
Abstract
The importance of reversing brain serotonin (5-HT) deficiency and promoting hippocampal neurogenesis in the mechanisms of action for antidepressants remain highly controversial. Here we examined the behavioral, neurochemical and neurogenic effects of chronic fluoxetine (FLX) in a mouse model of congenital 5-HT deficiency, the tryptophan hydroxylase 2 (R439H) knock-in (Tph2KI) mouse. Our results demonstrate that congenital 5-HT deficiency prevents a subset of the signature molecular, cellular and behavioral effects of FLX, despite the fact that FLX restores the 5-HT levels of Tph2KI mice to essentially the levels observed in wild-type mice at baseline. These results suggest that inducing supra-physiological levels of 5-HT, not merely reversing 5-HT deficiency, is required for many of the antidepressant-like effects of FLX. We also demonstrate that co-administration of the 5-HT precursor, 5-hydroxytryptophan (5-HTP), along with FLX rescues the novelty suppressed feeding (NSF) anxiolytic-like effect of FLX in Tph2KI mice, despite still failing to induce neurogenesis. Thus, our results indicate that brain 5-HT deficiency reduces the efficacy of FLX and that supplementation with 5-HTP can restore some antidepressant-like responses in the context of 5-HT deficiency. Our findings also suggest that feeding latency reductions in the NSF induced by chronic 5-HT elevation are not mediated by drug-induced increments in neurogenesis in 5-HT-deficient animals. Overall, these findings shed new light on the impact of 5-HT deficiency on responses to FLX and may have important implications for treatment selection in depression and anxiety disorders.
Similar content being viewed by others


Introduction
Major depression and anxiety disorders are highly prevalent diseases that rank among the leading causes of disability worldwide.1, 2, 3 The negative impact of these disorders is exacerbated by the poor remission rates obtained with standard treatments.4 Most antidepressants acutely increase the extracellular levels of serotonin (5-HT), but mood improvements typically do not emerge until after weeks of treatment.5 Consequently, the clinical effects of antidepressants have been hypothesized to result from long-term adaptive responses to antidepressant administration, such as increased neurogenesis.6, 7, 8
Antidepressant effects have also been hypothesized to result from the correction of endogenous 5-HT deficiency,9 but recent studies have shown that mutant forms of the 5-HT synthesis gene, tryptophan hydroxylase 2 (Tph2),10, 11 which could result in impaired 5-HT synthesis,12 are associated with poor antidepressant treatment responses.13, 14, 15 These observations suggest that congenital brain 5-HT deficiency might reduce antidepressant efficacy. Here we used a genetically engineered mouse line, the Tph2 (R439H) knock-in (Tph2KI) mouse, which exhibits ∼60–80% reductions in brain 5-HT16 throughout postnatal development,17 to examine the effects of 5-HT deficiency on responses to fluoxetine (FLX) in the novelty suppressed feeding (NSF) and tail suspension tests (TSTs). The NSF is a particularly interesting preclinical measure of antidepressant-like effects in that the efficacy of antidepressants in this test requires chronic administration and has been reported to be neurogenesis dependent.18, 19 However, the neurogenesis dependence of antidepressant-like effects in the NSF have been challenged,20 and the role of hippocampal neurogenesis in the etiology and treatment of depression and anxiety disorders remains highly controversial.21
Our data indicate that 5-HT deficiency impairs NSF responses to FLX and prevents the induction of neurogenesis by FLX. However, we also show that FLX reduces immobility time in the TST in both wild-type (WT) and Tph2KI mice, despite the fact that the magnitude of the FLX-induced increase in 5-HT is markedly reduced in Tph2KI compared with that of WT animals. In addition, we demonstrate that supplementation with 5-hydroxytryptophan (5-HTP), the 5-HT precursor, can restore the anxiolytic-like effects of FLX in the NSF in Tph2KI mice, despite failing to restore FLX’s pro-neurogenic effects in these animals. Thus, our results suggest that 5-HT deficiency can impair a subset of antidepressant effects and indicate that antidepressant-like effects in the NSF do not require antidepressant-induced increases in hippocampal neurogenesis, at least under conditions of congenital 5-HT deficiency.
