
Abstract
Background:
Hypoxia can activate autophagy, a self-digest adaptive process that maintains cell turnover. Mammalian target of rapamycin (mTOR) inhibitors are used to treat cancer but also stimulate autophagy.
Methods:
Human mammary cancer cells and derived xenografts were used to examine whether hypoxia could exacerbate autophagy-mediated resistance to the mTOR inhibitor rapamycin.
Results:
Rapamycin exerted potent antitumour effects in MCF-7 and MDA-MB-231 mammary tumours through a marked inhibition of angiogenesis, but the autophagy inhibitor chloroquine (CQ) failed to further sensitise tumours to mTOR inhibition. Rapamycin treatment actually led to tumour reoxygenation, thereby preventing the development of autophagy. Chloroquine alone, however, blocked the growth of MCF-7 tumours and in vitro blunted the hypoxia-induced component of autophagy in these cells. Finally, when initiating CQ treatment in large, hypoxic tumours, a robust antitumour effect could be observed, which also further increased the antiproliferative effects of rapamycin.
Conclusion:
The mTOR inhibitor rapamycin significantly contributes to tumour growth inhibition and normalisation of the tumour vasculature through potent antiangiogenic effects. The resulting reduction in hypoxia accounts for a lack of sensitisation by the autophagy inhibitor CQ, except if the tumours are already at an advanced stage, and thus largely hypoxic at the initiation of the combination of rapamycin and CQ treatment.
Similar content being viewed by others


Main
Rapidly proliferating tumours are characterised by structural and functional abnormalities in vascular network, preventing homogeneous delivery of O2 to tumour cells (Feron, 2004; Vaupel et al, 2005; Bouzin and Feron, 2007; Pries et al, 2010). Furthermore, transient variations in tumour blood flow lead to cycling changes in the O2 concentration in specific tumour areas (Pries et al, 2010). This stressful environment induces adaptative mechanisms in cancer cells contributing to the selection of more aggressive cells that are also better armed to resist therapies (Harris, 2002; Bertout et al, 2008; Bristow and Hill, 2008; Vaupel, 2008). In the clinics, the extent of tumour hypoxia is thus not surprisingly associated with poor prognosis (Milani and Harris, 2008; Semenza, 2012).
Multiple reports have demonstrated that hypoxia can activate autophagy, a ‘self-digest’ process, characterised by the formation of double membrane vesicles, called autophagosomes, containing damaged organelles and long-lived proteins, which further fuse with lysosomes to form autolysosomes (Rouschop et al, 2009; Dalby et al, 2010; Mazure and Pouyssegur, 2010; Mehrpour et al, 2010; Rouschop et al, 2010). The content of autolysosomes is digested, maintaining the cell turnover and generating energy to help cells to adapt to stressful conditions (Eng and Abraham, 2011). Among the multiple actors regulating autophagy, the mammalian target of rapamycin (mTOR) kinase has a key role (Easton and Houghton, 2006; Amaravadi et al, 2011). Inhibition of mTOR mediates, for instance, nutrient starvation-induced autophagy, thereby coordinately regulating the balance between cell growth and autophagy in response to stress signals (Wouters and Koritzinsky, 2008; Janku et al, 2011). Indeed, physiologically activated mTOR protein phosphorylates different proteins, such as the ribosomal protein S6 kinase and the eukaryotic initiation factor 4E binding protein 1, leading to stimulation of the mRNA translation and ribosomal biogenesis. Inhibitors of mTOR, although initially developed as immunosuppressors (Thomson et al, 2009), are therefore used for their antiproliferative potential to treat cancer (Easton and Houghton, 2006; Dancey, 2010). Whether their effects are attenuated by the concomitant induction of autophagy remains poorly explored. Interestingly, mTOR inhibitors evaluated in the treatment of breast cancer, have achieved modest response rates, suggesting resistance or adaptative mechanisms developed by tumour cells (Chan et al, 2005; Tabernero et al, 2008; Baselga et al, 2009; Andre et al, 2010; O’Regan and Hawk, 2011; Fleming et al, 2012).
In this study, we investigated the influence of hypoxia on rapamycin toxicity in breast cancer. Our preliminary data indicated that the two extremes of the human mammary cancer phenotypes, namely the triple-negative MDA-MB-231 and the oestrogen-dependent MCF-7 tumour cells, were both in vitro largely resistant to rapamycin. The search for a general mechanism such as autophagy supporting a defect in the response to mTOR inhibition therefore prompted us to use these two cell lines to evaluate in vivo the effects of rapamycin in mouse xenograft models. We actually found that rapamycin was equally active on both tumour types mostly through antiangiogenic effects. We further identified hypoxia as a major determinant of the activity of the autophagy inhibitor chloroquine (CQ) in the MCF-7 tumour models. Indeed, reduction in hypoxia due to rapamycin-induced normalisation of the tumour vasculature prevented the benefit of CQ coadministration, whereas the initiation of the treatment combination (rapamycin+CQ) in more advanced and thus more hypoxic tumours led to a more pronounced tumour response.
