
Abstract
Background:
Docetaxel and irinotecan chemotherapy have shown good efficacy in the treatment of advanced oesophago-gastric cancer. This randomised phase II study evaluated the efficacy and toxicity profile of two non-platinum docetaxel-based doublet regimens in advanced oesophago-gastric cancer.
Methods:
Chemotherapy-naïve patients with advanced oesophago-gastric cancer were randomised to receive either 3-weekly DI (docetaxel 60 mg m−2 plus irinotecan 250 mg m−2 (Day 1)) or 3-weekly DF (docetaxel 85 mg m−2 (Day 1) followed by 5-fluorouracil 750 mg m−2 per day as a continuous infusion (Days 1–5)).
Results:
A total of 85 patients received DI (n=42) or DF (n=43). The primary endpoint was overall response rate (ORR). The ORR and time to progression (TTP) in the evaluable population (n=65) were 37.5% (DI) vs 33.3% (DF), and 4.2 months vs 4.4 months, respectively. In the intent-to-treat population, the observed ORR, TTP and median overall survival were similar between the two groups. Grade 3–4 neutropenia, febrile neutropenia and diarrhoea were more frequent in the DI arm as compared with the DF arm (83.3% vs 69.8%, 40.5% vs 18.6%, and 42.9% vs 16.3%, respectively).
Conclusion:
Both docetaxel-based doublet regimens show comparable efficacy; however, the DF regimen was associated with a better toxicity profile and is an alternative treatment option for patients in whom platinum-based regimens are unsuitable.
Similar content being viewed by others


Main
Oesophago-gastric cancer remains a major cause of cancer-related mortality worldwide (Jemal et al, 2011). In patients with incurable disease, palliative chemotherapy improves survival as compared with best supportive care (Wagner et al, 2010). Despite several studies over the past decades to define an optimal first-line regimen, to date, no chemotherapy combination has been internationally accepted as standard first-line therapy for advanced oesophago-gastric cancer.
The standard chemotherapy backbone for advanced oesophago-gastric cancer patients is fluropyrimidine-/platinum-based. The combination of epirubicin, oxaliplatin and capecitabine (EOX) represents an optimal triplet regimen having demonstrated favourable toxicity and efficacy when compared with the standard epirubicin, cisplatin, 5-fluorouracil (5-FU) (ECF) regimen in the REAL-2 study. In this study, EOX demonstrated improved overall survival (OS) as compared with the reference ECF regimen (hazard ratio for death, 0.80; 95% confidence interval (CI), 0.66 to 0.97; P=0.02) with a median OS of 11.2 months (Cunningham et al, 2008). Other regimens, such as FOLFOX, using biweekly oxaliplatin and continuous infusional 5-FU/folinic acid has demonstrated better safety profile compared with 5-FU/cisplatin in several phase II trials (Van Cutsem et al, 2011b). Doublet regimens containing oral fluropyrimidine compound, S1, is considered to be the standard regimen in Japan and many Asian countries (Boku et al, 2009).
Docetaxel, a semi-synthetic taxoid has shown activity in gastric cancer, and has been examined as a single agent and in combination therapy. Docetaxel in combination with cisplatin has shown encouraging activity with objective response rate ranging between 31 and 56%, and OS between 9 months and 10.5 months (Ajani et al, 2005). The TAX325 study, which evaluated docetaxel, cisplatin and 5-FU regimen in advanced oesophago-gastric cancer demonstrated significantly improved OS (risk reduction 23%) and increased time to progression (TTP) compared with cisplatin and 5-FU (Van Cutsem et al, 2006). Similarly, the activity of irinotecan has been established in advanced gastric cancer. In first-line treatment, irinotecan monotherapy (350 mg m−2) is tolerable and provides modest response rates of 20%, and a median OS of 7.1 months, whereas combination therapy with irinotecan, and 5-FU and folinic acid is associated with a response rate of 42.4%, and median OS of 10.7 months (Kohne et al, 2003; Pozzo et al, 2004).