Materials and methods
Animals and drug treatments
The Tph2KI mouse line is on a mixed background (c57BL6/J—129S6/SvEv) and has been described previously.16 Homozygous Tph2 R439H KI mice and WT littermate controls were derived from heterozygous breeding pairs and were housed two to five per cage in a facility maintained at 23±2 °C on a 12 h light–dark cycle. Eight- to 10-week-old age-matched littermates were used for all experiments. Male mice were exclusively used for all behavioral analyses and the subsequent FLX/neurogenesis studies. The baseline neurogenesis experiments (Figure 2) included balanced numbers of males and females, but the study was not sufficiently powered to reveal any significant sex differences. FLX (Spectrum Chemical Corporation, New Brunswick, NJ, USA) and desipramine (DES; Sigma, St Louis, MO, USA) were administered to mice via their drinking water (155 mg l−1) for a total of 4 weeks. Behavioral testing was conducted after 3 weeks of FLX administration, and mice were killed and processed for immunohistichemistry (IHC) analysis 1 week after behavioral testing. In our hands, this treatment regimen results in a dose equivalent of FLX to ∼20 mg kg−1 per day for both WT and Tph2KI mice.22 An analogous treatment paradigm has been shown to be effective for DES.23
Supplementation with 5-HTP was performed by injecting WT and Tph2KI mice intraperitoneally twice daily (at 0900 and 1700 h) with 5-HTP (20 mg kg−1) for 4 weeks, while administering FLX as described above. Again, behavioral testing was performed after 3 weeks (2 h following the first daily injection) and the mice were killed for IHC analysis 1 week later (2 h after the final 5-HTP injection). This treatment paradigm partially restores tissue levels of 5-HT in Tph2KI mice 2 h after administration.22 Antidepressant-containing drinking water was administered via opaque bottles and was replaced twice weekly. Chlordiazepoxide (7.5 mg kg−1, Sigma) was administered intraperitoneally 20 min before performing the NSF. For proliferation studies, bromodeoxyuridine (BrdU; Sigma) solutions were prepared fresh daily by dissolution in saline (10 mg ml−1) and were intraperitoneally injected (100 mg kg−1) into mice 4, 18 and 24 h before killing. For survival and double-labeling studies, BrdU was administered via the drinking water (1 g l−1) in opaque bottles that were replaced twice weekly. All experiments were conducted in accordance with an animal protocol that was approved by the Duke University Institutional Animal Care and Use Committee.
Microdialysis
Surgery
Mice were anesthetized using isoflurane and placed in a Kopf (Tujunga, CA, USA) stereotaxic frame equipped with a mouse adapter (Stoelting Mouse and Neonatal Rat Adaptor, Wood Dale, IL, USA). A guide cannula (catalog number 5-300004; Brainlink, Groningen, The Netherlands) was implanted into the hippocampus (HIP; anterior-posterior: 3.3 mm, medial-lateral: 3.0 mm, dorsal-ventral: 1.5 mm), according to the Franklin and Paxinos mouse brain atlas. Each cannula was fixed in place with two anchor screws (CMA, Chelmsford, MA, USA) and carboxylate dental cement (CMA). Operated mice were single housed, treated with antibiotics (1.2 mg sulfamethoxazole per ml and 0.24 mg trimethoprim per ml) in the drinking water (with or without FLX) and allowed to recover 48–96 h after implantation.
Dialysate collection
Sterile artificial cerebrospinal fluid (147 mM NaCl, 2.7 mM KCl, 0.85 mM MgCl2, 1.2 mM CaCl2; CMA) was delivered from a CMA 400 syringe pump at a flow rate of 0.45 μl min−1. Sixteen to 24 h before the start of sample collection, each mouse was gently restrained and a microdialysis probe (catalog number 5-140040, 2 mm membrane; Brainlink) was inserted into the guide cannula. Each mouse was then placed in a circular cage with bedding, chow and water (with or without FLX) available ad libitum. The probe tubing was stiffened with laboratory paper tape to avoid biting. A two-channel swivel (catalog number 375/D/22QM; Instech, Plymouth Meeting, PA, USA) allowed for unimpeded movement of the mouse. Between 0900 and 1100 h, one baseline dialysate (30 min duration) was collected on ice, shielded from light, immediately frozen on dry ice and stored at −80 °C.
High-performance liquid chromatography–electrochemical detection analysis
The high-performance liquid chromatography system consisted of a BASi (West Lafayette, IN, USA) LC-4C detector coupled to a BASi LCEC radial flow cell. The potential was set at +650 mV. Flow was provided by a Shimadzu (Columbia, MD, USA) LC-20AD solvent delivery module. The pump was preceded by an online degasser series 1100 from Agilent (Santa Clara, CA, USA). The chromatograms were analyzed using PowerChrom software (eDAQ, Colorado Springs, CO, USA). Ten microliters of dialysate was separated on a 1 × 100-mm UniJet microbore 3 μm octadecylsilyl column (BASi, West Lafayette, IN, USA) at a flow rate of 80 μl min−1. The mobile phase consisted of 24 mM Na2HPO4, 3 mM octanesulfonic acid, 27.4 mM citric acid, 107 μM EDTA and 17–18.5% (v/v) MeOH, pH adjusted to 4.8 with NaOH, 5-HT eluted at 11–13 min.
Behavioral analyses
The TST and NSF were performed as described previously.16, 24 Immediately following the NSF, mice were returned to the home cage, where the latency to feed and quantity of food consumed within 5 min were measured.