Materials and Methods
Cell culture
Human breast cancer cell lines MDA-MB-231 and MCF-7 were acquired in the past 3 years from the ATCC (American Type Culture Collection, Manassas, VA, USA) where they are regularly authenticated. Cells were stored according to the supplier’s instructions and used within 6 months after resuscitation of frozen aliquots. MCF-7 and MDA-MB-231 cells were routinely cultured in DMEM supplemented with 10% fetal bovine serum and antibiotics. In some experiments, cells were cultured under 1% O2 conditions in a Ruskin Invivo 500 workstation (Pencoed, UK). Cell proliferation was determined in 96-well plates using Crystal violet after treatment or not with rapamycin (LC laboratory, Woburn, MA, USA) or CQ (Sigma, St Louis, MO, USA).
Mouse and in vivo treatments
All the experiments involving mice received the approval of the Comité d'Ethique Facultaire of the Université catholique de Louvain (UCL) (approval ID 2012/UCL/MD005) and all the in vivo procedures were carried out according to National Animal Care Regulations that meet the standards required by the UKCCCR guidelines (Workman et al, 2010). Eight-week–old female NMRI nude mice (Elevage Janvier, LeGenest-St-Isle, France) were injected subcutaneously with 3 × 106 tumour cells in the mammary fat pad (10 mice per group). In the experiments with MCF-7 tumours, 17β-oestradiol pellets (0.36 mg, 60-day release; Innovative Research of America, Sarasota, FL, USA) were preimplanted in the cervical region of mice. Tumours were allowed to reach a minimal diameter of 5 or 10 mm and animals were then daily injected (5 days a week) with either DMSO (vehicle), rapamycin (1 mg kg−1 per day) diluted in DMSO and/or CQ (50 mg kg−1 per day) diluted in saline solution (10 mice per group). Tumour sizes were tracked with an electronic caliper, and at the time of being killed, tumour samples were frozen in liquid nitrogen.
Immunoblotting and immunostaining
For immunoblotting, cells extracts were separated on SDS–PAGE and transferred onto PVDF membranes before incubation with antibodies against LC3 (Novus Biological, Littleton, CO, USA) and p62 (Cell Signaling, Danvers, MA, USA); gel loading was normalised with a β-actin antibody (Sigma).
For immunocytochemistry, cells were seeded in 8-chambers slides (Labtek, ThermoFischer, Waltham, MA, USA), treated and fixed before staining with antibodies directed against LC3 (Novus Biological) or LAMP-1 (Abcam, Cambridge, UK) followed by secondary antibodies coupled to Alexa Fluor 568 (Invitrogen, Carlsbad, CA, USA) or Dylight 488 (Jackson Immunoresearch, West Grove, PA, USA), as described previously (Vegran et al, 2011; Boidot et al, 2012; Seront et al, 2013). To track autophagy, cells were also incubated with fluorogenic protease substrate DQ Green BSA (Invitrogen) for 1 h at 37 °C before fixation with 4% paraformaldehyde. In the above experiments, nuclei were counterstained in blue with DAPI.
For immunohistochemistry, tumours were cryosliced and sections were probed with primary antibodies against CD31 (BD PharMingen, Lexington, KY, USA), LC3 (Novus Biological) and carbonic anhydrase IX (CAIX) (R&D System, Minneapolis, MN, USA) followed by secondary antibodies coupled to an Alexa Fluor 568 or Alexa Fluor 488, as described previously (Vegran et al, 2011; Boidot et al, 2012); nuclei were counterstained in blue with DAPI. In some experiments, tumour sections were were fixed with 4% paraformaldehyde and stained with a primary antibody against Ser 235/236 phospho-S6 ribosomal protein (P-S6RP) (Cell Signalling) followed by a secondary antibody coupled to 3,3′-diaminobenzidine (Sigma); sections were counterstained with haematoxylin–eosin.
In vivo tumour pO2 measurement
In vivo tumour pO2 was monitored using EPR oximetry as described previously (Sonveaux et al, 2004; Crokart et al, 2005). Electron paramagnetic resonance (EPR) spectra were recorded using an EPR spectrometer (Magnettech, Berlin, Germany) with a low-frequency microwave bridge operating at 1.1 GHz and an extended loop resonator. A charcoal (Charcoal wood powder, CX0670-1; EM Science, Gibbstown, NJ, USA) was used as the oxygen-sensitive probe in all experiments. Calibrations curves were made by measuring the EPR line width as a function of the pO2.