This randomised phase II study was designed to assess the efficacy of docetaxel in combination with either irinotecan or 5-FU in advanced oesophago-gastric cancer.
Patients and methods
Study design
Eligible patients were aged 18–75 years with measurable and/or evaluable metastatic, histologically proven gastric adenocarcinoma (including adenocarcinoma of the oesophago-gastric junction). Other key eligibility criteria were the following: Karnofsky performance status (KPS)⩾70%, life expectancy >12 weeks, and adequate haematological, renal and hepatic function. Previous adjuvant (and/or neoadjuvant) chemotherapy was allowed, provided a period of 12 months had elapsed since the end of therapy and first relapse. No prior palliative chemotherapy was permitted. Patients must have had ⩾6 weeks since prior radiotherapy and ⩾3 weeks since major surgery. Written informed consent was obtained from all patients. The protocol was approved by an independent ethics committee/institutional review board, and the study was conducted in accordance with the amended Declaration of Helsinki.
Dosing and administration
Patients were randomised to receive either docetaxel 60 mg m−2 (1-h IV infusion, Day 1) followed by irinotecan 250 mg m−2 (30- to 90-min IV infusion, Day 1) every 3 weeks (DI), or docetaxel 85 mg m−2 (1-h IV infusion, day 1) followed by 5-FU 750 mg m−2 per day (continuous infusion, days 1 to 5) every 3 weeks (DF) until disease progression, unacceptable toxicity or withdrawal of consent. Dose reductions were required in both treatment arms in case of severe toxicities (graded according to the National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG)-expanded common toxicity (CTC) criteria version 2). Chemotherapy was dosed according to the haematological function on the day of treatment. If the neutrophil count was<1.5 × 109 l−1 or the platelet count was <100 × 109 l−1 treatment was delayed by 1 week (maximum of 2 weeks). During the second and/or subsequent cycles in cases of febrile neutropenia or grade 3/4 neutropenia lasting more than 7 days, G-CSF was recommended. Dose reductions were recommended if despite the use of G-CSF, grade 3/4 neutropenia occurred with subsequent cycles. Treatment was delayed for 1 week for patients with ⩾grade 1 diarrhoea (maximum of 2 weeks delay). Protocol-specified dose reduction was recommended if despite the use of loperamide, ⩾grade 3 diarrhoea occurred. A maximum of two dose reductions or two treatment delays were permissible, if despite dose reductions, the same complication-persisted treatment was terminated unless anti-neoplastic efficacy justified continuation. Both arms received standard steroid pre-medication consisting of six doses of dexamethasone 8 mg for 3 days. Patients in the DI arm received anti-emetic pre-medications with 5HT3 antagonists.
Study endpoints
The primary endpoint was a radiological response rate as assessed by the external response review committee. Overall response rates (ORR) was assessed by a CT scan and was defined as the percentage of patients who achieved a complete response (CR) or a partial response (PR). A CR or PR had to be confirmed by two evaluations of the disease taken ⩾4 weeks apart, and all responses were reviewed according to World Health Organization criteria (Miller et al, 1981). The CT response assessments were performed every two cycles.
Secondary endpoints included TTP, time to treatment failure (TTF), duration of response, OS, treatment toxicities and clinical benefit. Clinical benefit was assessed in the intention-to-treat (ITT) population in terms of time to definitive worsening of KPS (a decrease by ⩾1 category compared with baseline without any further improvement); time to definitive weight loss (definitive decrease in weight by ⩾5% compared with baseline); time to definitive worsening of appetite (deterioration of appetite by ⩾1grade on a scale of 1 to 5, where 1=very poor and 5=excellent) and pain-free survival (time from randomisation to first appearance of ⩾grade 1 cancer pain in patients with NCIC-CTG-expanded CTC, version 2, grade 0 cancer pain at baseline).
Adverse events (AEs) and laboratory values were graded according to the NCIC-CTG-expanded CTC, version 2.