Immunohistochemistry
Tissue was processed for IHC analysis as described previously.24 Primary antibodies used were rabbit anti-doublecortin (DCX; Abcam, Cambridge, MA, USA; 1:200), rabbit anti-cleaved caspase-3 (1:500; Cell Signaling, Danvers, MA, USA), mouse anti-NeuN (Millipore, Temecula, CA, USA; 1:200) and rat anti-BrdU (Accurate Chemical Corporation, Westbury, NY, USA; 1:200). Alexafluor 488- or Alexafluor 568-conjugated secondary antibodies (Life Technologies, Carlsbad, CA, USA) diluted 1:500 in 5% bovine serum albumin in phosphate-buffered saline-t were used. Coverslipping was performed using SlowFade Gold Antifade Reagent with 4',6-diamidino-2-phenylindole (Life Technologies). For assessments of granule cell layer (GCL) and medial habenula size, images of 4',6-diamidino-2-phenylindole-stained sections (six sections per mouse for the GCL and four sections per mouse for the medial habenula) were taken, and the GCL and medial habenula of each hemisphere was outlined and measured using Axiovision software (Zeiss, Oberkochen, Germany).
For confocal analysis, sections were stained according to the above protocol, except that they were counterstained with TOTO-3, a nuclear marker, for 15 min (1:500 dilution, Life Technologies) before coverslipping with SlowFade Gold Antifade Reagent without 4',6-diamidino-2-phenylindole (Life Technologies).
Non-overlapping images of the entire GCL and subgranular zone were taken on a fluorescence microscope (Zeiss, Oberkochen, Germany) by an individual unaware of genotype and treatment condition. The numbers of BrdU+, BrdU+/NeuN+, DCX+ or activated caspase-3+ cells were counted by an observer unaware of genotype and treatment condition. For proliferation studies, only BrdU+ cells within two cell widths of the subgranular zone were counted. However, for survival and double-labeling studies, BrdU+ cells were counted throughout the entire GCL, as they are known to migrate.25 For BrdU/NeuN double-labeling experiments, at least 600 BrdU+ cells were examined in each group.
Real-time PCR
Mice were killed by cervical dislocation, decapitated and heads were rapidly cooled by submersion in liquid nitrogen for ∼6 s. Brains were removed and a 1.5-mm diameter punch of dorsal HIP (primarily dentate gyrus) was obtained from a 1-mm coronal section, snap-frozen in liquid nitrogen and stored at −80 °C until further use. RNA was extracted with Trizol in combination with RNeasy minikits according to the manufacturer’s protocol (Qiagen, Valencia, CA, USA). RNA was reverse transcribed using the iScript cDNA synthesis kit according to the manufacturer’s protocol (Bio-Rad, Hercules, CA, USA), and real-time PCR was performed using a LightCycler (Roche Applied Science, Indianapolis, IN, USA). The glyceraldehydes 3-phosphate dehydrogenase primers used were: 5'-CAT GTT CCA GTA TGA CTC CAC TC-3′ and 5'-GGC CTC ACC CCA TTT GAT GT-3'. The brain-derived neurotrophic factor (BDNF) primers used were: 5'-CAA TGC CGA ACT ACC CAA-3' and 5'-AAC ATA AAT CCA CTA TCT TCC CC-3' The cAMP response element-binding (CREB) primers used were: 5'-AGC CGG GTA CTA CCA TTC TAC-3' and 5'-GCA GCT TGA ACA ACA ACT TGG-3'.
Statistical analysis
Data were analyzed using Student’s t-tests or two-way analysis of variances with Tukey’s post-hoc tests, where appropriate. In some cases (for example,, microdialysis experiments), data were transformed (for example, log-transformed) before performing statistical analyses. Statistical analyses were performed using JMP software (SAS, Cary, NC, USA).
Results
FLX increases extracellular 5-HT levels in the HIP of 5-HT-deficient mice
Consistent with our previous results,16, 17 microdialysis revealed that Tph2KI mice have reduced extracellular 5-HT (5-HTEXT) in the HIP compared with WT controls (main effect of genotype: F(1,25)=135.8074, P<0.0001, Figure 1a). Chronic treatment with FLX increased 5-HTEXT in both genotypes (main effect of FLX: F(1,25)=88.8577, P<0.0001, Figure 1a). However, the magnitude of the FLX-induced increase in 5-HTEXT in Tph2KI mice (∼2.25-fold, P=0.0326) was markedly less than that observed in WT animals (∼6.4-fold, genotype by drug interaction: F(1,25)=28.3908, P<0.0001, Figure 1a). Importantly, the levels of 5-HTEXT in Tph2KI mice after chronic FLX treatment were not significantly different from those in untreated WT mice (P=0.3963), but they were only 12% of the levels achieved in FLX-treated WT animals. These results suggest that although chronic FLX treatment essentially reverses hippocampal 5-HT deficiency in Tph2KI mice, congenital 5-HT deficiency can significantly blunt the neurochemical effects of selective serotonin reuptake inhibitors (SSRIs).

