
Abstract
We developed a novel integrative genomic tool called GRANITE (Genetic Regulatory Analysis of Networks Investigational Tool Environment) that can effectively analyze large complex data sets to generate interactive networks. GRANITE is an open-source tool and invaluable resource for a variety of genomic fields. Although our analysis is confined to static expression data, GRANITE has the capability of evaluating time-course data and generating interactive networks that may shed light on acute versus chronic treatment, as well as evaluating dose response and providing insight into mechanisms that underlie therapeutic versus sub-therapeutic doses or toxic doses. As a proof-of-concept study, we investigated lithium (Li) response in bipolar disorder (BD). BD is a severe mood disorder marked by cycles of mania and depression. Li is one of the most commonly prescribed and decidedly effective treatments for many patients (responders), although its mode of action is not yet fully understood, nor is it effective in every patient (non-responders). In an in vitro study, we compared vehicle versus chronic Li treatment in patient-derived lymphoblastoid cells (LCLs) (derived from either responders or non-responders) using both microRNA (miRNA) and messenger RNA gene expression profiling. We present both Li responder and non-responder network visualizations created by our GRANITE analysis in BD. We identified by network visualization that the Let-7 family is consistently downregulated by Li in both groups where this miRNA family has been implicated in neurodegeneration, cell survival and synaptic development. We discuss the potential of this analysis for investigating treatment response and even providing clinicians with a tool for predicting treatment response in their patients, as well as for providing the industry with a tool for identifying network nodes as targets for novel drug discovery.
Similar content being viewed by others

Introducing the problem: large complex data sets for human disorders are difficult to analyze
Bipolar disorder (BD) is a complex psychiatric disease, affecting 1–4% of the population1, 2, 3, 4 worldwide, and characterized by recurrence of depressive, hypomanic or manic episodes alternating with intervals of full remission.5 Lithium (Li) represents the mainstay for the management of BD, however, there is only ~30% of patients in long-term cohorts showing excellent response.5, 6, 7 Nevertheless, the mechanism underlying the mood-stabilizing effect of Li is still not completely understood. Early studies have shown that Li directly inhibits two enzymes of the inositol pathway, inositol-monophosphatase and inositol polyphosphate 1-phosphatase, in addition to glycogen synthase kinase-3 (GSK-3), a key kinase involved in the regulation of transcription, apoptosis, mood state, circadian rhythm and neurotransmission.8 However, pharmacogenetic studies focusing on known or putative targets of Li have so far provided little evidence for a major role of single genes predisposing patients to clinically respond to Li.9
Li is also known to indirectly interfere with a large number of molecular processes. This complexity, in conjunction with the heterogeneity of BD and wide phenotypic response to Li, significantly contributes to the lack of conclusive findings from studies based on the candidate gene approach. By interrogating the whole genome, transcriptomic analysis represents a promising approach with great potential for untangling the molecular underpinnings of complex phenotypes. However, high-throughput approaches produce large complex data sets contributing to the difficulty in interrogation and analysis. Therefore, the benefit of applying whole-genome exploration approaches to complex phenotypes is unrealized, unless a network-based approach is used to interrogate and interpret molecular networks and interactions.
To this aim, we applied GRANITE (Genetic Regulatory Analysis of Networks Investigational Tool Environment), an integrative genomic tool that provides visualization of complex data sets and generates interactive networks. GRANITE is unique in its attention to a data-processing pipeline for producing microRNA (miRNA)/messenger RNA (mRNA) graphs. Discovering miRNA networks was the proof-of-principle problem for GRANITE, and as a result, GRANITE makes it easy to produce not just the graphs, but also downstream measures, such as the rank-order charts, that are particularly useful for analyzing miRNA networks. We applied GRANITE to genome-wide mRNA and miRNA expression data from lymphoblasts derived from BD patients classified as excellent responders or non-responders to Li treatment, as described previously.10, 11 Lymphoblasts were cultured with either a therapeutic dose of Li (1.0 mM l−1) or vehicle in order to highlight genetic networks differentially influenced by the treatment in the responder and non-responder patients. The network analysis algorithms and visualizations implemented in GRANITE may be instrumental in biomarker identification that potentially could aid in predicting Li responsiveness in patients, as well as providing insights in other similarly complex phenotypes.
