
Abstract
Irinotecan and 5-fluorouracil (5-FU) are used to treat metastatic colorectal cancer. Irinotecan's active metabolite is inactivated by UDP-glucuronosyltransferase 1A1 (UGT1A1), which is deficient in Gilbert's syndrome. Irinotecan and metabolites are transported by P-glycoprotein, encoded by ABCB1. 5-FU targets folate metabolism through inhibition of thymidylate synthase (TYMS). Methylenetetrahydrofolate reductase (MTHFR) generates active folate necessary for haematopoiesis. We retrospectively genotyped 140 Swedish and Norwegian irinotecan and 5-FU-treated colorectal cancer patients from the Nordic VI clinical trial for selected variants of UGT1A1, ABCB1, TYMS and MTHFR. We found an increased risk of clinically relevant early toxicity in patients carrying the ABCB1 3435 T/T genotype, Odds ratio (OR)=3.79 (95% confidence interval (CI)=1.09–13.2), and in patients carrying the UGT1A1*28/*28 genotype, OR=4.43 (95% CI=1.30–15.2). Patients with UGT1A1*28/*28 had an especially high risk of neutropenia, OR=6.87 (95% CI=1.70–27.7). Patients who had reacted with toxicity during the first two cycles were in total treated with fewer cycles (P<0.001), and less often responded to treatment (P<0.001). Genetic variation in ABCB1 was associated with both early toxicity and lower response to treatment. Carriers of the ABCB1 1236T-2677T-3435T haplotype responded to treatment less frequently (43 vs 67%, P=0.027), and survived shorter time, OR=1.56 (95% CI=1.01–2.45).
Similar content being viewed by others

Introduction
Irinotecan is a camptothecin analogue used for the treatment of metastatic colorectal cancer, either alone or in combination with 5-fluorouracil (5-FU) and folic acid (FA). The use of irinotecan, also called CPT-11, is burdened by unpredictable severe toxicity.1 The main adverse reactions to irinotecan that frequently result in dose reduction or discontinuation of treatment are severe diarrhoea and neutropenia.2 Other common side effects are alopecia, nausea, vomiting and acute cholinergic-like syndrome.
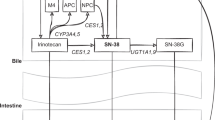
Irinotecan is activated to a cytotoxic metabolite, SN-38, that inhibits the nuclear enzyme topoisomerase 1 (TOP1), thereby blocking DNA replication. SN-38 is primarily inactivated in the liver by the bilirubin metabolizing enzyme UDP-glucuronosyltransferase 1A1.3 Mild hereditary deficiency of this enzyme leads to Gilbert's syndrome, which is characterized by intermittent hyperbilirubinaemia. During phase I trials of irinotecan, a number of patients with Gilbert's syndrome experienced severe toxicity, suggesting a genetic component of the reaction.4 Subsequently, the pharmacogenetics of irinotecan toxicity has been the focus of several studies.5, 6, 7
Gilbert's syndrome is commonly caused by homozygosity for a thymine-adenine (TA) insertion in the TATA element of the UDP-glucuronosyltransferase 1A1 (UGT1A1) promoter.8 This leads to seven TA repeats (the *28 allele) instead of six, but five and eight TA repeats also occur, although rarely in Caucasians. Inactivation of the irinotecan metabolite SN-38 is inversely correlated with the number of TA repeats, and is significantly reduced in individuals homozygous for UGT1A1*28.9, 10, 11 Thus, these patients are exposed to higher concentrations of the active metabolite, and may respond better to treatment.12
In an early study on the pharmacogenetics of irinotecan, it was observed that the UGT1A1*28 allele increased the risk of leukopenia and/or diarrhoea.13 Although subsequent studies have been partly conflicting,14 most of them find a correlation between UGT1A1*28 and haematologic toxicity such as neutropenia.9, 15, 16, 17, 18, 19, 20, 21 Medium-to-high doses of irinotecan increase the risk of haematologic toxicity in *28/*28 patients according to a meta-analysis.22 A few studies find a correlation between UGT1A1*28 and diarrhoea9, 17, 23, 24 or severe toxicity in general,24, 25 whereas others find no correlation with toxicity at all.26, 27
As UGT1A1*28 only explains part of the toxic reactions to irinotecan, interest has turned to other polymorphisms.6, 12 In East Asia, single nucleotide polymorphisms (SNPs) in UGT1A1 (for example *6, *27 and *60) are more prevalent than the TA insertion and these SNPs partly explain irinotecan toxicity.28, 29, 30 Genes of other glucuronidating enzymes (UGT1A7 and UGT1A9) also contribute to irinotecan toxicity according to a few studies.13, 26, 29
Transport proteins that excrete irinotecan and metabolites into the bile and urine have, to some extent, been investigated for association with response and adverse reactions to irinotecan.6, 31 P-glycoprotein, encoded by the ATP-binding cassette gene B1 (ABCB1 or MDR1), has rendered inconsistent results in irinotecan studies. There is some evidence that variant genotypes and haplotypes of ABCB1 increase the bioavailability of irinotecan and SN-38,32, 33 while other data point in the opposite direction.31, 34 In Korean's, wild-type ABCB1 was associated with neutropenia,34 but not in an American study.31 Neither was any difference in toxicity or disease-free survival observed in a French study of ABCB1 and irinotecan.21