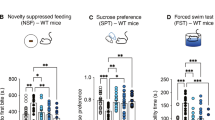
Neurochemical and behavioral responses to chronic fluoxetine (FLX) in wild-type (WT) and tryptophan hydroxylase 2 (R439H) knock-in (Tph2KI) mice. (a) Levels of extracellular 5-HT (5-HTEXT) in the hippocampus (HIP) were determined by microdialysis. (b) Feeding latency in the novelty suppressed feeding (NSF) test after chronic FLX treatment. (c) Home-cage feeding latency in WT and Tph2KI mice following chronic FLX. (d) Feeding latency in the NSF after acute chlordiazepoxide administration. (e) Feeding latency in the NSF following chronic desipramine (DES) treatment in WT and Tph2KI mice. (f) Immobility time in the tail suspension test (TST) following chronic FLX. *Significant main effect of treatment by two-way analysis of variance (ANOVA; P<0.05). **P<0.05 compared with WT control by Tukey’s post-hoc test. ^P<0.05 compared with WT FLX by Tukey’s post-hoc test. @P<0.05 by Tukey’s post-hoc test compared with control Tph2KI mice. ‘X’ denotes a significant genotype by treatment interaction by two-way ANOVA (P<0.05) and ‘#’ denotes a significant main effect of genotype (P<0.05 by two-way ANOVA); n=7–8 per group for a, n=19–21 per group for b, n=10 per group for c, n=9 per group for d, n=8–11 per group for e and n=22–27 per group for f.
Chronic FLX fails to induce an antidepressant-like effect in the NSF in Tph2KI mice
In the NSF, chronic FLX significantly reduced feeding latency (main effect of treatment: F(1,68)=6.0055, P=0.0168, Figure 1b), but a significant genotype by treatment interaction was also observed (F(1, 68)=10.4593, P=0.0019, Figure 1b). Indeed, FLX reduced feeding latency in WT mice (P=0.0012) but not in Tph2KI animals. Importantly, FLX-treated WT mice exhibited significantly shorter feeding latencies than FLX-treated Tph2KI animals (P=0.0414), suggesting that the lack of effect in Tph2KI mice was not due to a floor effect. A significant main effect of FLX on home-cage feeding latency was also observed (F(1, 76)=4.1378, P=0.0454, Figure 1c), but the genotype by treatment interaction was not significant (P=0.3142), thus suggesting that differential effects of FLX on appetitive drive in WT and Tph2KI animals were not responsible for the observed differential responses to FLX. In contrast to chronic FLX, acute treatment with chlordiazepoxide, a benzodiazepine, reduced feeding latency in both genotypes (main effect of treatment: F(1,32)=15.809, P=0.0004, Figure 1d). Similarly, chronic treatment with DES, a tricyclic antidepressant that preferentially inhibits norepinephrine reuptake, reduced feeding latency in both genotypes (main effect of treatment: F(1,35)=11.3246, P=0.0019, Figure 1e), suggesting that brain 5-HT deficiency does not impair anxiolytic-like responses to non-5-HT-specific drugs.
Throughout the NSF experiments, control Tph2KI mice exhibited a tendency towards reduced feeding latencies compared with that of WT animals (Figures 1 b–e). To determine the basis for this, we examined home-cage food consumption and feeding latency, and weight loss following a 24-h food-deprivation period. No significant genotype differences were observed in home-cage food consumption or weight loss (BDS, unpublished observations).
Similar to our previous reports,16, 26 Tph2KI mice exhibited increased immobility in the TST (main effect of genotype: F(1, 97)=4.9953, P=0.0277, Figure 1f). In contrast to the NSF, chronic treatment with FLX significantly reduced immobility time in the TST (main effect of treatment: F(1,97)=21.3464, P<0.0001, Figure 1f) with no significant genotype by treatment interaction.
Brain 5-HT deficiency does not impair baseline neurogenesis
Because of the reported importance of hippocampal neurogenesis in the NSF,18, 19 we hypothesized that the lack of effect of FLX in Tph2KI mice in the NSF might result from a defect in the neurogenic response to FLX. As we have shown previously,24 there are no significant baseline differences between WT and Tph2KI mice in BrdU incorporation in the subgranular zone (Figures 2a–c), but a detailed analysis of baseline neurogenesis in Tph2KI mice has not been performed previously. Interestingly, IHC analysis for DCX, a marker of immature neurons, revealed a significant 37% increase in the number of DCX+ neurons in Tph2KI mice compared with that in WT controls (Student’s t-test: P=0.0025, degrees of freedom=31, Figures 2d–f).