Introducing the solution: an integrative genomic tool for complex data analysis
The solution for making sense of large complex data sets is to use an integrative genomic tool. We developed GRANITE as a software workbench for representing, combining and interpreting biological models, particularly network models. GRANITE allows related models, for example, patient versus control or responder versus non-responder, to be composed using the logical operators AND/NOT/OR. Each unique combination of these operators results in a distinct way to partition the set of all available relationships into those that are part of the network and those that are not. For instance, responder AND non-responder yields the ‘common’ or ‘intersection’ network made up of the relationships that are common to both responder and non-responder groups. GRANITE supports six different methods to partition a graph using these logical operators:
-
1
Responder: relationships significant to the responder group;
-
2
Non-responder: relationships significant to the non-responder group;
-
3
Responder-only: responder AND NOT non-responder;
-
4
Non-responder-only: NOT responder AND non-responder;
-
5
Common network: responder AND non-responder;
-
6
Union network: responder OR non-responder;
The exclusion networks, responder-only and non-responder-only, are often the most useful because they capture relationships that are unique to treatment responders or non-responders, making those relationships particularly interesting research targets.
The relevant network models are constructed by inducing a subgraph on larger, more universal networks. For our exemplar, GRANITE begins with the TargetScan (http://www.targetscan.org/) Predicted Pairs network, which is a large graph database defining predicted regulatory relationships between miRNAs and mRNAs. The miRNAs and mRNAs are the nodes in that network, and a link exists between a miRNA and mRNA where a regulatory relationship is predicted. Note that this is a bipartite graph (miRNAs link only to mRNAs and mRNAs link only to miRNAs). The TargetScan graph is the superset of all the graphs we investigate, and for our purposes, TargetScan is the source of all network links. If there is a regulatory relationship between a miRNA and a mRNA, we assume that TargetScan captures that relationship. Importantly, this database is not specific to any disease or treatment. Expression data that is disease/treatment specific is used to select the set of nodes on which the subgraph will be induced. The graph induction process is deterministic. If, say, we induce on the responder mRNA expression data (responder-only), the induced graph contains exactly the edges from the TargetScan graph that are incident on the nodes representing mRNAs that are significantly differentially expressed by responder cells under Li treatment. For a complete workflow of this process that illustrates the data inputs, please see Supplementary Figure 1, and for additional information on GRANITE, please see Supplementary Table 1.
For our example, data were collected from patients with bipolar I disorder divided into responder (N=8) and non-responder (N=8) groups based on their clinical response to Li. Supplementary Figure 2 illustrates the experimental design and methods. Briefly, two cohorts of BD patients (see Supplementary Table 2 for demographic and clinical data)—excellent responders and non-responders to Li treatment were chosen. All patients were recruited through a specialty mood disorders program and followed prospectively. Diagnostic and Statistical Manual of Mental Disorders-IV and Research Diagnostic Criteria diagnoses were based on all available information including Schedule for Affective Disorders and Schizophrenia-Lifetime interviews carried out in blind fashion and independently reviewed by a panel of senior clinical researchers. Li response was evaluated using prospective follow-up data and quantified using a rating scale previously validated.5 The responders had to have a minimum of three illness episodes before Li treatment (4.13±1.55 on average) and remained stable for an average of 12.13±7.26 years of Li monotherapy at therapeutic levels (Li plasma level of 0.6 mEq l−1 or higher). The non-responders were treated with Li monotherapy for a minimum of 2 years and experienced at least two episodes while on Li monotherapy with documented compliance (Li plasma levels of 0.6 mEq l−1 or higher).
Blood samples from all subjects were obtained for DNA, and Epstein–Barr virus-transformed B-lymphoblastoid cell lines (LCLs) were prepared using standard procedures as described previously.12, 13 The transformation protocol was uniform for cells from all subjects; these were transformed and grown in a single passage prior to being frozen. All transformed cell lines were stored in liquid nitrogen after Epstein–Barr virus transformation, for each sample according to ‘LCL frozen storage’ time until the samples were selected for the experiment, thawed and regrown (three passages for each sample) and then processed in a sequential fashion as described below. The patient-derived LCLs from each of these patient groups were grown in Iscove’s modified Dulbecco's medium supplemented with 15% fetal bovine serum, 1% Fungizone and 1% penicillin/streptomycin/L-glutamine (Invitrogen) in a 5% CO2 humidified incubator at 37 °C, in the continuous presence of LiCl (1.0 mM l−1) in the vehicle or vehicle alone for 7 days after which cell pellets were collected and frozen at −80 °C. Seven days of treatment with 1 mM l−1 Li is considered in the literature to mimic chronic exposure and treatment concentrations of Li in patients’ brains.14, 15, 16 Furthermore, all samples were matched for patient age and sex. Thus samples from individual patients were chronically (7 days) treated independently in cell culture (in triplicate experiments), as well as for RNA and miRNA extractions. Per-individual replicates were pooled at the high-throughput microarray (miRNA chips 3.0 and U133-Plus 2.0 Human gene profiling mRNA arrays) level yielding N=8 Li-treated and N=8 vehicle-treated independent data points per responder and non-responder group, respectively.