Statistics
Results are expressed as mean±s.d. Student’s t-test and (two-tailed) ANOVA tests were used where indicated. *P<0.05, **P<0.01 or ***P<0.001 was considered statistically significant in the different experiments.
Results
CQ does not exert additive effects on the growth inhibition of mammary tumours by rapamycin
MDA-MB-231 and MCF-7 mammary tumour cells were subcutaneously implanted in mice. When tumours reached a diameter of 5 mm, mice were treated with rapamycin and/or CQ, a compound known to inhibit the final step of autophagy; untreated tumour-bearing mice were used as controls. Rapamycin inhibited both MDA-MB-231 and MCF-7 tumour growth (Figure 1A, red curves). When combined with CQ, rapamycin did not further alter tumour progression in either model (Figure 1A, blue curves), suggesting that potential rapamycin-induced autophagy was not playing a critical role in these tumours. However, while in MDA-MB-231 xenografts, CQ alone had no significant effect on tumour growth, this single treatment led to a late inhibition of MCF-7 tumour growth (detectable after 8 days of treatment) (Figure 1A, green curves).

Chloroquine does not exert additive effects on the inhibition of tumour growth and angiogenesis by rapamycin but prevents the progression of late-stage MCF-7 tumours. (A) Graphs represent the effects of intraperitoneal administration of rapamycin (1 mg kg−1 per day), CQ (50 mg kg−1 per day) or both on the growth of MDA-MB-231 (left) or MCF-7 (right) tumours injected in the mammary fat pad. Treatment was initiated when tumours reached a diameter of 5 mm; mice treated with vehicle were used as controls (**P<0.01, ***P<0.001, n=10 mice per group). Representative phospho-S6RP (P-S6RP) immunohistochemical (B) and CD31 immunofluorescence (C) staining of MDA-MB-231 (left) and MCF-7 (right) tumour sections harvested after 16 days of treatment as described above. In (C), bar graphs show the quantification of CD31 immunofluorescent signal (***P<0.001, n=3–4). A.u., arbitrary unit; NS, nonsignificant.
To verify the inhibition of mTOR by rapamycin in our experimental conditions, we examined the extent of the phosphorylation of S6RP, a downstream target of S6K itself under the control of mTOR. We found that in both MDA-MB-231 and in MCF-7 xenografts, rapamycin markedly inhibited P-S6RP (Figure 1B). Also, using an anti-CD31 antibody to probe the tumour vasculature, we identified a robust antiangiogenic effect of rapamycin in both cell types (Figure 1C). Of note, CQ alone did not lead to inhibition of P-S6RP and did not alter tumour vasculature (Figure 1B and C).
Rapamycin treatment reduces hypoxia in MCF-7 tumours
The late effects of CQ as a single treatment in MCF-7 tumour-bearing mice (Figure 1A, right panel) suggested that at a given tumour size, autophagy could support tumour growth. This observation, together with the potent antiangiogenic effects of rapamycin, led us to examine the extent of hypoxia (a well-known inducer of autophagy in tumours) in MCF-7 tumour xenografts in the absence and in the presence of rapamycin. We first confirmed that CD31 staining mirrored the pattern of both CAIX, a hypoxia-inducible protein used as a marker of low pO2 areas in tumours, and LC3 punctate staining known to reflect autophagy (Figure 2A). We then showed that the extent of CAIX expression in MCF-7 xenografts was markedly reduced following rapamycin treatment, while LC3 staining was not significantly altered (Figure 2B and C for quantification). To more definitely evaluate the effects of rapamycin on tumour oxygenation, we used EPR technology based on the oxygen-dependent broadening of the EPR line width of a paramagnetic oxygen sensor to directly measure pO2 in control and treated tumours. These experiments documented a lower pO2 in >10-mm-diameter MCF-7 tumours (day 10) than in 5-mm-diameter tumours (day 0), and, importantly, confirmed a paradoxical increase in tumour oxygenation in rapamycin-treated mice (Figure 2D). Altogether, these data strongly suggested that (i) hypoxia progression was associated with autophagy in MCF-7 tumours (and could account for the late response to CQ as single treatment) and (ii) that rapamycin-induced tumour reoxygenation (due to tumour vessel normalisation) could prevent the stimulation of autophagy (and thus opposed the effects of CQ cotreatment).