Statistical analyses
The primary objective of the study was to rank the two test arms on the basis of their efficacy. No formal statistical comparison was planned to compare the treatment groups. The trial design was based on the assumption that there is a true ORR difference of 15% between the two test arms (40% vs 55%); 37 evaluable patients per arm were necessary to ensure a 90% probability to rank correctly the two test arms according to their observed response rates. With this rationale, the test arm with the highest observed response rate was to be identified as the most efficacious test arm. Taking into account a rate of 10% of non-evaluable patients, it was estimated that 84 patients (42 per arm) in total were needed to be enrolled in the trial.
Efficacy was evaluated in two populations. The ITT population was defined as all randomised patients who received treatment in the arm to which they were assigned. The per protocol population (PPP), a subset of the ITT, was defined as patients who were eligible/evaluable for response, and who did not experience any major protocol deviation during the study. The PPP was used for the primary endpoint analysis. Safety analyses included all treated patients.
Patients were evaluable for response if they were considered to be eligible and had at least one complete tumour assessment after receiving two cycles of study drugs (unless early progression occurred). For TTP, death was considered an event if it occurred within 9 weeks of the last evaluable tumour assessment. Incidence, severity (worst grade), relation to study treatment and seriousness of toxicity were described by patient and cycle. Worst-grade toxicity of laboratory values was recorded by the patient and by the cycle.
Results
Patient characteristics
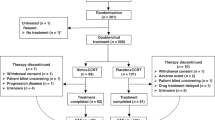
Between August 1999 and August 2000, 86 patients were enrolled and randomised from 19 centres in 6 countries (43 to each group) (Figure 1). One patient in the DI group was not treated and discontinued the study because of jaundice; therefore, the ITT included 85 patients (Figure 1). The participant flow during the study is summarised in Figure 1. Gender, race and disease characteristics (except for anatomic site of the primary tumour) were well balanced between the two groups (Table 1). A between-group difference of more than 10% points was noted for KPS 70, patient age groups and >5% weight loss (Table 1). The majority of patients (94.1%) had metastatic disease; 36.5% of patients had three or more organs involved. Approximately a third of patients (36.5%) had prior palliative or curative gastrectomy.

Consort diagram. Total analysed for primary endpoint=85 (ITT), 65 (PPP). Total analysed for safety=85. Abbreviations: DF=docetaxel plus 5-fluorouracil; DI=docetaxel plus irinotecan; ITT=intention-to-treat; PPP=per protocol population; LFT=liver function test.
Exposure to study medication
Overall, both groups received a similar number of treatment cycles (median (range): 5 (1 to 21) with DI, 6 (1 to 14) with DF). Both groups received similar overall duration of study treatment (median, 16 and 18 weeks in the DI and DF groups, respectively). The median relative dose intensity was high for all drugs (>0.90), showing that the administered dose was close to the planned dose.
A similar proportion of patients had dose reductions (47.6% and 46.5% of patients receiving DI and DF, respectively). The main reason for dose reduction was haematological toxicity with DI (23.8%) and non-haematological toxicity with DF (27.9%). All AEs leading to dose reduction were related to the study medication, except for one. These AEs included diarrhoea (26.2%), fever (21.4%) and neutropenia (11.9%) in the DI group, and diarrhoea (11.6%), stomatitis (11.6%) and fever (9.3%) in the DF group. The proportion of patients with at least one cycle delay was comparable (45.2% and 44.2% of patients receiving DI and DF, respectively), with the main reason for delay being ‘other reasons’ (e.g., patients’ holidays, patients’ requests, or administrative logistical reasons) in both groups (23.8% and 32.6% of patients receiving DI and DF, respectively).
The main reason for treatment discontinuation in both groups was progressive disease (DI: 57.1%; DF: 48.8%), followed by AEs, with a difference of more than 10% between groups (DI: 14.3%; DF: 27.9%).
Efficacy
The ORR in the PPP was comparable between groups: 37.5% (95% CI, 21.1 to 56.3) for DI and 33.3% (95% CI, 18.0 to 51.8) for DF (Table 2). Similar ORR was demonstrated for the ITT population (DI: 31.0% (95% CI, 17.6 to 47.1) and DF: 25.6% (95% CI, 13.5 to 41.2)). The median duration of response in the PPP was similar between groups (Table 2). The median TTF (PPP) was 3.8 months (95% CI, 2.1 to 4.8) with DI and 3.9 months (95% CI, 2.8 to 4.4) with DF. The median TTF in the ITT population was 2.8 months (95% CI, 1.8 to 4.2) with DI and 3.9 months (95% CI, 2.6 to 4.3) with DF.