The rationale for this experimental design is based on our hypothesis that miRNA and mRNA changes in patient-derived peripheral blood cells may have clinical utility as biomarkers for BD. Our expectation is that comparing baseline vs Li treatment will identify a Li-responsive miRNA–mRNA signature that could be used clinically in the future for (a) investigating the Li-regulated miRNA networks that are differentially expressed in responders vs non-responders, and determining the genetic mechanisms that may underlie these responses (that is, single-nucleotide polymorphisms (SNPs)); (b) predicting a naive BD patient’s response to Li; and (c) identifying network nodes for novel drug discovery. We anticipate that an effective biomarker will not come from simply profiling a patient-derived cell type at baseline. A more effective biomarker is to challenge the patient-derived cells in such a way that it reveals a potential endophenotype. In this case, we are investigating the response to Li in a peripheral cell line, namely LCL, because they are readily available, whereas neuronal cells from live patients are difficult to acquire. LCLs are Epstein–Barr-transformed B-cells, thus it is expected that there will be many changes in gene expression caused by the virus and culturing. Nonetheless, as we are comparing the biological effects of exposure to Li in the same cell lines, most biases should be eliminated. Future studies using patient-derived neuronal cultures differentiated from induced pluripotent stem cells (iPSCs) should be conducted to generate Li response networks in this more disease-relevant cell type. However, several studies have been successful in defining blood gene signatures associated with brain disorder.17, 18, 19, 20
The BD LCL expression data following this experimental design is shown in Figure 1, which highlights both the miRNA (Figures 1a and b) and mRNA (Figures 1c and d) data sets. Note that the patient code designation follows R=responder and NR=non-responder, which is then followed by age and then by F for female and M for male. A full list of this data in a table format is also included (Supplementary Table 3). These data highlight differentially expressed miRNA and mRNA in both responders and non-responders, but fail to provide a comprehensive network that one can visualize to gain access to complex interactions that cannot readily be ascertained. We employed GRANITE to visualize this genome-wide data set, which integrated over 1000 mRNAs and almost 300 miRNAs (see Supplementary Table 7 for relative graph sizes) into different networks.

Bipolar disorder lymphoblastoid cell expression data in vehicle versus chronic lithium treatment. Affymetrix arrays (miRNA chips 3.0 for a and b; U133-Plus 2.0 Human gene profiling messenger RNA (mRNA) arrays for c and d) are shown depicting microRNAs (miRNAs) or mRNAs differentially expressed between lithium vs vehicle in responders (a and c) and non-responders (b and d) and then subject to hierarchical clustering. Significantly regulated miRNAs (determined by meeting these criteria ±1.5-fold regulation and unadjusted P-value <0.05) are shown for both groups (a and b). Note that in the responder group (a) when R31F-lithium (orange) is placed in a separate group, it then clusters with responder vehicle group rather than responder lithium group. Also, in the non-responder group (b) when 18M_veh (purple) is placed in the outlier group, it clusters with the non-responder lithium group. Also note the increased heterogeneity in the non-responder group where the vehicle group shows two sets of clusters. Significantly regulated mRNAs (determined by meeting these criteria ±1.2-fold regulation and unadjusted P-value <0.05) are shown for both groups (c and d). Note that in the responder group (c) when R39F-lithium and 45M_lithium (red) are placed in a separate group, they cluster with responder vehicle treatment. Also, in the non-responder group (d) 10F_Veh (gray) shows abnormal hybridization and is placed in a new group. This greatly enhances clustering in non-responder groups (lithium vs vehicle). There are fewer genes that pass the same significance/fold filter in the non-responder group (d) compared with the responder group (c). These results illustrate a greater heterogeneity in the non-responder group than the responder group, and this may be indicative of an intrinsic lithium influence on transcriptional targets in the responder group.
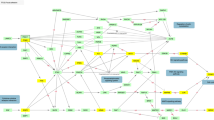
Three networks are visualized in Figure 2 pertaining to a responder network (Figure 2a), a non-responder network (Figure 2b) and a common network (Figure 2c). The visualizations created are based on clustering the miRNAs and mRNAs based on the number of connections (degrees). High-degree entities are clustered near the center and are color-coded blue, whereas low-degree entities surround the periphery and are color-coded red. Intermediate-degree entities are in the middle and are color-coded yellow.