Effects of brain serotonin (5-HT) deficiency on neurogenesis in the subgranular zone (SGZ). (a) Quantification of bromodeoxyuridine (BrdU)+ cell number in the SGZ. Representative BrdU images from wild-type (WT) (b) and tryptophan hydroxylase 2 (R439H) knock-in (Tph2KI) (c) animals are shown with BrdU in red. (d) Quantification of the number of doublecortin (DCX)+ immature neurons is shown. Representative DCX images from WT (e) and Tph2KI (f) animals are shown with DCX staining in green. (g) Quantification of BrdU+ cell number one day after BrdU administration. (h) Quantification of BrdU+ cell number 3 weeks after BrdU administration. (i) Quantification of activated caspase-3 staining. (j) Quantification of BrdU+ cell number 3 weeks after BrdU administration in WT and Tph2KI mice treated with fluoxetine (FLX) for 3 weeks. (k) Quantification of granule layer size in control and FLX-treated WT and Tph2KI mice. (l) Quantification of medial habenula size in control and FLX-treated WT and Tph2KI mice. TOTO-3+ nuclei are shown in blue. ‘$’ Indicates P<0.05 by Student’s t-test. ‘#’ Denotes a significant main effect of genotype by two-way analysis of variance, P<0.05; n=12 per group for a–c; n=16–17 per group for d–f; n=9 per group for g, n=11–12 per group for h, n=10–11 per group for i, n=6 per group for j, n=8–9 per group in k and l. The scale bar indicates 20 μm. Arrows denote BrdU+ cells, and arrowheads indicate DCX+ cells.
To determine whether the increased numbers of DCX+ neurons resulted from increased survival of adult-generated neural progenitor cells, Tph2KI and WT mice were administered BrdU for 1 week and were killed either 1 or 21 days later. Importantly, no significant differences in the number of BrdU+ cells were observed in WT and Tph2KI mice killed on day 1 (Figure 2g). However, Tph2KI mice killed on day 21 had significantly more BrdU+ cells than WT controls (Student’s t-test: P=0.0351, degrees of freedom=21, Figure 2h). In addition, IHC analysis for activated caspase-3, a marker of apoptosis, revealed decreased numbers of apoptotic cells within the GCL of Tph2KI animals when compared with that in WT controls (Student’s t-test: P=0.0142, degrees of freedom=19, Figure 2i). We did not observe any statistically significant effects of 3 weeks of FLX treatment (beginning after the cessation of BrdU administration) on the survival of BrdU+ cells in either genotype (Figure 2j), although Tph2KI animals again exhibited an overall increase in survival compared with that of WT controls (main effect of genotype: F(1,20)=4.446, P=0.0478, Figure 2j). To evaluate whether this increased survival of adult-generated neurons might lead to a larger GCL, we compared GCL size in WT and in Tph2KI mice under baseline conditions and following chronic FLX administration. Tph2KI mice were observed to have a significantly larger GCL than WT controls (main effect of genotype: F(1,31)=5.8218, P=0.0219, Figure 2k). Although chronic FLX administration led to a slight increase in GCL size in WT mice, this effect did not reach significance. As a control, no significant genotype or treatment differences were observed in the size of the medial habenula (Figure 2l).
Brain 5-HT deficiency prevents the neurogenic effects of FLX
After observing no significant effects of FLX on cell survival, we next examined the effects of FLX on cell proliferation. No significant main effects of chronic FLX treatment or genotype were observed on BrdU incorporation. However, a significant genotype by treatment interaction was observed (F(1,37)=5.5383, P=0.024, Figures 3a–e). As expected,6 chronic treatment with FLX before BrdU administration increased the number of BrdU+ cells in WT mice (P=0.0259, Figures 3a, c and e). However, this treatment had no effect on BrdU incorporation in Tph2KI animals (Figures 3b, d and e).

Effects of chronic fluoxetine (FLX) treatment on neurogenesis in wild-type (WT) and tryptophan hydroxylase 2 (R439H) knock-in (Tph2KI) mice.Representative bromodeoxyuridine (BrdU)-stained micrographs of WT control (a), Tph2KI control (b), WT FLX (c) and Tph2KI FLX hippocampus (HIP) (d) are shown with quantifications shown in panel e. Arrows denote BrdU+ cells. Representative doublecortin (DCX)-stained micrographs of WT control (f), Tph2KI control (g), WT FLX (h) and Tph2KI FLX (i). Quantification of immunohistochemistry (IHC) for DCX is shown (j). Data were analyzed by two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc tests to compare individual differences. ‘X’ denotes interaction between treatment and genotype (P<0.05) by two-way ANOVA. *Significant main effect of genotype (P<0.05). **P<0.05 compared with WT control by Tukey’s post-hoc test; n=10–11 per group for a–e and n=12–14 per group for f–j. The scale bar indicates 20 μm.