The median OS (ITT) was 8.6 months (95% CI, 6.1 to 12.2) with DI and 9.4 months (95% CI, 7.7 to 11.0) with DF. A total of 15 out of 42 patients in the DI group and 11 out of 43 patients in DF group were living 1 year after randomisation (Figure 2); 6 and 2 patients, respectively, remained alive after 2 years. The median TTP in the ITT group was 3.8 months (95% CI, 2.2 to 6.0) with DI and 4.4 months (95% CI, 2.7 to 6.8) with DF (Table 2).

Kaplan–Meier curve of overall survival in the ITT population. Abbreviations: DF=docetaxel plus 5-fluorouracil; DI=docetaxel plus irinotecan; ITT=intention-to-treat population.
Clinical benefit
Clinical benefit was measured in the ITT population. The median time to definitive deterioration of KPS was 2.6 months (95% CI, not reached) with DI and 4.9 months (95% CI, 1.9 to 11.2) with DF. Median time to definitive worsening of appetite was identical in both groups at 4.9 months (95% CI, not reached). Median time to definitive 5% weight loss was 7.6 months with DI and was not reached with DF. Median pain-free survival was not reached in either group.
Second-line therapy
A total of 18 (43%) patients in the DI group and 21 (49%) patients in the DF group received further second-line chemotherapy. Majority of these patients in both arms received a platinum-containing regimen.
Toxicity
The AEs are summarised in Table 3. Diarrhoea was the most common non-haematological toxicity seen in both arms (DI: 42.9%, DF: 16.3%). There was a higher incidence of grade 3–4 stomatitis in the DF arm (14.0%). Myelotoxicity was more frequent in the DI arm with grade 3–4 neutropenia occurring in 83.3% of patients in this group and 69.8% in the DF arm. This translated into a higher rate of febrile neutropenia/neutropenic infection in the DI arm (40.5% vs 18.6%, P=0.03). The G-CSF for secondary prophylaxis was administered to a similar proportion of patients in both groups (DI: 19.0% of patients and 15.2% of cycles; DF: 20.9% of patients and 13.4% of cycles). Among these patients, two DI-treated patients and one DF-treated patient experienced febrile neutropenia or neutropenic infection despite G-CSF administration. No study treatment-related deaths were reported.
A total of 6 patients in the DI arm and 12 patients in the DF arm discontinued study treatment because of AEs. In all but two of these cases (both DF-treated patients), AEs were considered to be related to study treatment and included gastrointestinal toxicities (DI: three patients; DF: four patients), neurological toxicities (DI: 0 patient; DF: five patients), flu-like symptoms (DI: two patients; DF: two patients), infection (DI: one patient; DF: one patient) and pleural effusion (DI: one patient; DF: one patient).
Overall, nine (10.6%) patients died within 30 days of the last infusion of chemotherapy. None of these deaths was considered to be treatment-related (five patients were attributed to progressive disease and four patients to other causes).
Discussion
This randomised phase II study evaluated the efficacy and safety of two docetaxel-based regimens in advanced oesophago-gastric cancer. The observed ORR and median duration of response were similar for the DI and DF groups (37.5% vs 33.0% and 8.3 months vs 8.0 months, respectively). In addition, the survival outcomes were comparable between the treatment arms with a median OS of 8.6 months and 9.4 months with DI and DF respectively.
Similar to other docetaxel studies, haematological toxicity was the most frequently observed AE. Treatment with DI was associated with a higher incidence of febrile neutropenia and/or neutropenic infection, which occurred in 40.5% of patients compared with 18.6% of patients in the DF arm. Additionally, a lower incidence of grade 3–4 treatment-related diarrhoea, anorexia, nausea and lethargy occurred in the DF arm. As compared with other studies evaluating the DI regimen in advanced gastro-oesophageal cancer, in our study, a higher dose of irinotecan was used, which could explain the higher incidence of grade 3–4 events.