MicroRNA (miRNA)–messenger RNA (mRNA) network visualization. Depicted are three miRNA–mRNA networks generated by GRANITE (Genetic Regulatory Analysis of Networks Investigational Tool Environment) using an mRNA filter (>1.2-fold regulation), where target scan is used to predict miRNAs that are targeted by lithium-regulated mRNAs taken from the arrays. Each dot represents either a miRNA (in blue) or mRNA (in yellow or red) node. The blue core represents high edge density miRNA nodes and the outer mantel clusters represent relatively lower-density nodes, with the red ones representing the lowest density. The responder (a) network shows the greatest gulf between the blue core and the mantle of visualization due to the high degree of miRNAs contained at the core, with many having over 500 connections. The non-responder (b) network has a less-pronounced core that may be due to more sample heterogeneity. There is a cluster of miRNAs in the Let-7 family as shown. The common (c) network was generated to identify common lithium networks that are conserved between responders and non-responders.
The node selection criteria involve the significance of differential expression (either mRNA expression or miRNA expression) with drug treatment verses a control. These selection criteria may involve statistical significance, that is, P<0.05, or fold change ratios, that is, |f|>1.2, or both. Typically, for this study, selection criteria filter only on the differential expression of one type of node (either miRNA or mRNA). GRANITE allows more complex filtering with multiple passes, but that has not proven critical. A common choice is to induce a subgraph on all miRNAs, plus their conjunction with those mRNAs whose differential expression satisfies a P<0.05 threshold criteria. The induced subgraph is the selected (unfiltered) subset of the nodes of the network together with any links from the universal network, whose end points are both contained in this subset. This gives a gene regulatory network that is disease/treatment specific, and which is also a subgraph of the universal network. GRANITE drops nodes of zero degree (spurious/no connections) in the induced subgraph. Network models are induced in this way for both the responder group and the non-responder group, and then analysis is performed on the partitions defined above through graph visualization and graph measures.
Although graph induction is deterministic, graph visualization is not. Graph layout is the most critical aspect of graph visualization, and layout algorithms tend to be heuristic and incremental. They are also computationally expressive, and so typically the heuristic runs for as long as we can afford to run, and then the layout stops although further improvements in the layout would be possible. Owing to the complexity of large graphs, visual cues are often the most important analytics available. Although there have been advances in the layout of large graphs in recent years, most of that work has focused on power-law graphs where there are many low-degree (low connectivity) nodes and only a few highly connected nodes. The miRNA graphs do not satisfy this description. They tend to be very dense graphs with many high-degree nodes and relatively few low-degree nodes. In fact, the low-degree nodes in miRNA graphs are of special interest. Further, clustering in bipartite graphs usually has a different interpretation than in general graphs, and these differences parallel the difference between a complete bipartite graph and a complete general graph. In general graphs, nodes in a cluster tend to link to one another forming cliques (complete general graphs that are subgraphs of the main graph). In a bipartite graph, nodes in the same set are forbidden to connect to one another. So clustering is more often decided by topology; two nodes in one set that link to the same nodes in the other set are usually considered to be in the same cluster (complete bipartite graphs that are subgraphs of the main graph). Although GRANITE supports a number of network layout algorithms, we have found the Fruchterman–Reingold algorithm,21 a force-directed layout algorithm, which dates from 1991, to be the most useful for miRNA graphs. Because the Fruchterman–Reingold algorithm minimizes link crossings, it tends to place nodes with similar topology together on the display, nodes with high connectivity at the center of the display and nodes with low connectivity at the perimeter. This yields a visualization pattern where similarly connected miRNAs, that is, in regulatory relationships with the same mRNAs, are usually placed very near to one another and vice versa. Clusters of miRNAs that form in these displays are likely to have similar biological function. Usually their underlying sequences are similar, but sometimes not. In any case, proximity and other such visual cues are important to establishing biological insight, and often we have found it useful to apply different layout algorithms—usually other force-directed algorithms—to the same graph, because each layout algorithm leads to different kinds of visual cues. We have also found chromaticity extremely useful in visualization. We use this arcane term to distinguish what we are doing from graph coloring, which has a very specific and mathematically precise meaning that is orthogonal to our purposes. We choose a drawing color for each node based on the degree (number of incident edges). Say we choose blue for high-degree nodes and red for low-degree nodes. We then draw each link in the color that is a blend of the two colors of the nodes it connects. In a network with hundreds of thousands of links, we can, for example, identify at a glance links that connect low-degree nodes, or low-degree nodes to high-degree nodes, high-degree nodes to high-degree nodes and so on.