The uracil analogue 5-FU is often given together with irinotecan for the treatment of metastatic colorectal cancer.2 Only a fraction of the administered 5-FU reaches its target cell and is transformed to active metabolites, whose mode of action is to both inhibit the enzyme thymidylate synthase (TYMS) and to be incorporated into RNA and DNA.35, 36 The 5′untranslated region of the TYMS gene contains a 28-base-pair tandem-repeat sequence in the promoter region, most commonly double (*2) and triple (*3) repeats.35 The *3 allele is associated with a two- to fourfold increased expression of TYMS compared with *2.37 In some studies, 5-FU-treated colorectal cancer patients carrying the *3 allele experience better response to treatment and less toxicity, but also the opposite has been seen.35, 38
Another enzyme implicated in the pharmacogenetics of 5-FU is methylenetetrahydrofolate reductase, encoded by MTHFR.35 This enzyme generates active folate necessary for normal haematopoiesis, and reduced activity has been associated with sensitivity to 5-FU.39 According to some studies, the MTHFR low activity variants 677T and 1298C predispose to severe bone marrow toxicity in patients treated with 5-FU.35, 38
In the randomized controlled phase III trial Nordic VI, we compared the effects of irinotecan with either bolus 5-FU/FA or bolus/infused 5FU/FA in patients with metastatic colorectal cancer.40, 41 These two schedules (FLIRI and Lv5FU2-IRI, see below) resulted in the same overall survival (OS) and progression-free survival (PFS, which was the primary endpoint), although response and toxicity were slightly more favourable with the Lv5FU2-IRI schedule. In this explorative study, we retrospectively genotyped patients from three Nordic VI study sites in Sweden and Norway for candidate genes in the irinotecan and 5-FU pathways and tested them for association with toxicity, response and survival.
Materials and methods
Patients
From June 2001 to March 2004, 182 mainly Caucasian patients with non-resectable metastatic histologically confirmed stage IV colorectal adenocarcinoma were included in Uppsala and Stockholm (Sweden) and Bergen (Norway) for the Nordic VI clinical trial.41 Results at these three sites were comparable to results from the 567 patients included in the entire Nordic VI clinical trial. DNA was retrospectively obtained from 77% of the patients from Uppsala, Stockholm and Bergen. Blood for DNA extraction was collected from 41 surviving patients and DNA was isolated from tissue blocks containing normal intestinal cells from 99 patients (+6 that were duplicates with 6 blood samples). These 140 patients were representative for the 182 patients from the three sites concerning pretreatment characteristics and treatment outcome (data not illustrated).
No prior chemotherapy other than adjuvant 5-FU-based chemotherapy completed at least 6 months before the study entry was allowed. All patients should have measurable disease according to the response evaluation criteria in solid tumours (RECIST),42 a WHO performance status of 0 to 2, adequate laboratory values, and be aged between 19 and 76 years. Elevated plasma bilirubin, ⩾1.25 × the upper normal limit (UNL) or ⩾1.5 × UNL if liver metastases, was an exclusion criterion. The study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. The clinical trial and this substudy were approved by the Ethical committees at each site/country. All patients provided informed consent for the clinical trial, and patients who donated blood provided a separate consent.
Treatment
Patients were randomly assigned to receive irinotecan according to the FLIRI regimen or the Lv5FU2-IRI regimen.41 The FLIRI regimen consisted of irinotecan (Campto, Sanofi-Aventis, Paris, France) 180 mg m−2 (initially 210 mg m−2, see Glimelius et al.40, 41) as a 60 mi intravenous (i.v.) infusion on day 1, followed immediately by 5-FU 500 mg m−2 as a bolus injection, followed by FA 60 mg m−2 i.v. bolus 30–40 min later. The Lv5FU2-IRI regimen consisted of irinotecan 180 mg m−2 i.v. infusion on day 1, followed by 5-FU bolus 400 mg m−2 and FA 200 mg m−2 as a 2 h infusion, and lastly a 22-h infusion of 5-FU 600 mg m−2. With both regimens, the 5-FU/FA administrations were repeated on day 2. All treatments were repeated every 2 weeks until disease progression or unacceptable toxicity. If toxicity grade 2 (except alopecia) remained after 2 weeks, the next cycle was either delayed for up to 2 weeks until the toxicity returned to grade 1 or the dose was reduced by 20%. In case of grade 3–4 toxicity, irinotecan and 5-FU doses were reduced by 20%. A second 20% reduction was allowed if toxicity recurred at a following cycle. If doses were reduced, they were not increased at subsequent cycles.
Concomitant medication included subcutaneous atropine (0.25 mg) as prophylaxis or treatment for severe cholinergic symptoms. As soon as the first liquid stool occurred, oral loperamide was administered (2 mg) every 2 h for at least 12 h. If diarrhoea persisted >48 h or in case of severe diarrhoea or diarrhoea associated with vomiting, fever or severe neutropenia, the antibiotic ciprofloxacin was administered. Patients with febrile neutropenia were hospitalized to receive i.v. antibiotics. Colony stimulating factor was not used.