Rapamycin treatment reduces hypoxia in MCF-7 tumours. (A) Representative immunofluorescence staining of consecutive MCF-7 tumour sections for CD31, CAIX and LC3 harvested at a tumour size of about 10 mm. (B) Representative immunofluorescence staining of CAIX in MCF-7 tumour sections harvested from mice exposed for 16 days to vehicle or rapamycin; 4′,6-diamidino-2-phenylindole (DAPI) counterstaining is also presented. (C) Bar graph shows the quantification of CAIX immunofluorescent signal (***P<0.001, n=3–4). (D) Bar graph shows the extent of intratumour mean pO2 as determined by EPR in vehicle- and in rapamycin-treated mice; Day 0 and Day 10 correspond to tumour diameter of about 5 and 10 mm in vehicle conditions, respectively (*P<0.05, **P<0.01, n=5 mice per group).
Hypoxia decreases the antiproliferative effects of rapamycin and stimulates autophagy in MCF-7 but not in MDA-MB-231 mammary tumour cells
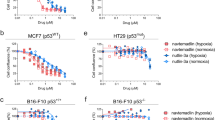
Next, to understand the antitumour effect of CQ as a single treatment in MCF-7 and not in MDA-MB-231 tumours, we examined the in vitro behaviour of both mammary cancer cell lines exposed to rapamycin (or not) under normoxia and hypoxia (1% O2). Although in MDA-MB-231 cells treatment with rapamycin slightly reduced cell proliferation to the same extent in both conditions (Figure 3A, left panel), the antiproliferative effects of rapamycin differed between normoxia and hypoxia in MCF-7 cells. Indeed, the inhibition of MCF-7 cell proliferation in response to rapamycin observed under normoxia was significantly attenuated in hypoxic MCF-7 cells (P<0.001) (Figure 3A, right panel). Trypan blue exclusion experiments confirmed that the observed reductions in cell density corresponded to direct inhibition of cell proliferation and not induction of cell death (not shown).

Hypoxia promotes rapamycin resistance and autophagy in MCF-7 but not in MDA-MB-231 cells. (A) Bar graphs represent the amounts of viable MDA-MB-231 and MCF-7 cells (expressed as % of control) after 72 h treatment with the indicated doses of rapamycin under normoxia or hypoxia (***P<0.001, n=3–4). (B) Representative LC3I and LC3II immunoblotting experiments are presented for MDA-MB-231 and MCF-7 cells exposed for 48 h to normoxia or hypoxia with or without 500 μ M leupeptin; a longer film exposure is provided to better show LC3II accumulation. Bar graphs represent the autophagic flux determined as the fold-induction LC3-II accumulation in the presence of leupeptin (**P<0.01, n=3). (C) Representative immunofluorescence detection of LC3 (green staining) and LAMP-1 (red staining) in MDA-MB-231 and MCF-7 cells after 48 h treatment with 100 nM rapamycin under normoxia or hypoxia; yellow–orange staining indicates the presence of autolysosomes. Bar graphs represent the proportion of cells (%) exhibiting autolysosomes (yellow punctate staining reflecting LC3/LAMP-1 colocalisation) (*P<0.05, **P< 0.01, ***P<0.001, NS, not significantly different, n=3).
We then checked whether basal and/or hypoxia-induced autophagy could account for the observed resistance to rapamycin in MDA-MB231 and MCF-7 cells. To determine the basal autophagic flux (i.e., the rate of formation and clearance of autophagosomes), we first used leupeptin to block lysosomal proteolysis in autophagosomes and measured the accumulation of LC3II by immunoblotting. In MDA-MB-231 cells, LC3II level was increased by ∼1.8-fold in both normoxia and hypoxia, reflecting an active basal autophagic activity in these cells independently of the O2 conditions (Figure 3B). In MCF-7 cells, basal autophagy was very low in normoxia as reflected by a limited LC3II accumulation, which, however, increased by ∼3-fold in response to hypoxia (Figure 3B). We also probed the autophagic flux through immunocytochemical costaining of autophagosomes with LC3 antibodies (green staining in Figure 3C) and lysosomes with LAMP-1 antibodies (red staining in Figure 3C). Autophagy could then be estimated by the extent of yellow–orange-coloured punctate staining resulting from the fusion of autophagosomes and lysosomes. In MDA-MB-231 cells, autolysosomes were detected in the same proportion of (non-treated) cells in both normoxic and hypoxic conditions. By contrast, in MCF-7 cells, only a very small proportion of cells showed autolysosomes under normoxia, confirming the low basal autophagy in these cells (see bar graphs in Figure 3C). Exposure of MCF-7 cells to hypoxia, however, induced an increase in the amount of cells exhibiting LC3 and LAMP-1 costaining. Interestingly, using the same methodology, we documented that rapamycin stimulated autophagy in both cell types, under normoxia and hypoxia (Figure 3C). Still, the proportion of MCF-7 cells with autolysosomes was more largely increased in response to hypoxia than following exposure to rapamycin, while in MDA-MB-231 cells, rapamycin did stimulate autophagy where hypoxia failed to do so (compare bars 2 and 3 in graphs of Figure 3C). Similar conclusions could be drawn from experiments using the fluorogenic protease substrate DQ Green BSA (to image the formation of the autolysosome) (see Supplementary Figure 1A) or the reduction in p62 immunoblotting signal (reflecting autophagic degradation) (see Supplementary Figure 1B).