We also use degree-based graph measures to gain insight into these networks. Again, the complexity of these networks can be daunting. But the low-degree (low connectivity) and high-degree nodes are of particular interest, although for different reasons. The high-degree nodes are important because they contribute to the regulation of many functions, whereas the low-degree nodes are important as the functions they affect may be the easiest to control and around which to build hypotheses/experiments.
From this analysis, there are many options on how one might proceed to interrogate the networks. We chose to focus on the high-degree miRNAs (Table 1). These are miRNAs with the most connections, which potentially have the most predominant global control of the network. Our strategy focuses on identifying these critical high-degree nodes, which are either common to both groups or different. The common nodes may reflect pathways that Li activates in BD patients that do not readily translate into therapeutic benefits, whereas the unique nodes may reflect pathways that are preferentially activated in responders, leading to a clinical response.
We also took another approach by focusing on a miRNA family that was implicated in our data set to be preferentially regulated by Li, the Let-7 family (Supplementary Table 8). The Let-7 family was consistently downregulated by Li in both groups, although preferentially more family members (11 versus only 6 in the non-responder group) were downregulated by Li in the responder group (Supplementary Table 8). The Let-7 family could represent a novel target for Li response, although further investigation is warranted. For instance, one could overlay responder and non-responder SNP data and determine whether there are Let-7 targets where these SNPs alter miRNA-binding sites. Figure 3 depicts genes controlled by Let-7 that may be implicated in the Li response. Interestingly, let-7e expression was found upregulated in schizophrenia patient dorsal lateral prefrontal cortex and temporal lobe epilepsy.22, 23 Let-7f is upregulated in newborn brains following maternal stress.24

Genes controlled by Let-7. Depicted is an illustration of genes that have microRNA (miRNA)-binding sites to be regulated by Let-7 in the non-responder (NR) GRANITE network.
In addition, microRNA let-7 is elevated in the cerebrospinal fluid from patients with Alzheimer’s disease, and has an unconventional role to cause neurodegeneration by activating Toll-like receptor 7.25 In addition, Let-7c was reported to suppress the expression of the major anti-apoptotic protein Bcl-xl.26 The Let-7 family has also been suggested to have effects on synaptic development and function,27 and its expression in the brain is regulated by sleep deprivation.28 Finally, let-7 miR expression is suppressed by the Wnt-β-catenin signaling pathway,29 thus being consistent with a role in conferring the responsiveness to Li treatment.
We also performed quantitative reverse transcription (qRT)-PCR validation on a subset of Li-downregulated mRNAs in the responder group (Supplementary Table 9). These genes include THRAP3, CDC27, TRP, TFAM and LARS. Although there was a trend for all the qRT-PCR data to confirm the array data, only THRAP3 and TFAM were nominally significant (paired T-test).
Finally, we computed a list of miRNAs for responders and non-responders based on the normalized weighted difference (Table 2). A glance at the degree rank tables (Table 1) reveals that the highest ranked responder miRNAs are also the highest ranked non-responder miRNAs. In fact, 18 miRNAs are common to both of the top 20 rank tables, and in both cases, the two miRNAs that are not present in the other table just barely missed the cutoff. To make sense of the degree rank information, we developed a measure of difference between responder and non-responder for each miRNA. That measure is computed in four steps: first, the degree of each miRNA in each graph is normalized. We call this the ‘normalized weight’. So if a miRNA is an end point of 0.40% of all graph edges in the responder graph, its normalized weight for that graph will be 0.0040. It will have a different normalized weight for the non-responder graph. Second, the magnitude of the difference in normalized weight between the responder and non-responder graph is computed. This is the ‘normalized difference’. So if a miRNA has a normalized weight of 0.0040 in the responder graph and 0.0038 in the non-responder graph, the normalized difference for this miRNA is 0.0002. As this is treated as a difference measure, its absolute value is used. Third, the mean (9.38E−5) and s.d. (6.11E−5) of the normalized differences for all miRNAs in the study are computed. Finally, a z-score is computed for each miRNA based on its normalized difference, and the mean and s.d. computed from step 3. So if the normalized difference for a miRNA is 0.0002, its z-score is (2E−4–9.38E−5)/6.11E−5 or 1.74. The normalized weights computed in step 1 constitute discrete nominal probability distributions. Tests do not indicate significant differences between the responder and non-responder distributions. This is consistent with the similarities in the degree rank tables. Instead, we expect the differences to be limited to a few miRNAs that become the targets of further investigation. Those miRNAs will have z-scores of high magnitude. For 95% confidence, |z|>1.96. The miRNAs satisfying these criteria are presented in Table 2.