Toxicity and response evaluation
Toxicity recorded in the case record forms was evaluated according to the National Cancer Institute common toxicity criteria, version 2 (http://ctep.cancer.gov/reporting/ctc.html).
-
Clinically relevant early toxicity (outcome A) was registered after one or two treatment cycles. It was either grade 3–4 toxicity except alopecia or ⩾20% dose reduction of the following cycle or toxicity leading to >5 days delay or discontinuation of treatment. Clinically relevant early toxicity was the primary endpoint.
-
Overall toxicity (outcome B) was any grade 3–4 toxicity except alopecia during the entire course of therapy.
Tumour response was assessed according to RECIST.42 Assessed outcomes were complete response (CR), partial response (PR), stable disease (SD) and progressive disease (PD). PFS was defined as the time from randomization to the date of progression or death. OS was the time to death.
DNA extraction
DNA from whole blood was extracted using the QIAamp DNA Blood Mini Kit (QIAGEN, Hinden, Germany). When whole blood was not available, DNA was extracted from paraffin-embedded tissue blocks containing normal intestine taken either far (10–15 cm) from the primary tumour or microdissected if in proximity to the tumour. Biopsies from Sweden were extracted using the boiling method according to Cao et al.43 Norwegian biopsies were extracted using the QIAamp DNA Mini Kit protocol for paraffin-embedded tissue (QIAGEN). Consistency of DNA quality was assessed by extracting DNA from both blood and intestine for six patients.
Genotyping
Analysis of TA-repeat variability in the promoter region of UGT1A1 (rs8175347) was performed using DNA fragment analysis. Primer sequences were according to Monaghan et al.,8 with addition of fluorescent labelling of the forward primer with D4 WellRed (Sigma-Aldrich, MO, USA). The amplification reaction mixture (20 μl) contained 50 ng DNA, 0.2 mmol l−1 each of deoxynucleoside triphoshate, 2,5 mmol l−1 MgCl2, 50 nmol l−1 of each primer, 0.4 U GoTaq Flexi DNA Polymerase (Promega, WI, USA), 1 × Colourless GoTaq Flexi Buffer (Promega) and 0.12 g l−1 Bovine serum albumin (Sigma-Aldrich). Polymerase chain reaction (PCR) was performed using the thermal cycler (M.J. Research, MN, USA) with the following temperatures: 95 °C 5 min, 30 × (95 °C 30 s, 58 °C 40 s, 72 °C 40 s), 72 °C 10 min, followed by an 8 °C cooling step. The amplified products were separated by capillary electrophoresis using the CEQ 8000 Genetic Analysis System (Beckman Coulter, CA, USA). A size marker was labelled with another fluorescent colour according to the CEQ 8000 manual. Results were analysed using the Beckman Coulter Fragment analysis software. Allelic discrimination of the upstream UGT1A1 -3279T>G (rs4124874) polymorphism named *60 was performed using the ABI 7500 FAST real-Time PCR system (Applied Biosystems, CA, USA) and the TaqMan Drug Metabolism Genotyping Assay kit containing primers and probes (C__1432134_10, Applied Biosystems).
Genotyping of three ABCB1 polymorphisms and two MTHFR polymorphisms was performed using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Genotyping of ABCB1 c.2677G>T/A (rs2032582), which leads to a change in amino acid 893 from serine to alanine or threonine, was performed according to Saito et al.44 Allelic discrimination of the synonymous ABCB1 polymorphisms c.1236C>T (rs1128503) and c.3435C>T (rs1045642) was performed using TaqMan SNP Genotyping Assay kits containing primers and probes (C__7586662_10 and C__7586657_1, Applied Biosystems). Genotyping of the c.677C>T locus of MTHFR (rs1801133), which leads to a change from amino acid alanine to valine at position 222, was performed using a TaqMan SNP Genotyping Assay kit containing primers and probes (C__1202883_20, Applied Biosystems). The c.1298A>C locus of MTHFR (rs1801131), which leads to a substitution from glutamic acid to alanine at residue 429, was genotyped according to Robien et al.45
The 28 bp microsatellite in the 5′ regulatory region of TYMS (rs45445694) was genotyped by PCR amplification according to Horie et al.37 To discriminate between two (*2) and three (*3) repeat sequences, PCR fragments were separated by electrophoresis on 3% agarose gels.
Statistical analyses
All genotypes were checked for deviation from Hardy–Weinberg equilibrium. Statistical analyses were performed using the statistical program package R.46 Associations between genes and toxicity/response were studied through contingency tables. Tests of homogeneity were performed by use of Fisher's exact test. Odds ratios (ORs) were calculated with logistic regression. Hazard ratios (HRs) were analysed by means of Cox regression. ABCB1 haplotypes were inferred using PHASE, a software for haplotype reconstruction, and recombination rate estimation from population data.47
Results
Patients and treatment outcomes
Patient characteristics of the 140 genotyped patients are given in Table 1. The two Nordic VI treatment groups FLIRI and Lv5FU2-IRI were well balanced at baseline. As in the entire Nordic VI study, PFS and OS did not differ significantly between the treatment groups, but the response to treatment was better in the Lv5FU2-Iri group (58 vs 39% responders, P=0.03). Toxicity did not differ according to randomization arm. The distribution of number of cycles given is shown in Table 2. The median number of cycles was 12 in both groups. The distribution of different genotypes is shown in Table 3. The concordance between the six duplicate DNAs from blood and intestine that were analysed for UGT1A1, ABCB1 and MTHFR was complete. Genotype distribution did not differ significantly between the two randomization arms. Thus, corrections for randomization did not change any of the results of the statistical analyses. No significant deviation from Hardy–Weinberg equilibrium was observed. DNA from paraffin-embedded tissues was frequently too degraded to be genotyped with the TYMS method that requires large DNA fragments. The number of patients successfully genotyped for TYMS was therefore fewer than for the other genes.