The efficacy and safety results for DF are consistent with those reported in previous phase II studies (Constenla et al, 2002; Thuss-Patience et al, 2005). These studies reported an ORR of 28% and 38%, and OS of 7.7 months and 9.5 months, respectively. Furthermore, DF regimen in these studies had a comparable safety profile to that observed in our study. Similarly, efficacy results for DI regimen were consistent with those reported in other studies, in which the response rate was 26% and 45.7%, respectively (Jatoi et al, 2002; Park et al, 2006). In general, the type of toxicity observed with the DI regimen was also comparable with those previously reported; however, the incidence of grade 3–4 events was higher in our study as compared with the studies by Park et al (2006) and Jatoi et al, 2002. We speculate that this is probably a result of the higher dose of irinotecan administered. It is of interest to note that the efficacy with DF in our study is comparable to doublet combination studies, which had substituted 5-FU with capecitabine or S-1 (ORR ranging between 39% and 52.1%; Giordano et al, 2006; Yamaguchi et al, 2006; Yoshida et al, 2006).
Platinum-containing triplet regimens are considered a standard of care for patients who have good performance status and whose tumour does not express human epidermal growth factor receptor-2 (HER-2). On the basis of the REAL-2 study, the EOX regimen has been widely adopted in the United Kingdom and in many centres across Europe, because of its efficacy, ease of administration and safety. The docetaxel-containing DCF regimen has been shown to provide a significant survival benefit over CF, reducing the risk of death by 23% (P=0.02), which resulted in a 2-year survival rate of 18% compared with 9% with CF alone (Van Cutsem et al, 2006). However, this benefit was associated with increased toxicity and has subsequently led to the recommendation to add docetaxel in patients with good KPS and organ function, using a modified regimen with either reduced dose, or as a weekly or biweekly regimen (Shah et al, 2010; Tebbutt et al, 2010; Van Cutsem et al, 2011b). Another potential docetaxel-containing regimen with promising results was recently reported in the phase II GATE study (Van Cutsem et al, 2011a). In this study, patients with advanced gastric cancer were randomised to receive TEF (docetaxel, oxaliplatin and infusional 5-FU q2w), TE (docetaxel and oxaliplatin q3w) and TEX (docetaxel, oxaliplatin q3w and capecitabine continuously). In the TEF regimen a biweekly lower dose of docetaxel was used, and this was associated with improved TTP, RR and OS, with a better safety profile compared with TE and TEX.
Current attempts to improve outcomes have included the combination of targeted agents with cytotoxic chemotherapy. With the successful addition of trastuzumab as a targeted agent to chemotherapy, present guidelines recommend testing of HER-2 in all patients who are candidates for first-line chemotherapy, and patients with a tumour overexpressing the HER-2 receptor should be treated with the cisplatin/fluoropyrimidine and trastuzumab combination (Bang et al, 2010; Van Cutsem et al, 2011b). However, a definitive role for other biological agents targeting mainly the EGFR, HER-2 or VEGF pathways is still evolving with the aim of identifying subgroups of patients who may potentially benefit from a particular approach.
Non-platinum-containing cytotoxic regimens have been previously tested in patients with advanced gastric cancer. Combinations such as irinotecan and 5-FU, and DF when compared with platinum-containing doublet or triplet regimens were associated with comparable efficacy (Dank et al, 2005; Thuss-Patience et al, 2005). Thus, because of toxicity concerns in patients who are deemed to be unsuitable for platinum-containing regimens, alternative doublet regimens may represent a viable treatment option.