We also performed pathway analysis of the targets of these miRNAs to understand their functional role. For this analysis, the targets of the miRNAs were obtained from miRWalk database30 and run through QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity) core analysis scheme, using the default parameters specific to human species. The top three enriched pathways for each of these miRNAs target genes along with their –log(P-value) are presented in Table 2. Some interesting candidate-enriched pathways for further investigation into Li’s beneficial effects include (a) semaphorin signaling in neurons, (b) sonic hedgehog signaling, (c) agrin interactions at neuromuscular junction and (d) serotonin signaling. Interestingly, one of Li’s known targets, GSK-3, has been implicated in modulating semaphorin signaling to influence cortical neuron migration and dendritic orientation,31 whereas knockouts of GSK-3 resulted in hyperproliferation of neural progenitors and resulting dysregulation of beta-catenin, sonic hedgehog, notch and fibroblast growth factor signaling.32 Although additional investigation is warranted, perhaps some of these GSK-3-mediated pathways including semaphorin or sonic hedgehog signaling may also be mediated by Li-regulated miRNAs listed in Table 2 to promote neuronal migration, dendritic orientation and neurogenesis. Overall, this methodology enables one to identify potentially critical miRNAs and associated pathways that are different between the responder and non-responder groups, which may be useful for interrogating treatment response.
There are several limitations that should be addressed properly with our current study. As regards to the fold change differences, we can explain this as follows. First, the Li concentration used in this experiment is chosen to mimic clinical treatment levels and thus is not expected to induce marked changes in cell function responses, but rather fine-tuning of the system. Other studies focusing on Li toxicity have used higher levels, and achieved higher fold changes, but these conditions would be toxic in patients33 or would lead to severe side effects and thus not be tolerated.33, 34 Second, the lower fold changes are a limitation of the hybridization probe design of microarrays and the intrinsic aspect of background noise. Likely, future studies using RNAseq will be able to achieve somewhat higher fold changes, although not markedly increased given the first reason mentioned.
Current and future applications to interrogate static and dynamic data sets
The current application of GRANITE is to integrate genome-wide miRNA and mRNA data sets following Li treatment. This application compares vehicle treatment (baseline) versus chronic Li treatment. More dynamic states are possible. For instance, GRANITE could be used to develop interactive networks for acute versus chronic treatment (for example, 0-, 1-, 3-, 7-, 14-, 21- and 28-day treatments) or also across different doses (for example, sub-therapeutic, therapeutic and toxic). Moving forward, we envision assembling a database for investigators to upload their data sets to create and share their interactive networks to gain insight into complex, large data sets.
With larger data sets, this tool may provide a foundation for clinicians to predict whether their patients will respond to certain medications. We envision in future applications, that with larger data sets, this tool could allow clinicians to predict Li response by simply genotyping their patients. The genotypes would then be coded for SNPs that either interfere or enhance miRNA-binding sites within nodes that are known to be correlated with either Li response or non-response. In this way, an algorithm could be used to calculate the likelihood that a patient would respond to Li. Further, this model could be transplanted to other phenotypically complex and heterogeneous disease processes, thus yielding predictions of most any response, and thereby revolutionizing the field of psychiatry and moving it towards personalized medicine.
This tool could also be used by industry to develop novel medications or repurpose existing Food and Drug Administration-approved medications in a more cost effective and efficient manner. In particular, with the advent of iPSCs-based technologies, disease-relevant cell types could be used for patients to construct interactive networks for safety and efficacy studies that could be used to find a correct drug dose, avoid toxicity and select for patients who have the highest probability of responding to a current new therapy.