Clinically relevant early toxicity
Clinically relevant early toxicity (outcome A) defined as grade 3–4 toxicity except alopecia or ⩾20% dose reduction of the following cycle or toxicity leading to >5 days delay or discontinuation of treatment after the first one–two cycles, was recorded in 35 patients (25%). Grade 3–4 toxicity (except alopecia) was noted in 16 patients, the dose was reduced for 25 patients, and treatment was delayed or discontinued in 10 patients. Eighteen of these patients had neutropenia, 10 had diarrhoea and 21 had other types of toxicity. Patients who experienced clinically relevant early toxicity in total received significantly fewer treatment cycles (P<0.001, Table 2). Among patients who received 1–8 cycles, the OR for outcome A was 9.37 (95% confidence interval (CI)=2.92–30.1) compared with patients who received >13 treatment cycles.
Correlations between genetic variability in UGT1A1, ABCB1, TYMS and MTHFR and clinically relevant early toxicity are shown in Figure 1a and Table 3. The risk of outcome A was significantly increased in patients carrying UGT1A1*28/*28 compared with patients without *28, OR=4.43 (95% CI=1.30–15.2). The individual outcome A toxicity that reached statistical significance in *28 homozygotes was neutropenia, OR=6.87 (95% CI=1.70–27.7, Table 4). A high risk of outcome A toxicity other than neutropenia and diarrhoea was also observed in *28 homozygotes, OR=3.87 (95% CI=1.05–14.2).

Odds ratios with 95% confidence limits for clinically relevant toxicity by genotype. (a) Outcome A: Grade 3–4 toxicity after cycle 1 or 2, delay or dose reductions of cycle 2 or 3. (b) Outcome B: Worst grade 3–4 toxicity, all cycles.
The risk of clinically relevant early toxicity was significantly increased in carriers of the ABCB1 3435 T/T genotype, OR=3.79 (95% CI=1.09–13.2, Table 3). Outcome A was experienced by 40% of the patients homozygous for the ABCB1 1236T–2677T–3435T haplotype, but only by 8% of the homozygotes for the ABCB1 1236C-2677G-3435C haplotype (P=0.036). The incidence of outcome A was decreased in MTHFR 677 heterozygotes compared with C/C and T/T (P=0.005, Table 3). Outcome A did not differ significantly between different MTHFR 677–1298 haplotypes (P=0.069). Outcome A was more frequent in patients heterozygous for TYMS*2/*3, OR=3.40 (95% CI=1.03–11.3, Table 3).
The risk of toxicity was increased in patients with low haemoglobin at baseline (<130 g l−1 in men, <120 g l−1 in women), P=0.015, OR=2.76 (95% CI=1.18–6.60). We did not detect any significant differences in outcome A according to age, gender, baseline alkaline phosphatase or bilirubin levels, WHO performance status, number of metastatic sites or whether the primary tumour was removed or not (data not shown).
Overall toxicity
Overall toxicity (outcome B) defined as any grade 3–4 toxicity except alopecia during the entire course of therapy was seen in 65 patients (46%), most commonly diarrhoea and neutropenia. In this Nordic VI substudy, neutropenia grade 3–4 was equally distributed among patients randomized to FLIRI and Lv5FU2-Iri (7% in both groups). There was no correlation between outcome B and the number of treatment cycles (Table 2). Nor did we detect any correlation between outcome B and the explored genetic variants (Figure 1b).
Response
Sixty-seven patients (48%) responded completely (CR) or partially (PR) to the treatment, whereas 73 patients (52%) had SD or PD. Patients who experienced clinically relevant early toxicity (outcome A) in total received fewer treatment cycles than others (P<0.001, Table 2). Naturally, patients who received fewer treatment cycles responded less well to treatment. Non-responders (SD+PD) had been given a median of 8 cycles, whereas responders (CR+PR) had been given a median of 14 cycles (mean difference 5.7 cycles, P<0.001). Of patients with outcome A, 28 (80%) became non-responders and 7 (20%) became responders, whereas 45 (43%) without outcome A became non-responders and 60 (57%) became responders (P<0.001). The OR for non-response in patients with outcome A was 5.33 (95% CI=2.14–13.3). In retrospect, outcome A had occurred among 38% of the non-responders and 10% of the responders (P<0.001). During the entire treatment, non-responders experienced toxicity ⩾grade 3 (outcome B) more frequently than responders (50 vs 39%, P=0.05). Nearly all patients experienced toxicity ⩾grade 2 (other than alopecia) at some time during treatment, and this distribution was equal between non-responders and responders (88 vs 87%, P=0.90).