Our trial shows comparable efficacy with both regimens as first-line treatment for patients with advanced oesophago-gastric cancer. We think irinotecan as monotherapy or in combination could be an appropriate second-line strategy on the basis of recent studies. In the second-line setting, a number of chemotherapy regimens, including a combination of irinotecan, continuous 5-FU and leucovorin (FOLFIRI; Assersohn et al, 2004; Wesolowski et al, 2009; Kim et al, 2010), and weekly docetaxel in combination with irinotecan have reported modest efficacy (Hawkes et al, 2011; OS benefit between 5 to 10.3 months). However, the toxicity with DI regimen at the doses used in this study is high and should be balanced against the potential benefit associated with it in the palliative setting (Sym et al, 2008). More recently, second-line chemotherapy with either docetaxel or irinotecan has demonstrated significant benefit in OS when compared with best supportive care in a randomised phase III trial (Thuss-Patience et al, 2011; Kang et al, 2012); however, it should be noted that only 20% of advanced gastric cancer patients may actually receive second-line therapy, and currently, no chemotherapy strategy has emerged as a clear winner in this setting. The activity and tolerability of DF regimen reported in our study provides additional evidence that this regimen could represent a viable first-line alternative for patients who cannot tolerate platinum-based chemotherapy or for patients who have specific contra-indications, such as renal and hearing impairment, and pre-existing peripheral neuropathy.
Change history
17 July 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ajani JA, Fodor MB, Tjulandin SA, Moiseyenko VM, Chao Y, Cabral Filho S, Majlis A, Assadourian S, Van Cutsem E (2005) Phase II multi-institutional randomized trial of docetaxel plus cisplatin with or without fluorouracil in patients with untreated, advanced gastric, or gastroesophageal adenocarcinoma. J Clin Oncol 23 (24): 5660–5667
Assersohn L, Brown G, Cunningham D, Ward C, Oates J, Waters JS, Hill ME, Norman AR (2004) Phase II study of irinotecan and 5-fluorouracil/leucovorin in patients with primary refractory or relapsed advanced oesophageal and gastric carcinoma. Ann Oncol 15 (1): 64–69
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Ruschoff J, Kang YK (2010) Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376 (9742): 687–697
Boku N, Yamamoto S, Fukuda H, Shirao K, Doi T, Sawaki A, Koizumi W, Saito H, Yamaguchi K, Takiuchi H, Nasu J, Ohtsu A (2009) Fluorouracil versus combination of irinotecan plus cisplatin versus S-1 in metastatic gastric cancer: a randomised phase 3 study. Lancet Oncol 10 (11): 1063–1069
Constenla M, Garcia-Arroyo R, Lorenzo I, Carrete N, Campos B, Palacios P (2002) Docetaxel, 5-fluorouracil, and leucovorin as treatment for advanced gastric cancer: results of a phase II study. Gastric Cancer 5 (3): 142–147
Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, Middleton G, Daniel F, Oates J, Norman AR (2008) Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med 358 (1): 36–46
Dank M, Zaluski J, Barone C, Valvere V, Peschel C, Wenczl M, Goker E, Risse M-L, Awad L, Bugat R (2005) Randomized phase 3 trial of irinotecan (CPT-11)+5FU/folinic acid (FA) vs CDDP+5FU in 1st-line advanced gastric cancer patients. ASCO Meeting Abstracts 23 (16_suppl): 4003
Giordano KF, Jatoi A, Stella PJ, Foster N, Tschetter LK, Alberts SR, Dakhil SR, Mailliard JA, Flynn PJ, Nikcevich DA (2006) Docetaxel and capecitabine in patients with metastatic adenocarcinoma of the stomach and gastroesophageal junction: a phase II study from the North Central Cancer Treatment Group. Ann Oncol 17 (4): 652–656