Chronic FLX significantly increased DCX immunoreactivity (main effect of treatment: F(1,55)=8.4018, P=0.0054, Figures 3f, h and j), but a significant genotype by treatment interaction was also observed (F(1,55)=21.3702, P<0.0001, Figure 3j). Indeed, the increased DCX was only apparent in WT mice (P<0.0001) and not in Tph2KI animals. Similar to what was observed above (Figure 2d), control Tph2KI animals exhibited a 30% increase in the number of DCX+ cells, but this effect did not reach statistical significance using Tukey’s post-hoc analysis, only with the less conservative Student’s t-test (P=0.0437). We did not observe a significant increase in BrdU or DCX immunoreactivity in response to chronic DES treatment in either genotype (BDS and TLT, unpublished observations).
We next performed double-labeling experiments to compare the percentage of BrdU+ cells that become NeuN+ neurons between the groups. In both WT and Tph2KI animals, ∼80% of the BrdU+ cells in the GCL were also immunopositive for NeuN 3 weeks after a 1-week exposure to BrdU, and FLX administration did not significantly affect the proportion of BrdU+/NeuN+ cells in either genotype (Figure 4a). We again observed a significant increase in the number of surviving BrdU+ cells in Tph2KI mice compared with WT animals (main effect of genotype: F(1,20)=5.3493, P=0.0315, Figure 4b). Similarly, the total number of BrdU+/NeuN+ neurons was greater in Tph2KI than in WT animals (main effect of genotype: F(1,20)=5.5470, P=0.0288, Figure 4c). Representative images are shown in Figures 4 d–i.

A quantification of double immunofluorescence for bromodeoxyuridine (BrdU) and NeuN in wild-type (WT) and tryptophan hydroxylase 2 (R439H) knock-in (Tph2KI) mice. (a) The average percentage of BrdU+ cells that are also NeuN+. (b) The average total number of BrdU+ and (c) the average total number of BrdU+/NeuN+ cells in control and fluoxetine (FLX)-treated WT and Tph2KI mice. Representative micrographs from control WT (d), control Tph2KI (e), FLX-treated WT (f) and FLX-treated Tph2KI (g) are shown. Higher magnification images of the indicated area in e reveal a BrdU+ and an adjacent BrdU+/NeuN+ cell (h, i). TOTO-3+ nuclei are shown in blue, NeuN is shown in green and BrdU is shown in red. The scale bar indicates 20 μm for d–g and 10 μm for h and i. *Significant main effect of genotype P<0.05, n=6 per group.
Chronic FLX treatment fails to increase hippocampal BDNF mRNA levels in Tph2KI mice
As expected,27 chronic FLX treatment led to significantly increased mRNA levels of BDNF (main effect of treatment: F(3,37)=4.4874, P=0.0409, Figure 5a); however, a significant genotype by treatment interaction was also observed (F(3,37)=4.5347, P=0.0399). Tukey’s post-hoc tests revealed that the effect of FLX was only significant in WT mice (P=0.0216), not in Tph2KI animals. Tukey’s post-hoc tests also revealed that Tph2KI animals exhibit increased hippocampal BDNF mRNA at baseline (P=0.0446). The effects of FLX on CREB mRNA expression in the HIP were dependent upon genotype (significant genotype by treatment interaction: F(3,36)=4.7420, P=0.0361, Figure 5b). However, Tukey’s post-hoc tests did not reveal any significant differences between the groups (although the less conservative Student’s t-test revealed a slight increase in CREB levels in FLX-treated WT mice, P=0.0367, as expected28). We did not observe any significant effects of DES on hippocampal levels of CREB or BDNF (Figures 5c and d).

mRNA analysis and effects of chronic 5-hydroxytryptophan (5-HTP). (a) The effects of chronic fluoxetine (FLX) treatment on hippocampal brain-derived neurotrophic factor (BDNF) mRNA in wild-type (WT) and tryptophan hydroxylase 2 (R439H) knock-in (Tph2KI) mice. (b) The effects of chronic FLX on hippocampal cAMP response element-binding (CREB) mRNA. (c) The effects of chronic desipramine (DES) on hippocampal BDNF mRNA in WT and Tph2KI mice. (d) The effects of chronic DES on hippocampal CREB mRNA. (e) The feeding latencies of WT and Tph2KI mice chronically treated with FLX+5-HTP are shown. Quantification of the number of bromodeoxyuridine (BrdU)+ (f) and doublecortin (DCX)+ (g) cells in FLX+5-HTP-treated WT and Tph2KI mice are shown. *Significant main effect of FLX by two way analysis of variance (ANOVA; P<0.05). **P<0.05 by Tukey’s post-hoc test compared with WT control. ‘X’ indicates significant genotype by treatment interaction by two-way ANOVA (P<0.05); n=10–11 mice per group for a; n=9–11 mice per group for b–d; n=9–10 per group for e, n=11 per group for f and n=11–15 per group for g.