Patients who carried variant alleles of ABCB1 tended to respond less well to treatment (Figure 2a). Carriers of at least one T/T genotype of ABCB1 1236, 2677 or 3435 were less likely to respond to treatment, OR=0.32 (95% CI=0.11–0.91). A post hoc analysis showed that fewer patients with at least one ABCB1 1236T-2677T-3435T haplotype responded to treatment compared with others (43 vs 67%, P=0.027). No other genes were associated with response. There was no difference in the number of treatment cycles between genotypes.

(a) Odds ratios with 95% confidence limits for non-response (SD+PD) by genotype. (b) Hazard ratios with 95% confidence limits for the risk of progression or death by genotype. (c) Hazard ratios with 95% confidence limits for the risk death by genotype.
OS and PFS
The mean follow-up time was 20 months. PFS was reduced in ABCB1 2677 heterozygotes, HR=1.65 (95% CI=1.08–2.53) and so was OS, HR=1.81 (95% CI=1.12–2.92, Figures 3b and c). OS was decreased in carriers of at least one T/T genotype of ABCB1 1236, 2677 or 3435 compared with others, HR=1.99 (95% CI=1.07–3.70). A post hoc analysis showed that OS was decreased in patients with at least one ABCB1 1236T-2677T-3435T haplotype compared with others, P=0.044, OR=1.56 (95% CI=1.01–2.45) Figure 3 illustrates that the probability of survival for each of the ABCB1 1236, 2677 and 3435 genotypes. No other genes correlated with PFS or OS (Figures 2b and c).

Kaplan–Meier survival curves showing the probability of survival for different ABCB1 genotypes. (a) ABCB1 1236C>T. (b) ABCB1 2677G>T/A. (c) ABCB1 3435C>T.
Discussion
Irinotecan is an important medication for the treatment of metastatic colorectal cancer, but its use is impeded by a comparably high incidence of unpredictable severe toxicity.1 A number of studies have attempted to explain this toxicity by analysing candidate genes in the irinotecan pathways.38 In an exploratory study, we tested whether variability in selected genes was associated with toxicity, response and survival in a clinical trial of irinotecan and 5-FU. Patients with metastatic colorectal cancer were recruited for the trial between 2001 and 2004, and genotyping of UGT1A1, ABCB1, TYMS and MTHFR was performed retrospectively.
In agreement with most earlier studies,22 we found an increased risk of early irinotecan-induced toxicity, and especially neutropenia, in patients homozygous for the UGT1A1 *28 variant.8 Patients carrying ABCB1 variant genotypes and haplotypes tended to have more toxic reactions, to respond less well to treatment and to die earlier. Bearing previous conflicting results concerning ABCB1 and irinotecan in mind,21, 31, 32, 33, 34 these findings need to be confirmed in larger studies. Other associations were weak, apart from the puzzling low toxicity in carriers of MTHFR 677 C/T, which would suggest a heterozygous advantage.
The most convincing results in our and earlier studies of the pharmacogenetics of irinotecan relate to homozygosity for UGT1A1*28, which is associated with risk of haematologic toxicity in patients treated with medium-to-high doses.22 This research has led the US Food and Drug Administration to change the label for irinotecan to encourage lower initial doses in patients homozygous for UGT1A1*28 (http://www.fda.gov/medwatch/SAFETY/2005). In the Nordic VI study, patients who reacted with early irinotecan toxicity at a certain level had their subsequent doses delayed or reduced. Rules for dose reductions and delays were defined in the clinical trial protocol and were similar to those in other first-line studies of metastatic colorectal cancer. These dose adjustments correct for a low clearance of irinotecan and probably explain why overall toxicity and response did not differ between UGT1A1 genotypes.
The association between clinically relevant early toxicity, fewer treatment cycles and worse prognosis found in this study indicates that it is important to avoid severe initial toxicity. The magnitude of this correlation surprised us, and emphasizes the importance of genotyping UGT1A1 to enable dose adjustments before starting irinotecan therapy. There is no consensus concerning dose adjustments in patients homozygous for UGT1A1*28/*28, but a reduction by 20–40% seems appropriate, with a rapid dose escalation if adverse effects above grade 1 are not seen. A similar dose escalation was tested in a randomized phase II trial using single-drug irinotecan as third-line treatment, albeit without knowledge of UGT1A1 genotype.48 An initial dose of 250 mg m−2 was given every third week, as opposed to 350 mg m−2, with an escalation to 350–500 mg m−2 in the absence of toxicity above grade 1. This lower dose schedule diminished early severe toxicity, but patients without the UGT1A1*28/*28 genotype may risk suboptimal dosing initially and lower response.