In advancing towards personalized medicine, Supplementary Figure 3 illustrates three representative Li networks derived from the top 20 Li-regulated miRNAs in responders, non-responders and common networks. The responder (A) and non-responder (B) networks are generated using a different force-directed graph layout algorithm, which organizes the heaviest or highest-degree items and separates them from the lower-degree items (blue nodes are mRNAs; red nodes are miRNAs). In both responder and non-responder networks, we find has-miR-491-5p to be the right red node and has-miR-544a to be the left red node. This analysis suggests that there may not be overwhelming differences in the high-degree miRNA networks between Li responders and non-responders. Rather, differences may be more apparent with low-degree miRNA networks. Nevertheless, we have created a visualization tool that enables one to interrogate these networks in new ways. One could imagine using a personalized medicine approach where each patient could have a ‘drug’ response network made that would be created based on a patient-specific cellular response to a given medication. This analysis could be applied toward prescribing a more efficacious dose initially to the patient. With further data sets and combining a patient’s genomic data, we envision that correlations can be made that will allow clinicians to predict treatment response with improved confidence.
Summary and conclusions
In summary, we have developed an integrative genomic systems biology-based tool, GRANITE, which can effectively analyze large complex data sets to generate interactive networks. This tool is open source and is available for use and collaboration (the link to the code is: http://exon.niaid.nih.gov/granite/). As a proof-of-concept study, we used GRANITE to interrogate Li response in BD by measuring a large data set of mRNAs and miRNAs, and found that the Let-7 miRNA family was consistently and preferentially downregulated by Li in the BD responder group. We anticipate that the dynamic networks created by GRANITE will lead to a more effective and reliable tool for clinical use in predicting patients’ response to medications, and will assist the industry in reducing costs and time during the drug development process.
References
Angst J . The emerging epidemiology of hypomania and bipolar II disorder. J Affect Disord 1998; 50: 143–151.
Goodwin F, Jamison K . Manic-Depressive Illness. Oxford University Press: Oxford, UK, 2007.
Merikangas KR, Akiskal HS, Angst J, Greenberg PE, Hirschfeld RM, Petukhova M et al. Lifetime and 12-month prevalence of bipolar spectrum disorder in the National Comorbidity Survey replication. Arch Gen Psychiatry 2007; 64: 543–552.
Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry 2011; 68: 241–251.
Garnham J, Munro A, Slaney C, Macdougall M, Passmore M, Duffy A et al. Prophylactic treatment response in bipolar disorder: results of a naturalistic observation study. J Affect Disord. 2007; 104: 185–190.
Baldessarini RJ, Tondo L . Does lithium treatment still work? Evidence of stable responses over three decades. Arch Gen Psychiatry 2000; 57: 187–190.
Coryell W . Maintenance treatment in bipolar disorder: a reassessment of lithium as the first choice. Bipolar Disord. 2009; 11: 77–83.
Quiroz JA, Gould TD, Manji HK . Molecular effects of lithium. Mol Interv 2004; 4: 259–272.
Severino G, Squassina A, Costa M, Pisanu C, Calza S, Alda M et al. Pharmacogenomics of bipolar disorder. Pharmacogenomics 2013; 14: 655–674.
Alda M, Grof P, Rouleau GA, Turecki G, Young LT . Investigating responders to lithium prophylaxis as a strategy for mapping susceptibility genes for bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29: 1038–1045.
Turecki G, Grof P, Grof E, D'Souza V, Lebuis L, Marineau C et al. Mapping susceptibility genes for bipolar disorder: a pharmacogenetic approach based on excellent response to lithium. Mol Psychiatry 2001; 6: 570–578.
Alda M, Keller D, Grof E, Turecki G, Cavazzoni P, Duffy A et al. Is lithium response related to G(s)alpha levels in transformed lymphoblasts from subjects with bipolar disorder? J Affect Disord 2001; 65: 117–122.
Sun X, Young LT, Wang JF, Grof P, Turecki G, Rouleau GA et al. Identification of lithium-regulated genes in cultured lymphoblasts of lithium responsive subjects with bipolar disorder. Neuropsychopharmacology 2004; 29: 799–804.
Sugawara H, Iwamoto K, Bundo M, Ishiwata M, Ueda J, Kakiuchi C et al. Effect of mood stabilizers on gene expression in lymphoblastoid cells. J Neural Transm 2010; 117: 155–164.
Asai T, Bundo M, Sugawara H, Sunaga F, Ueda J, Tanaka G et al. Effect of mood stabilizers on DNA methylation in human neuroblastoma cells. Int J Neuropsychopharmacology 2013; 16: 2285–2294.
Cruceanu C, Alda M, Grof P, Rouleau GA, Turecki G . Synapsin II is involved in the molecular pathway of lithium treatment in bipolar disorder. PloS One 2012; 7: e32680.
Glatt SJ, Chandler SD, Bousman CA, Chana G, Lucero GR, Tatro E et al. Alternatively spliced genes as biomarkers for schizophrenia, bipolar disorder and psychosis: a blood-based spliceome-profiling exploratory study. Curr Pharmacogenomics Person Med 2009; 7: 164–188.