Inhibition of autophagy by CQ sensitises hypoxic MCF-7 cells to rapamycin in vitro
To evaluate the contribution of autophagy in the differential in vitro sensitivity of MCF-7 and MDA-MB-231 cells to rapamycin, we next used CQ. We first showed that in CQ-treated MDA-MB-231 cells, LC3 staining was similarly increased under normoxia and hypoxia (Figure 4A, left panels). By contrast, in MCF-7 cells, 5 μ M CQ markedly increased LC3 staining under hypoxia but had only a marginal effect on LC3II accumulation under normoxia (Figure 4A, right panels). These results were confirmed by immunoblotting, with a similar increase in LC3II upon CQ in normoxic and hypoxic MDA-MB-231 cells and a much higher LC3 accumulation in MCF-7 cells under hypoxia than normoxia (Figure 4B); the inhibitory effects of CQ on autophagy in both cell lines were also documented using the fluorogenic protease substrate DQ Green BSA (see Supplementary Figure 2A). Interestingly, we also found that while MDA-MB-231 cell proliferation was only slightly altered at the highest dose of CQ, a dose-dependent reduction in MCF-7 cell density was observed with CQ under hypoxia (Figure 4C). Similar conclusions could be drawn from experiments using bafilomycin as another autophagy inhibitor (see Supplementary Figure 2B). In parallel, we showed that CQ treatment of MCF-7 cells for 72 h further increased the anti-proliferative effects obtained with rapamycin under low pO2 but not under normoxia (Figure 4C, right graph). By contrast, CQ did not further affect the viability of rapamycin-treated MDA-MB-231 cells, neither in normoxia nor in hypoxia (Figure 4C, left graphs). For the latter experiments, we used CQ at the concentration of 5 μ M to exclude an effect on tumour cell cycle progression as confirmed by flow cytometry analysis (data not shown). Finally, to document that autophagy could have a functional role in MDA-MB-231 cells in another context than hypoxia, we also exposed these cells to an amino-acid-deprived medium (minimal medium) and found that autophagy was stimulated in these conditions (Supplementary Figure 3A) and importantly contributed to MDA-MB-231 cell survival in a CQ-sensitive manner (Supplementary Figure 3B).

Chloroquine inhibits autophagy and reduces the proliferation of hypoxic MCF-7 cells but not MDA-MB231 cells. (A) Representative immunofluorescence detection of LC3 (green staining) in MDA-MB-231 and MCF-7 cells after 48 h treatment with or without 5 μ M CQ under normoxia or hypoxia; note that slide illumination was reduced (vs Figure 1C) 1 to avoid signal saturation in CQ conditions. Bar graphs represent the extent of LC3 staining per cell number (arbitrary units (a.u.) (*P<0.05, **P<0.01, n=3). (B) Representative LC3I and LC3II immunoblotting experiments (n=3) are presented for MDA-MB-231 and MCF-7 cells exposed for 48 h to normoxia or hypoxia with or without 5 μ M CQ. Bar graphs represent the extent of MDA-MB-231 and MCF-7 cell proliferation (expressed as % of untreated cells) (C) after 48 h treatment with increasing concentrations of CQ and (D) after 72 h treatment with either 5 μ M CQ, 100 nM rapamycin or both (*P<0.05, **P<0.01, ***P<0.001, n=3). NS, non-significant.
CQ exerts additive effects on the growth inhibition of large, hypoxic MCF-7 tumours by rapamycin
The above in vitro experiments strongly suggested that hypoxia-induced autophagy as observed in MCF-7 cells could account for the antitumour effects of CQ (as a single treatment) observed when MCF-7 tumours reached a size more likely to be associated with hypoxia (see Figure 1A, right panel, green curve). It would also explain how rapamycin treatment, by reducing the extent of hypoxia in MCF-7 tumours, could render small tumours less dependent on autophagy (and thus less sensitive to CQ addition).