Hawkes E, Okines AF, Papamichael D, Rao S, Ashley S, Charalambous H, Koukouma A, Chau I, Cunningham D (2011) Docetaxel and irinotecan as second-line therapy for advanced oesophagogastric cancer. Eur J Cancer 47 (8): 1146–1151
Jatoi A, Tirona MT, Cha SS, Alberts SR, Rowland KM, Morton RF, Nair S, Kardinal CG, Stella PJ, Mailliard JA, Sargen D, Goldberg RM (2002) A phase II trial of docetaxel and CPT-11 in patients with metastatic adenocarcinoma of the esophagus, gastroesophageal junction, and gastric cardia. Int J Gastrointest Cancer 32 (2-3): 115–123
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61 (2): 69–90
Kang JH, Lee SI, Lim do H, Park KW, Oh SY, Kwon HC, Hwang IG, Lee SC, Nam E, Shin DB, Lee J, Park JO, Park YS, Lim HY, Kang WK, Park SH (2012) Salvage chemotherapy for pretreated gastric cancer: a randomized phase iii trial comparing chemotherapy plus best supportive care with best supportive care alone. J Clin Oncol 30 (13): 1513–1518
Kim SH, Lee GW, Go SI, Cho SH, Kim HJ, Kim HG, Kang JH (2010) A phase II study of irinotecan, continuous 5-fluorouracil, and leucovorin (FOLFIRI) combination chemotherapy for patients with recurrent or metastatic gastric cancer previously treated with a fluoropyrimidine-based regimen. Am J Clin Oncol 33 (6): 572–576
Kohne CH, Catane R, Klein B, Ducreux M, Thuss-Patience P, Niederle N, Gips M, Preusser P, Knuth A, Clemens M, Bugat R, Figer I, Shani A, Fages B, Di Betta D, Jacques C, Wilke HJ (2003) Irinotecan is active in chemonaive patients with metastatic gastric cancer: a phase II multicentric trial. Br J Cancer 89 (6): 997–1001
Miller AB, Hoogstraten B, Staquet M, Winkler A (1981) Reporting results of cancer treatment. Cancer 47 (1): 207–214
Park SR, Chun JH, Yu MS, Lee JH, Ryu KW, Choi IJ, Kim CG, Lee JS, Kim YW, Bae JM, Kim HK (2006) Phase II study of docetaxel and irinotecan combination chemotherapy in metastatic gastric carcinoma. Br J Cancer 94 (10): 1402–1406
Pozzo C, Barone C, Szanto J, Padi E, Peschel C, Bukki J, Gorbunova V, Valvere V, Zaluski J, Biakhov M, Zuber E, Jacques C, Bugat R (2004) Irinotecan in combination with 5-fluorouracil and folinic acid or with cisplatin in patients with advanced gastric or esophageal-gastric junction adenocarcinoma: results of a randomized phase II study. Ann Oncol 15 (12): 1773–1781
Shah MA, Shibata S, Stoller RG, Kemeny M, Ritch PS, Krishnamurthi SS, Su YB, Janjigian YY, Capanu M, Kelsen DP, MSKCC Gastric Cancer Consortium (2010) Random assignment multicenter phase II study of modified docetaxel, cisplatin, fluorouracil (mDCF) versus DCF with growth factor support (GCSF) in metastatic gastroesophageal adenocarcinoma (GE). ASCO Meeting Abstracts 28 (15_suppl): 4014
Sym SJ, Chang HM, Kang HJ, Lee SS, Ryu MH, Lee JL, Kim TW, Yook JH, Oh ST, Kim BS, Kang YK (2008) A phase II study of irinotecan and docetaxel combination chemotherapy for patients with previously treated metastatic or recurrent advanced gastric cancer. Cancer Chemother Pharmacol 63 (1): 1–8
Tebbutt NC, Cummins MM, Sourjina T, Strickland A, Van Hazel G, Ganju V, Gibbs D, Stockler M, Gebski V, Zalcberg J (2010) Randomised, non-comparative phase II study of weekly docetaxel with cisplatin and 5-fluorouracil or with capecitabine in oesophagogastric cancer: the AGITG ATTAX trial. Br J Cancer 102 (3): 475–481
Thuss-Patience PC, Kretzschmar A, Bichev D, Deist T, Hinke A, Breithaupt K, Dogan Y, Gebauer B, Schumacher G, Reichardt P (2011) Survival advantage for irinotecan versus best supportive care as second-line chemotherapy in gastric cancer--a randomised phase III study of the Arbeitsgemeinschaft Internistische Onkologie (AIO). Eur J Cancer 47 (15): 2306–2314