Co-administration of 5-HTP restores the anxiolytic ability of FLX in Tph2KI mice in the NSF
Unlike FLX alone, chronic co-administration of 5-HTP+FLX reduced feeding latency in both Tph2KI and WT animals (significant main effect of treatment: F(3,34)=16.3484, P=0.0003, Figure 5e). Co-administration of 5-HTP+FLX also led to an increase in the number of BrdU+ (P=0.0474, Figure 5f) and DCX+ cells (P=0.0187, Figure 5g) in WT mice. However, 5-HTP+FLX treatment failed to increase the number of BrdU+ (genotype by treatment interaction: F(3,41)=4.1731, P=0.0475, Figure 5f) or DCX+ cells (genotype by treatment interaction: F(3,63)=9.8198, P=0.0027, Figure 5g) in Tph2KI mice. Similar to what was observed above (Figures 2d and 3j), Tph2KI mice exhibited a 55% increase in the number of DCX+ neurons, but this effect did not achieve statistical significance. Chronic 5-HTP+FLX administration also failed to induce a significant increase in BDNF or CREB expression in Tph2KI animals (BDS, unpublished observations).
Discussion
Our results suggest that 5-HT deficiency could reduce the efficacy of FLX by limiting FLX-induced increases in 5-HTEXT, thus blocking downstream cellular and molecular responses. This would be consistent with prior work that has implicated variants in Tph2 in antidepressant sensitivity in humans14, 15 and with prior preclinical work showing that acute pharmacologic inhibition of 5-HT synthesis blocks the acute effects of SSRIs in the TST29 and forced swim test30, 31, 32 in rodents. Although only FLX was examined here, it is likely that other SSRIs would be impacted by 5-HT deficiency as well. A previous report demonstrated that acute 5-HTP administration can restore antidepressant-like responses to acute SSRI treatment in otherwise SSRI-insensitive NMRI mice, suggesting that combined 5-HTP+SSRI therapy could represent an antidepressant augmentation strategy,33 a hypothesis that is further supported by our finding that NSF behavior can be modified in Tph2KI mice by chronic combined 5-HTP+FLX treatment.
Although the specific mutation expressed by Tph2KI mice is extremely rare, 5-HT deficiency could result from many different mutations in 5-HT system genes.34 As such, we hypothesize that the current results will be relevant for a wide range of genetic insults leading to 5-HT deficiency. Although we feel that studies using Tph2KI mice may be highly informative for psychiatric conditions, such as depression and anxiety, we do not claim that these animals completely recapitulate any disorder. Rather, we view these animals as a model of 5-HT deficiency, not of depression or anxiety per se. Similarly, we have utilized the TST and the NSF because of their strong predictive validity for antidepressant action, not on the basis of their face validity or relevance to depression- or anxiety-like behavior. Future studies examining the effects of 5-HT deficiency on responses to chronic stressors may be useful in determining the importance of 5-HT deficiency in regulating susceptibility to stress, which could, in turn, have implications for our understanding of the gene by environment interactions that lead to aberrant emotional behavior.
The observed trend towards a reduction in feeding latency in the NSF in Tph2KI animals compared with that in WT controls is consistent with a role for 5-HT in anxiety-like behavior and is similar to the phenotypes reported in other transgenic models of 5-HT deficiency35, 36 and an acute rat model of 5-HT depletion.37 We hypothesize that the variance in baseline feeding latencies in WT and Tph2KI mice (compare Figure 1 with Figure 5) may be associated with the varying levels of physiological arousal associated with different drug administration paradigms (that is, dietary vs injections). Indeed, previous reports from several groups, including our own, have shown that performance in the NSF test is sensitive to stress.19, 24
The importance of adult hippocampal neurogenesis in depression- and anxiety-like behavior and in responses to antidepressants has been widely debated.21, 38, 39 The reported relationships between neurogenesis and stress,40, 41, 42, 43, 44 along with the fact that completely inhibiting neurogenesis prevents some of the behavioral effects of antidepressants, have suggested a role for hippocampal neurogenesis in the development and treatment of mood disorders.18, 19, 44, 45, 46, 47 However, numerous studies, including the current study, have found neurogenesis to be of limited importance in depression-related behavior and/or in antidepressant-like responses.20, 38, 48, 49, 50
Our finding that chronic 5-HTP+FLX (or DES) administration, which does not increase neurogenesis in Tph2KI animals, reduces feeding latency in Tph2KI mice demonstrates that antidepressant-induced increases in neurogenesis are not required for this effect, at least not in 5-HT-deficient animals. These current results are distinct from previous studies that used X-ray irradiation to ablate all dividing cells, which revealed that antidepressants are ineffective when neurogenesis has been completely inhibited.18, 19 Interestingly, it has been shown that promoting neurogenesis is not sufficient to induce antidepressant-like effects in the NSF,51 and the effects of several classes of experimental antidepressants, such as corticotropin-releasing factor 1 and vasopressin 1b antagonists, reportedly do not require adult hippocampal neurogenesis in animal models.52 Taken together, these data suggest that increased neurogenesis is neither required nor sufficient for feeding latency reductions in the NSF.