To explore this hypothesis, we first confirmed that hypoxia and LC3 punctate staining increased with MCF-7 tumour growth by monitoring the expression patterns of LC3 and CAIX, respectively, according to the tumour diameters (5–7, 7–10 and >10 mm) (Figure 5A). We then examined the capacity of CQ to influence the growth of large MCF-7 tumours (i.e., diameter >10 mm). We found that CQ blocked tumour growth when used alone (without delay as observed in Figure 1A) and importantly further increased the antitumour effects of rapamycin (Figure 5B, blue curves). Moreover, in these tumours, after treatment, the extent of hypoxia as probed with CAIX immunostaining was found to be directly proportional to the tumour size (Figure 5C) and histological analyses of treated MCF-7 tumours revealed a net increase in central necrosis areas in tumours treated with the combination of rapamycin and CQ (vs rapamycin alone) (Figure 5D).

Chloroquine and rapamycin exert additive effects on the inhibition of large MCF-7 tumour growth. (A) Representative immunofluorescence CAIX (green) and LC3 (red) staining of MCF-7 tumour sections harvested from the vehicle group at different tumour diameters (5–7, 7–10 and >10 mm); 4′,6-diamidino-2-phenylindole (DAPI) counterstaining (blue) is also shown. Insets show magnification of LC3 punctate staining. (B) Graphs represent the effects of intraperitoneal administration of rapamycin (1 mg kg−1 per day), CQ (50 mg kg−1 per day) or both on the growth of MCF-7 tumours injected in the mammary fat pad. Treatment was initiated when tumours reached a diameter of 10 mm; mice treated with vehicle were used as controls (*P<0.05, ***P<0.001, n=10 mice per group). (C) Bar graphs show the quantification of CAIX immunofluorescent signals in the different conditions (*P<0.05, **P<0.01, ***P<0.001, n=5). (D) Typical haematoxylin colouration of MCF-7 tumour sections harvested from the rapamycin and the rapamycin-CQ groups as described above. Bar graphs represent the extent of the central necrosis area (*P<0.05, n=4).
Discussion
This study documents that (i) hypoxia is not necessarily associated with autophagy in mammary cancer cells, (ii) when it is the case, drugs like CQ can delay tumour growth, (iii) mTOR inhibition can per se lead to in vivo tumour reoxygenation, thereby abrogating this possible source of resistance. Our work also provides several insights on the optimal framework to combine mTOR and autophagy inhibitors such as CQ. First, our in vivo data demonstrate that when CQ treatment is initiated as single treatment in large, hypoxic tumours (and not early stage tumours), a robust antitumour effect is observed and importantly, an additive effect can be obtained with rapamycin. Second, our comparison of two breast cancer cell lines indicates that a differential induction of autophagy in response to hypoxia but not following exposure to mTOR inhibitors is critical to predict such capacity to respond to CQ.
A major finding of this study was actually the paradoxical absence of therapeutic benefits observed with the association rapamycin+CQ compared with rapamycin alone when treating MCF-7 and MDA-MB-231 tumours at early stage of growth (i.e., 5 mm diameter) (see Figure 1A). This was unexpected with regard to the antiangiogenic properties of rapamycin, which were primarily supposed to lead to a decrease in tumour oxygenation and thereby to an increase in autophagy for survival. Instead, we showed that rapamycin treatment resulted in a better oxygenation of the tumour, consistent with previous studies reporting that rapamycin could normalise tumour perfusion and facilitate delivery of chemotherapeutic agents (Xue et al, 2008; Zhang et al, 2011; Fokas et al, 2012). As a consequence, the lack of development of highly hypoxic areas rendered rapamycin-treated tumours less dependent on autophagy and thus less sensitive to CQ. By contrast, we showed that by initiating treatment later, MCF-7 tumours had the opportunity to grow and become hypoxic, the prerequisite to induce autophagy and promote CQ action (see Figure 5B).
The role of autophagy remains controversial in cancer, as to whether autophagy acts as a cell survival or a cell death pathway (Mazure and Pouyssegur, 2010; Eskelinen, 2011). Some recent reports tip the balance in favour of autophagy as a mode of resistance towards chemotherapy and inhibition of autophagy as a strategy to sensitise tumours to anticancer treatments (Amaravadi et al, 2007, 2011). Other reports have also indicated that hypoxia, a common hallmark of solid tumours, could activate the autophagic process as a protective mechanism (Rouschop et al, 2009, 2010; Eng and Abraham, 2011; Hu et al, 2012). Our work indicates that the prosurvival role of autophagy in response to hypoxia is cell-specific as it was observed in MCF-7 cells but not in MDA-MB-231 tumour cells. Yi et al (2013) recently reported a similar differential autophagic response of MCF-7 and MDA-MB-231 to radiotherapy, autophagy being stimulated and inhibited, respectively, because of an opposite effect of irradiation on the expression of miR199a-5p. Whether hypoxia induced a similar regulation or whether the different hormone-dependency or p53 status of either cell line has a role warrants further investigation. Still, it is interesting to note that a high basal autophagic activity, as observed in MDA-MB-231 cells, cannot predict CQ sensitivity in vivo (contrary to the net hypoxia inducibility of the phenomenon observed in MCF-7 cells). Interestingly, Bellot et al (2009) previously reported that rapamycin was a weak activator of the prosurvival component of autophagy compared to hypoxia and that hypoxia-induced autophagy was driven by BNIP3 in a manner largely independent of mTOR inhibition. Taken together with our data, this observation supports a model in which the nature of the stress determines the mode of autophagy, and thereby the positive or negative impact on cell survival.