Thuss-Patience PC, Kretzschmar A, Repp M, Kingreen D, Hennesser D, Micheel S, Pink D, Scholz C, Dorken B, Reichardt P (2005) Docetaxel and continuous-infusion fluorouracil versus epirubicin, cisplatin, and fluorouracil for advanced gastric adenocarcinoma: a randomized phase II study. J Clin Oncol 23 (3): 494–501
Van Cutsem E, Boni C, Tabernero J, Massuti B, Richards DA, Prenen H, Steinberg I, Rougier P (2011a) Randomized phase II study (GATE study) of docetaxel plus oxaliplatin with or without fluorouracil or capecitabine in metastatic or locally recurrent gastric cancer. ASCO Meeting Abstracts 29 (15_suppl): 4018
Van Cutsem E, Dicato M, Geva R, Arber N, Bang Y, Benson A, Cervantes A, Diaz-Rubio E, Ducreux M, Glynne-Jones R, Grothey A, Haller D, Haustermans K, Kerr D, Nordlinger B, Marshall J, Minsky BD, Kang YK, Labianca R, Lordick F, Ohtsu A, Pavlidis N, Roth A, Rougier P, Schmoll HJ, Sobrero A, Tabernero J, Van de Velde C, Zalcberg J (2011b) The diagnosis and management of gastric cancer: expert discussion and recommendations from the 12th ESMO/World Congress on Gastrointestinal Cancer, Barcelona, 2010. Ann Oncol 22 (suppl 5): v1–v9
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, Rodrigues A, Fodor M, Chao Y, Voznyi E, Risse ML, Ajani JA (2006) Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol 24 (31): 4991–4997
Wagner AD, Unverzagt S, Grothe W, Kleber G, Grothey A, Haerting J, Fleig WE (2010) Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev 3: CD004064
Wesolowski R, Lee C, Kim R (2009) Is there a role for second-line chemotherapy in advanced gastric cancer? Lancet Oncol 10 (9): 903–912
Yamaguchi K, Shimamura T, Hyodo I, Koizumi W, Doi T, Narahara H, Komatsu Y, Kato T, Saitoh S, Akiya T, Munakata M, Miyata Y, Maeda Y, Takiuchi H, Nakano S, Esaki T, Kinjo F, Sakata Y (2006) Phase I/II study of docetaxel and S-1 in patients with advanced gastric cancer. Br J Cancer 94 (12): 1803–1808
Yoshida K, Ninomiya M, Takakura N, Hirabayashi N, Takiyama W, Sato Y, Todo S, Terashima M, Gotoh M, Sakamoto J, Nishiyama M (2006) Phase II study of docetaxel and S-1 combination therapy for advanced or recurrent gastric cancer. Clin Cancer Res 12 (11 part 1): 3402–3407
Acknowledgements
We would like to acknowledge the participating patients and their families, and in addition, to the co-investigators, surgeons and their research staff at recruiting centres. Dr Amitesh Roy and Professor David Cunningham acknowledge NHS funding from the NIHR Biomedical Research Centre and the Peter Stebbings Memorial Charity. This work was partially supported by Sanofi-Aventis Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Roy, A., Cunningham, D., Hawkins, R. et al. Docetaxel combined with irinotecan or 5-fluorouracil in patients with advanced oesophago-gastric cancer: a randomised phase II study. Br J Cancer 107, 435–441 (2012). https://doi.org/10.1038/bjc.2012.286
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.286
Keywords
This article is cited by
-
Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics
Clinical Pharmacokinetics (2018)
-
A network meta-analysis of the short-term efficacy of five chemotherapy regimens based on cisplatin and fluorouracil for esophagogastric junctional adenocarcinoma
Experimental & Molecular Medicine (2017)
-
Second- and third-line systemic therapy in patients with advanced esophagogastric cancer: a systematic review of the literature
Cancer and Metastasis Reviews (2016)
-
Shenqi Fuzheng Injection (参芪扶正注射液) for advanced gastric cancer: A systematic review of randomized controlled trials
Chinese Journal of Integrative Medicine (2015)