Greiner HM, Horn PS, Holland K, Collins J, Hershey AD, Glauser TA . mRNA blood expression patterns in new-onset idiopathic pediatric epilepsy. Epilepsia 2013; 54: 272–279.
Han G, Wang J, Zeng F, Feng X, Yu J, Cao HY et al. Characteristic transformation of blood transcriptome in Alzheimer's disease. J Alzheimers Dis 2013; 35: 373–386.
Sabatino G, Rigante L, Minella D, Novelli G, Della Pepa GM, Esposito G et al. Transcriptional profile characterization for the identification of peripheral blood biomarkers in patients with cerebral aneurysms. J Biol Regul Homeost Agents 2013; 27: 729–738.
Fruchterman TMJ, Reingold EM . Graph drawing by force-directed placement. Softw Pract Exp 1991; 21: 1129–1164.
Beveridge NJ, Gardiner E, Carroll AP, Tooney PA, Cairns MJ . Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol Psychiatry 2010; 15: 1176–1189.
Song YJ, Tian XB, Zhang S, Zhang YX, Li X, Li D et al. Temporal lobe epilepsy induces differential expression of hippocampal miRNAs including let-7e and miR-23a/b. Brain Res 2011; 1387: 134–140.
Zucchi FC, Yao Y, Ward ID, Ilnytskyy Y, Olson DM, Benzies K et al. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PloS One 2013; 8: e56967.
Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J et al. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci 2012; 15: 827–835.
Qin B, Xiao B, Liang D, Li Y, Jiang T, Yang H . MicroRNA let-7c inhibits Bcl-xl expression and regulates ox-LDL-induced endothelial apoptosis. BMB Rep 2012; 45: 464–469.
Corbin R, Olsson-Carter K, Slack F . The role of microRNAs in synaptic development and function. BMB Rep 2009; 42: 131–135.
Davis CJ, Bohnet SG, Meyerson JM, Krueger JM . Sleep loss changes microRNA levels in the brain: a possible mechanism for state-dependent translational regulation. Neurosci Lett 2007; 422: 68–73.
Cai WY, Wei TZ, Luo QC, Wu QW, Liu QF, Yang M et al. The Wnt-beta-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J Cell Sci 2013; 126: 2877–2889.
Dweep H, Sticht C, Pandey P, Gretz N . miRWalk—database: prediction of possible miRNA binding sites by ‘walking’ the genes of three genomes. J Biomed Inform 2011; 44: 839–847.
Morgan-Smith M, Wu Y, Zhu X, Pringle J, Snider WD . GSK-3 signaling in developing cortical neurons is essential for radial migration and dendritic orientation. Elife 2014; 3: e02663.
Kim WY, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci 2009; 12: 1390–1397.
Oruch R, Elderbi MA, Khattab HA, Pryme IF, Lund A . Lithium: a review of pharmacology, clinical uses, and toxicity. Eur J Pharmacol 2014; 740C: 464–473.
Carter L, Zolezzi M, Lewczyk A . An updated review of the optimal lithium dosage regimen for renal protection. Can J Psychiatry 2013; 58: 595–600.
Acknowledgements
We would like to acknowledge the support of the Intramural Research Program of the NIMH, and the Office of Cyber Infrastructure and Computational Biology of NIAID, NIH. We would also like to acknowledge the technical support kindly provided by Weiwei Wu for array processing, and Peter Leeds for editorial suggestions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hunsberger, J., Chibane, F., Elkahloun, A. et al. Novel integrative genomic tool for interrogating lithium response in bipolar disorder. Transl Psychiatry 5, e504 (2015). https://doi.org/10.1038/tp.2014.139
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.139
This article is cited by
-
Transcriptomics and miRNomics data integration in lymphoblastoid cells highlights the key role of immune-related functions in lithium treatment response in Bipolar disorder
BMC Psychiatry (2022)
-
Deficient LEF1 expression is associated with lithium resistance and hyperexcitability in neurons derived from bipolar disorder patients
Molecular Psychiatry (2021)
-
Exemplar scoring identifies genetically separable phenotypes of lithium responsive bipolar disorder
Translational Psychiatry (2021)
-
Prediction of lithium response using genomic data
Scientific Reports (2021)
-
Predicted Cellular and Molecular Actions of Lithium in the Treatment of Bipolar Disorder: An In Silico Study
CNS Drugs (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}