Severe irinotecan toxicity was observed in a few patients with Gilbert's syndrome already in 1997.4 With the intention to avoid participants with Gilbert's syndrome, patients with elevated bilirubin were excluded from the Nordic VI trial. In our study, 13 (9%) of the patients had UGT1A1*28/*28, which corresponds with the 7–10% background incidence of Gilbert's syndrome in Norway and Sweden.49, 50 Evidently, Gilbert's syndrome was not avoided by excluding patients with elevated baseline bilirubin in this study, nor was there any association between baseline bilirubin and toxicity. The only clinical baseline laboratory value that predicted toxicity was low haemoglobin. The exclusion of patients with markedly pathological laboratory values or poor performance status from the Nordic VI trial may explain why no other correlation was found.51
We recognize limitations of our study. First, the sample size is too small to drive final conclusions. Second, we did not correct for multiple comparisons, and all results should be considered as exploratory. Third, there was a non-random pattern of missing genotypes of TYMS, because of a low success rate when genotyping DNA from biopsies. This lead to a bias for patients that were alive and could provide blood samples, in essence, responders. Considering these limitations, it is not possible to make any firm conclusions from this study alone. We therefore look forward to participating in the International Severe Irinotecan Neutropenia Consortium (http://www.pharmgkb.org/views/project.jsp?pId=69). Similarly to the International Warfarin Pharmacogenetics Consortium,52 this consortium will pool data on patients from several studies into a single repository, to increase sample sizes for investigating the genetic bases of outcomes.
In conclusion, we found a novel association between ABCB1 variant genotypes and early toxicity, less response and shorter survival. We confirmed an increased risk of early irinotecan toxicity, particularly neutropenia, in colorectal cancer patients carrying UGT1A1*28/*28. The association between early toxicity, fewer treatment cycles and worse prognosis indicates that it is important to avoid severe initial toxicity. However, to find out whether it is beneficial to follow the FDA recommendation to adjust irinotecan doses according to UGT1A1 genotype, a prospective trial needs to be conducted, with a regular dosing arm and an intervention arm.

Abbreviations
- ABCB1:
-
ATP-binding cassette gene B1
- CI:
-
confidence interval
- CPT-11:
-
7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxy-camptothecin
- CR:
-
complete response
- FA:
-
folic acid
- FLIRI:
-
bolus 5-FU/FA schedule plus irinotecan
- 5-FU:
-
5-fluorouracil
- HR:
-
hazard ratio
- i.v.:
-
intravenous
- Lv5FU2-IRI:
-
bolus/infused de Gramont 5FU/FA schedule plus irinotecan
- MDR1:
-
alternate name for ATP-binding cassette gene B1
- MTHFR :
-
methylenetetrahydrofolate reductase gene
- OR:
-
odds ratio
- OS:
-
overall survival
- PD:
-
progressive disease
- PFS:
-
progression-free survival
- PR:
-
partial response
- RECIST:
-
response evaluation criteria in solid tumours
- Q1:
-
1st quartile
- Q3:
-
3rd quartile
- SD:
-
stable disease
- SN-38:
-
7-ethyl-10-hydroxy-camptothecin
- SNP:
-
single nucleotide polymorphism
- TOP1:
-
topoisomerase 1
- TYMS :
-
thymidylate synthase (TS) gene
- UGT1A1 :
-
the UDP-glucuronosyltransferase 1A1 gene
- UGT1A7 :
-
the UDP-glucuronosyltransferase 1A7 gene
- UGT1A9 :
-
the UDP-glucuronosyltransferase 1A9 gene
- UNL:
-
upper normal limit
- Wt:
-
wild type
References
Ratain MJ . Irinotecan dosing: does the CPT in CPT-11 stand for “Can’t Predict Toxicity”? J Clin Oncol 2002; 20: 7–8.
Glimelius B . Benefit-risk assessment of irinotecan in advanced colorectal cancer. Drug Saf 2005; 28: 417–433.
Iyer L, Hall D, Das S, Mortell MA, Ramirez J, Kim S et al. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther 1999; 65: 576–582.
Wasserman E, Myara A, Lokiec F, Goldwasser F, Trivin F, Mahjoubi M et al. Severe CPT-11 toxicity in patients with Gilbert's syndrome: two case reports. Ann Oncol 1997; 8: 1049–1051.
Innocenti F, Ratain MJ . “Irinogenetics” and UGT1A: from genotypes to haplotypes. Clin Pharmacol Ther 2004; 75: 495–500.
Kim TW, Innocenti F . Insights, challenges, and future directions in irinogenetics. Ther Drug Monit 2007; 29: 265–270.
Kweekel D, Guchelaar HJ, Gelderblom H . Clinical and pharmacogenetic factors associated with irinotecan toxicity. Cancer Treat Rev 2008; 34: 656–669.
Monaghan G, Ryan M, Seddon R, Hume R, Burchell B . Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet 1996; 347: 578–581.
Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2002; 2: 43–47.
Sai K, Saeki M, Saito Y, Ozawa S, Katori N, Jinno H et al. UGT1A1 haplotypes associated with reduced glucuronidation and increased serum bilirubin in irinotecan-administered Japanese patients with cancer. Clin Pharmacol Ther 2004; 75: 501–515.
Paoluzzi L, Singh AS, Price DK, Danesi R, Mathijssen RH, Verweij J et al. Influence of genetic variants in UGT1A1 and UGT1A9 on the in vivo glucuronidation of SN-38. J Clin Pharmacol 2004; 44: 854–860.