In conclusion, the results of our study underscore a new rationale to use mTOR inhibitors in the clinics and to combine them with autophagy inhibitors. First, the therapeutic action of mTOR inhibitors can derive from their antiangiogenic effects in tumours even though the proliferation of cancer cells is per se barely sensitive to these drugs. Second, the sensitivity of late-stage tumours to mTOR inhibitors may be reduced under hypoxia because of autophagic activity. In these tumours, administration of inhibitors of autophagy such as CQ has the potential to enhance the antiproliferative effects of mTOR inhibitors with the caveat that only voluminous, hypoxic tumours will respond.
Change history
12 November 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17 (4): 654–666.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB (2007) Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 117 (2): 326–336.
Andre F, Campone M, O'Regan R, Manlius C, Massacesi C, Sahmoud T, Mukhopadhyay P, Soria JC, Naughton M, Hurvitz SA (2010) Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol 28 (34): 5110–5115.
Baselga J, Semiglazov V, van DP, Manikhas A, Bellet M, Mayordomo J, Campone M, Kubista E, Greil R, Bianchi G, Steinseifer J, Molloy B, Tokaji E, Gardner H, Phillips P, Stumm M, Lane HA, Dixon JM, Jonat W, Rugo HS (2009) Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol 27 (16): 2630–2637.
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29 (10): 2570–2581.
Bertout JA, Patel SA, Simon MC (2008) The impact of O2 availability on human cancer. Nat Rev Cancer 8 (12): 967–975.
Boidot R, Vegran F, Meulle A, Le BA, Dessy C, Sonveaux P, Lizard-Nacol S, Feron O (2012) Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res 72 (4): 939–948.
Bouzin C, Feron O (2007) Targeting tumor stroma and exploiting mature tumor vasculature to improve anti-cancer drug delivery. Drug Resist Updat 10 (3): 109–120.
Bristow RG, Hill RP (2008) Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 8 (3): 180–192.
Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, Eiermann W, Hess D, Morant R, Semiglazov V, Borner M, Salzberg M, Ostapenko V, Illiger HJ, Behringer D, Bardy-Bouxin N, Boni J, Kong S, Cincotta M, Moore L (2005) Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 23 (23): 5314–5322.
Crokart N, Radermacher K, Jordan BF, Baudelet C, Cron GO, Gregoire V, Beghein N, Bouzin C, Feron O, Gallez B (2005) Tumor radiosensitization by antiinflammatory drugs: evidence for a new mechanism involving the oxygen effect. Cancer Res 65 (17): 7911–7916.
Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B (2010) Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 6 (3): 322–329.
Dancey J (2010) mTOR signaling and drug development in cancer. Nat Rev Clin Oncol 7 (4): 209–219.
Easton JB, Houghton PJ (2006) mTOR and cancer therapy. Oncogene 25 (48): 6436–6446.
Eng CH, Abraham RT (2011) The autophagy conundrum in cancer: influence of tumorigenic metabolic reprogramming. Oncogene 30 (47): 4687–4696.
Eskelinen EL (2011) The dual role of autophagy in cancer. Curr Opin Pharmacol 11 (4): 294–300.
Feron O (2004) Targeting the tumor vascular compartment to improve conventional cancer therapy. Trends Pharmacol Sci 25 (10): 536–542.
Fleming GF, Ma CX, Huo D, Sattar H, Tretiakova M, Lin L, Hahn OM, Olopade FO, Nanda R, Hoffman PC, Naughton MJ, Pluard T, Conzen SD, Ellis MJ (2012) Phase II trial of temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat 136 (2): 355–363.
Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, Hackl W, Maira SM, Bernhard EJ, McKenna WG, Muschel RJ (2012) Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res 72 (1): 239–248.
Harris AL (2002) Hypoxia – a key regulatory factor in tumour growth. Nat Rev Cancer 2 (1): 38–47.
