Sen S (2010) Aurora kinase inhibitors as anticancer molecules. Biochim Biophys Acta 1799 (10–12): 829–839.
Kelly RJ, Robey RW, Chen CC, Draper D, Luchenko V, Barnett D, Oldham RK, Caluag Z, Frye AR, Steinberg SM, Fojo T, Bates SEA (2012) Pharmacodynamic study of the P-glycoprotein antagonist CBT-1® in combination with paclitaxel in solid tumors. Oncologist 17 (4): 512–523.
Lens SM, Voest EE, Medema RH (2010) Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer 10 (12): 825–841.
Li Y, Zhang ZF, Chen J, Huang D, Ding Y, Tan MH, Qian CN, Resau JH, Kim H, Teh BT (2010) VX680/MK-0457 a potent and selective aurora kinase inhibitor targets both tumor and endothelial cells in clear cell renal cell carcinoma. Am J Transl Res 2 (3): 296–308.
Messerschmitt PJ, Rettew AN, Brookover RE, Garcia RM, Getty PJ, Greenfield EM (2008) Specific tyrosine kinase inhibitors regulate human osteosarcoma cells in vitro. Clin Orthop Relat Res 466 (9): 2168–2175.
Molenaar JJ, Koster J, Ebus ME, van Sluis P, Westerhout EM, de Preter K, Gisselsson D, Øra I, Speleman F, Caron HN, Versteeg R (2012) Copy number defects of G1-cell cycle genes in neuroblastoma are frequent and correlate with high expression of E2F target genes and a poor prognosis. Genes Chromosomes Cancer 51 (1): 10–19.
Mountzios G, Terpos E, Dimopoulos MA (2008) Aurora kinases as targets for cancer therapy. Cancer Treat Rev 34 (2): 175–182.
Nair JS, Ho AL, Schwartz GK (2012) The induction of polyploidy or apoptosis by the aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle 11 (4): 807–817.
Ottaviano L, Schaefer KL, Gajewski M, Huckenbeck W, Baldus S, Rogel U, Mackintosh C, de Alava E, Myklebost O, Kresse SH, Meza-Zepeda LA, Serra M, Cleton-Jansen AM, Hogendoorn PC, Buerger H, Aigner T, Gabbert HE, Poremba C (2010) Molecular characterization of commonly used cell lines for bone tumor research A trans-European EuroBoNet effort. Genes Chromosomes Cancer 49 (1): 40–51.
Pasello M, Michelacci F, Scionti I, Hattinger CM, Zuntini M, Caccuri AM, Scotlandi K, Picci P, Serra M (2008) Overcoming glutathione S-transferase P1-related cisplatin resistance in osteosarcoma. Cancer Res 68 (16): 6661–6668.
Robey RW, Shukla S, Finley EM, Oldham RK, Barnett D, Ambudkar SV, Fojo T, Bates SE (2008) Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor CBT-1®. Biochem Pharmacol 75 (6): 1302–1312.
Saeki T, Ouchi M, Ouchi T (2009) Physiological and oncogenic Aurora-A pathway. Int J Biol Sci 5 (7): 758–762.
Serra M, Reverter-Branchat G, Maurici D, Benini S, Shen JN, Chano T, Hattinger CM, Manara MC, Pasello M, Scotlandi K, Picci P (2004) Analysis of dihydrofolate reductase and reduced folate carrier gene status in relation to methotrexate resistance in osteosarcoma cells. Ann Oncol 15 (1): 151–160.
Serra M, Scotlandi K, Manara MC, Maurici D, Lollini PL, De Giovanni C, Toffoli G, Baldini N (1993) Establishment and characterization of multidrug-resistant human osteosarcoma cell lines. Anticancer Res 13 (2): 323–329.
Shahzad MM, Lopez-Berestein G, Sood AK (2009) Novel strategies for reversing platinum resistance. Drug Resist Updat 12 (6): 148–152.
Ulisse S, Arlot-Bonnemains Y, Baldini E, Morrone S, Carocci S, Di Luigi L, D'Armiento M (2010) Inhibition of the aurora kinases suppresses in vitro NT2-D1 cell growth and tumorigenicity. J Endocrinol 204 (2): 135–142.
Wan XB, Fan XJ, Chen MY, Xu J, Long ZJ, Hua YJ, Ji H, Liu L, Hong MH, Zeng YX, Liu Q (2009) Inhibition of Aurora-A results in increased cell death in 3-dimensional culture microenvironment reduced migration and is associated with enhanced radiosensitivity in human nasopharyngeal carcinoma. Cancer Biol Ther 8 (15): 1500–1506.
Yamaguchi U, Honda K, Satow R, Kobayashi E, Nakayama R, Ichikawa H, Shoji A, Shitashige M, Masuda M, Kawai A, Chuman H, Iwamoto Y, Hirohashi S, Yamada T (2009) Functional genome screen for therapeutic target of osteosarcoma. Cancer Sci 100 (12): 2268–2274.
Acknowledgements
This study was supported by grants from: Associazione Italiana per la Ricerca sul Cancro (AIRC, grant IG10171 to Massimo Serra); Istituto Ortopedico Rizzoli (5‰ contributions to Rizzoli Institute); the European Project ‘Kids Cancer Kinome’ (KCK; grant No.037390; http://www.kidscancerkinome.org/). Dr Elisa Tavanti received a fellowship from the Associazione Italiana per la Ricerca sul Cancro (AIRC) to perform the research project ‘Pre-clinical validation of approaches targeting protein kinases in osteosarcoma’. We would like to thank CBA Research, Inc. (Lexington, KY, USA) that kindly provided us the CBT-1. We would like to thank Dr Andrea Grilli and Dr Cristina Ferrari (Laboratory of Experimental Oncology, Orthopaedic Rizzoli Institute, Bologna, Italy), respectively, for their help in analysing the motility assay data and for clinical data updating. We thank Dr Peter van Sluis and Dr Jan Koster (Academic Medical Center, University of Amsterdam, The Netherlands) for the profiling data handling and the assistance with the R2 software.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Tavanti, E., Sero, V., Vella, S. et al. Preclinical validation of Aurora kinases-targeting drugs in osteosarcoma. Br J Cancer 109, 2607–2618 (2013). https://doi.org/10.1038/bjc.2013.643
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.643
Keywords
This article is cited by
-
MicroRNA-mRNA networks define translatable molecular outcome phenotypes in osteosarcoma
Scientific Reports (2020)
-
The aurora kinase inhibitor VX-680 shows anti-cancer effects in primary metastatic cells and the SW13 cell line
Investigational New Drugs (2016)
-
ABCG2 impairs the activity of the aurora kinase inhibitor tozasertib but not of alisertib
BMC Research Notes (2015)
-
Alisertib (MLN8237), a selective Aurora-A kinase inhibitor, induces apoptosis in human tongue squamous cell carcinoma cell both in vitro and in vivo
Tumor Biology (2015)
-
Translational biology of osteosarcoma
Nature Reviews Cancer (2014)

{kind=link}
{kind=link}
{kind=link}
{kind=link}