Cecchin E, Innocenti F, D’Andrea M, Corona G, De Mattia E, Biason P et al. Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J Clin Oncol 2009; 27: 2457–2465.
Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H et al. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res 2000; 60: 6921–6926.
Deeken JF, Slack R, Marshall JL . Irinotecan and uridine diphosphate glucuronosyltransferase 1A1 pharmacogenetics: to test or not to test, that is the question. Cancer 2008; 113: 1502–1510.
Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol 2004; 22: 1382–1388.
Ruzzo A, Graziano F, Loupakis F, Santini D, Catalano V, Bisonni R et al. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFIRI chemotherapy. Pharmacogenomics J 2008; 8: 278–288.
Liu CY, Chen PM, Chiou TJ, Liu JH, Lin JK, Lin TC et al. UGT1A1*28 polymorphism predicts irinotecan-induced severe toxicities without affecting treatment outcome and survival in patients with metastatic colorectal carcinoma. Cancer 2008; 112: 1932–1940.
Kweekel DM, Gelderblom H, Van der Straaten T, Antonini NF, Punt CJ, Guchelaar HJ . UGT1A1*28 genotype and irinotecan dosage in patients with metastatic colorectal cancer: a Dutch Colorectal Cancer Group study. Br J Cancer 2008; 99: 275–282.
Toffoli G, Cecchin E, Corona G, Russo A, Buonadonna A, D’Andrea M et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol 2006; 24: 3061–3068.
Rouits E, Boisdron-Celle M, Dumont A, Guerin O, Morel A, Gamelin E . Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin Cancer Res 2004; 10: 5151–5159.
Cote JF, Kirzin S, Kramar A, Mosnier JF, Diebold MD, Soubeyran I et al. UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin Cancer Res 2007; 13: 3269–3275.
Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL . UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 2007; 99: 1290–1295.
Marcuello E, Altes A, Menoyo A, Del Rio E, Gomez-Pardo M, Baiget M . UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br J Cancer 2004; 91: 678–682.
Massacesi C, Terrazzino S, Marcucci F, Rocchi MB, Lippe P, Bisonni R et al. Uridine diphosphate glucuronosyl transferase 1A1 promoter polymorphism predicts the risk of gastrointestinal toxicity and fatigue induced by irinotecan-based chemotherapy. Cancer 2006; 106: 1007–1016.
Fakih MG, Ross ME, Starostik P . Increased frequency of uridine diphosphate glucuronosyltransferase 1A1 7/7 in patients experiencing severe irinotecan-induced toxicities. Clin Colorectal Cancer 2007; 6: 583–587.
Carlini LE, Meropol NJ, Bever J, Andria ML, Hill T, Gold P et al. UGT1A7 and UGT1A9 polymorphisms predict response and toxicity in colorectal cancer patients treated with capecitabine/irinotecan. Clin Cancer Res 2005; 11: 1226–1236.
Romero RZ, Morales R, Garcia F, Huarriz M, Bandres E, De la Haba J et al. Potential application of GSTT1-null genotype in predicting toxicity associated to 5-fluouracil irinotecan and leucovorin regimen in advanced stage colorectal cancer patients. Oncol Rep 2006; 16: 497–503.
Ando M, Hasegawa Y, Ando Y . Pharmacogenetics of irinotecan: a promoter polymorphism of UGT1A1 gene and severe adverse reactions to irinotecan. Invest New Drugs 2005; 23: 539–545.
Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE et al. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol 2006; 24: 2237–2244.
Saito Y, Sai K, Maekawa K, Kaniwa N, Shirao K, Hamaguchi T et al. Close association of UGT1A9 IVS1+399C>T with UGT1A1*28, *6, or *60 haplotype and its apparent influence on 7-ethyl-10-hydroxycamptothecin (SN-38) glucuronidation in Japanese. Drug Metab Dispos 2009; 37: 272–276.
Innocenti F, Kroetz DL, Schuetz E, Dolan ME, Ramirez J, Relling M et al. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol 2009; 27: 2604–2614.
Mathijssen RH, Marsh S, Karlsson MO, Xie R, Baker SD, Verweij J et al. Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin Cancer Res 2003; 9: 3246–3253.
Sai K, Kaniwa N, Itoda M, Saito Y, Hasegawa R, Komamura K et al. Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecan. Pharmacogenetics 2003; 13: 741–757.
Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY et al. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer 2007; 110: 138–147.
Robien K, Boynton A, Ulrich CM . Pharmacogenetics of folate-related drug targets in cancer treatment. Pharmacogenomics 2005; 6: 673–689.
Iyer L, Ratain MJ . 5-fluorouracil pharmacokinetics: causes for variability and strategies for modulation in cancer chemotherapy. Cancer Invest 1999; 17: 494–506.
Horie N, Aiba H, Oguro K, Hojo H, Takeishi K . Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5′-terminal regulatory region of the human gene for thymidylate synthase. Cell Struct Funct 1995; 20: 191–197.
Funke S, Brenner H, Chang-Claude J . Pharmacogenetics in colorectal cancer: a systematic review. Pharmacogenomics 2008; 9: 1079–1099.
Jakobsen A, Nielsen JN, Gyldenkerne N, Lindeberg J . Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphism in normal tissue as predictors of fluorouracil sensitivity. J Clin Oncol 2005; 23: 1365–1369.