Hu YL, Delay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, Aghi MK (2012) Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 72 (7): 1773–1783.
Janku F, McConkey DJ, Hong DS, Kurzrock R (2011) Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 8 (9): 528–539.
Mazure NM, Pouyssegur J (2010) Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 22 (2): 177–180.
Mehrpour M, Esclatine A, Beau I, Codogno P (2010) Overview of macroautophagy regulation in mammalian cells. Cell Res 20 (7): 748–762.
Milani M, Harris AL (2008) Targeting tumour hypoxia in breast cancer. Eur J Cancer 44 (18): 2766–2773.
O’Regan R, Hawk NN (2011) mTOR inhibition in breast cancer: unraveling the complex mechanisms of mTOR signal transduction and its clinical implications in therapy. Expert Opin Ther Targets 15 (7): 859–872.
Pries AR, Hopfner M, le NF, Dewhirst MW, Secomb TW (2010) The shunt problem: control of functional shunting in normal and tumour vasculature. Nat Rev Cancer 10 (8): 587–593.
Rouschop KM, Ramaekers CH, Schaaf MB, Keulers TG, Savelkouls KG, Lambin P, Koritzinsky M, Wouters BG (2009) Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother Oncol 92 (3): 411–416.
Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 120 (1): 127–141.
Semenza GL (2012) Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 33 (4): 207–214.
Seront E, Pinto A, Bouzin C, Bertrand L, Machiels JP, Feron O (2013) PTEN deficiency is associated with reduced sensitivity to mTOR inhibitor in human bladder cancer through the unhampered feedback loop driving PI3K/Akt activation. Br J Cancer 109 (6): 1586–1592.
Sonveaux P, Dessy C, Martinive P, Havaux X, Jordan BF, Gallez B, Gregoire V, Balligand JL, Feron O (2004) Endothelin-1 is a critical mediator of myogenic tone in tumor arterioles: implications for cancer treatment. Cancer Res 64 (9): 3209–3214.
Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, Martinelli E, Cajal S, Jones S, Vidal L, Shand N, Macarulla T, Ramos FJ, Dimitrijevic S, Zoellner U, Tang P, Stumm M, Lane HA, Lebwohl D, Baselga J (2008) Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol 26 (10): 1603–1610.
Thomson AW, Turnquist HR, Raimondi G (2009) Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol 9 (5): 324–337.
Vaupel P (2008) Hypoxia and aggressive tumor phenotype: implications for therapy and prognosis. Oncologist 13 (Suppl 3): 21–26.
Vaupel P, Mayer A, Briest S, Hockel M (2005) Hypoxia in breast cancer: role of blood flow, oxygen diffusion distances, and anemia in the development of oxygen depletion. Adv Exp Med Biol 566: 333–342.
Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O (2011) Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 71 (7): 2550–2560.
Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson S, Wedge SR, Eccles SA (2010) Guidelines for the welfare and use of animals in cancer research. Br J Cancer 102 (11): 1555–1577.
Wouters BG, Koritzinsky M (2008) Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 8 (11): 851–864.
Xue Q, Hopkins B, Perruzzi C, Udayakumar D, Sherris D, Benjamin LE (2008) Palomid 529, a novel small-molecule drug, is a TORC1/TORC2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer Res 68 (22): 9551–9557.
Yi H, Liang B, Jia J, Liang N, Xu H, Ju G, Ma S, Liu X (2013) Differential roles of miR-199a-5p in radiation-induced autophagy in breast cancer cells. FEBS Lett 587 (5): 436–443.
Zhang Q, Bindokas V, Shen J, Fan H, Hoffman RM, Xing HR (2011) Time-course imaging of therapeutic functional tumor vascular normalization by antiangiogenic agents. Mol Cancer Ther 10 (7): 1173–1184.
Acknowledgements
This work was supported by grants from the Fonds de la Recherche Scientifique FRS-FNRS, the Télévie, the Foundation against cancer, the J Maisin Foundation, an Action de Recherche Concertée (ARC 09/14-020) and the interuniversity attraction pole (IUAP program P7.03).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Seront, E., Boidot, R., Bouzin, C. et al. Tumour hypoxia determines the potential of combining mTOR and autophagy inhibitors to treat mammary tumours. Br J Cancer 109, 2597–2606 (2013). https://doi.org/10.1038/bjc.2013.644
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.644
Keywords
This article is cited by
-
SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway
British Journal of Cancer (2017)
-
Synergistic chemopreventive effects of curcumin and berberine on human breast cancer cells through induction of apoptosis and autophagic cell death
Scientific Reports (2016)
-
Cytotoxic activities and metabolic studies of new combretastatin analogues
Medicinal Chemistry Research (2015)