Glimelius B, Ristamaki R, Kjaer M, Pfeiffer P, Skovsgaard T, Tveit KM et al. Irinotecan combined with bolus 5-fluorouracil and folinic acid Nordic schedule as first-line therapy in advanced colorectal cancer. Ann Oncol 2002; 13: 1868–1873.
Glimelius B, Sorbye H, Balteskard L, Bystrom P, Pfeiffer P, Tveit KM et al. A randomized phase III multicenter trial comparing irinotecan in combination with the Nordic bolus 5-FU and folinic acid schedule or the bolus/infused de Gramont schedule (LV5FU2) in patients with metastatic colorectal cancer. Ann Oncol 2008; 19: 909–914.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–216.
Cao W, Hashibe M, Rao JY, Morgenstern H, Zhang ZF . Comparison of methods for DNA extraction from paraffin-embedded tissues and buccal cells. Cancer Detect Prev 2003; 27: 397–404.
Saito K, Miyake S, Moriya H, Yamazaki M, Itoh F, Imai K et al. Detection of the four sequence variations of MDR1 gene using TaqMan MGB probe based real-time PCR and haplotype analysis in healthy Japanese subjects. Clin Biochem 2003; 36: 511–518.
Robien K, Ulrich CM, Bigler J, Yasui Y, Gooley T, Bruemmer B et al. Methylenetetrahydrofolate reductase genotype affects risk of relapse after hematopoietic cell transplantation for chronic myelogenous leukemia. Clin Cancer Res 2004; 10: 7592–7598.
Ihaka R, Gentleman R . R: A language for data analysis and graphics. J Comput Graph Stat 1996; 5: 299–314.
Stephens M, Donnelly P . A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 2003; 73: 1162–1169.
Van Cutsem E, Dirix L, Van Laethem JL, Van Belle S, Borner M, Gonzalez Baron M et al. Optimisation of irinotecan dose in the treatment of patients with metastatic colorectal cancer after 5-FU failure: results from a multinational, randomised phase II study. Br J Cancer 2005; 92: 1055–1062.
Holthe M, Klepstad P, Zahlsen K, Borchgrevink PC, Hagen L, Dale O et al. Morphine glucuronide-to-morphine plasma ratios are unaffected by the UGT2B7 H268Y and UGT1A1*28 polymorphisms in cancer patients on chronic morphine therapy. Eur J Clin Pharmacol 2002; 58: 353–356.
Mercke Odeberg J, Andrade J, Holmberg K, Hoglund P, Malmqvist U, Odeberg J . UGT1A polymorphisms in a Swedish cohort and a human diversity panel, and the relation to bilirubin plasma levels in males and females. Eur J Clin Pharmacol 2006; 62: 829–837.
Sorbye H, Kohne CH, Sargent DJ, Glimelius B . Patient characteristics and stratification in medical treatment studies for metastatic colorectal cancer: a proposal for standardization of patient characteristic reporting and stratification. Ann Oncol 2007; 18: 1666–1672.
International warfarin pharmacogenetics consortium. Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360: 753–764.
Acknowledgements
We gratefully acknowledge the medical staff and participants of the study for their valuable contribution. This work was supported by the Swedish Cancer Society, the Norwegian Cancer Society, the Swedish Science Council (Medicine), the Swedish Society of Medicine, the Stockholm Cancer Society, Uppsala University Hospital's Cancer fund and Lion's Cancer fund, Uppsala University's Clinical Research Support (ALF) and Biomedical analyst project. The NORDIC VI clinical trial was partly supported by research funding from Aventis Pharma AB, later Sanofi-Aventis Pharma AB to BG and HS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
B Glimelius received research funding from Merck, and H Sørbye is on the Sanofi-Aventis Advisary Board, has Honoraria from Roche, Amgen and Novartis, and received research funding from Schering-Plough. The authors have no other conflict of interest. The funding organizations and sponsors did not influence the design and conduct of the study; data collection, analysis and interpretation of the data; preparation or approval of the manuscript.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Glimelius, B., Garmo, H., Berglund, Å. et al. Prediction of irinotecan and 5-fluorouracil toxicity and response in patients with advanced colorectal cancer. Pharmacogenomics J 11, 61–71 (2011). https://doi.org/10.1038/tpj.2010.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tpj.2010.10
Keywords
This article is cited by
-
Irinotecan-Induced Toxicity: A Pharmacogenetic Study Beyond UGT1A1
Clinical Pharmacokinetics (2023)
-
Supportive oncology care at home interventions: protocols for clinical trials to shift the paradigm of care for patients with cancer
BMC Cancer (2022)
-
Integration of DNA sequencing with population pharmacokinetics to improve the prediction of irinotecan exposure in cancer patients
British Journal of Cancer (2022)
-
Evaluation of pharmacogenomics and hepatic nuclear imaging–related covariates by population pharmacokinetic models of irinotecan and its metabolites
European Journal of Clinical Pharmacology (2022)
-
Pharmacogenomics and functional imaging to predict irinotecan pharmacokinetics and pharmacodynamics: the predict IR study
Cancer Chemotherapy and Pharmacology (2021)