Although 5-HT elevation has been repeatedly shown to increase the proliferation of adult hippocampal neural progenitor cells, the reported effects of 5-HT on the survival of neural progenitor cells have been inconsistent. Several groups have reported that chronic FLX administration increases the survival of newly born neurons in vivo,19, 53, 54 but other studies have suggested that chronic FLX increases both apoptosis and cell turnover in the HIP.55, 56 Our results did not reveal a significant effect of FLX on cell survival but did demonstrate an unexpected increase in cell survival in 5-HT-deficient animals compared with that in WT controls. It is possible that the improved survival of adult-generated neurons in Tph2KI mice is related to their increased levels of hippocampal BDNF, which has been shown to have an important role in the survival (but not proliferation or maturation) of adult neural progenitor cells.57 It is likely that the increased size of the GCL observed in Tph2KI mice will have important implications for hippocampal function and hippocampal-dependent behaviors, but future research will be required to evaluate this possibility.
Overall, our data indicate that chronic treatment with FLX can reverse brain 5-HT deficiency in Tph2KI mice but that FLX fails to induce several of its key molecular, cellular and behavioral effects in 5-HT-deficient animals. Importantly, several non-5-HTergic agents (that is, DES and chlordiazepoxide) appear to retain their efficacy in 5-HT-deficient animals, and behavioral responses to FLX can be restored in 5-HT-deficient animals by cotreatment with 5-HTP. These results suggest that 5-HT deficiency may contribute to insensitivity to SSRIs by limiting the magnitude of SSRI-induced 5-HT increments. In addition, the observed decrease in feeding latency induced by combined 5-HTP+FLX treatment in the absence of increased neurogenesis further refines our understanding of the importance of hippocampal neurogenesis in mediating the effects of antidepressants.
References
Merikangas KR, He JP, Burstein M, Swanson SA, Avenevoli S, Cui L et al. Lifetime prevalence of mental disorders in U.S. adolescents: results from the National Comorbidity Survey Replication—Adolescent Supplement (NCS-A). J Am Acad Child Adolesc Psychiatry 2010; 49: 980–989.
Bromet E, Andrade LH, Hwang I, Sampson NA, Alonso J, de Girolamo G et al. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med 2011; 9: 90.
Prina AM, Ferri CP, Guerra M, Brayne C, Prince M . Prevalence of anxiety and its correlates among older adults in Latin America, India and China: cross-cultural study. Br J Psychiatry 2011; 199: 485–491.
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163: 28–40.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM . Neurobiology of depression. Neuron 2002; 34: 13–25.
Malberg JE, Eisch AJ, Nestler EJ, Duman RS . Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 2000; 20: 9104–9110.
Duman RS, Malberg J, Thome J . Neural plasticity to stress and antidepressant treatment. Biol Psychiatry 1999; 46: 1181–1191.
Jacobs BL, van Praag H, Gage FH . Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry 2000; 5: 262–269.
Delgado PL . Depression: the case for a monoamine deficiency. J Clin Psychiatry 2000; 61 (Suppl 6): 7–11.
Walther DJ, Peter JU, Bashammakh S, Hortnagl H, Voits M, Fink H et al. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science 2003; 299: 76.
Zhang X, Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG . Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science 2004; 305: 217.
Zhang X, Gainetdinov RR, Beaulieu JM, Sotnikova TD, Burch LH, Williams RB et al. Loss-of-function mutation in tryptophan hydroxylase-2 identified in unipolar major depression. Neuron 2005; 45: 11–16.
Peters EJ, Slager SL, McGrath PJ, Knowles JA, Hamilton SP . Investigation of serotonin-related genes in antidepressant response. Mol Psychiatry 2004; 9: 879–889.
Tzvetkov MV, Brockmoller J, Roots I, Kirchheiner J . Common genetic variations in human brain-specific tryptophan hydroxylase-2 and response to antidepressant treatment. Pharmacogenet Genomics 2008; 18: 495–506.
Tsai SJ, Hong CJ, Liou YJ, Yu YW, Chen TJ, Hou SJ et al. Tryptophan hydroxylase 2 gene is associated with major depression and antidepressant treatment response. Prog Neuropsychopharmacol Biol Psychiatry 2009; 33: 637–641.
Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC et al. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci USA 2008; 105: 1333–1338.
Jacobsen JP, Siesser WB, Sachs BD, Peterson S, Cools MJ, Setola V et al. Deficient serotonin neurotransmission and depression-like serotonin biomarker alterations in tryptophan hydroxylase 2 (Tph2) loss-of-function mice. Mol Psychiatry 2012; 17: 694–704.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003; 301: 805–809.
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 2009; 62: 479–493.
Holick KA, Lee DC, Hen R, Dulawa SC . Behavioral effects of chronic fluoxetine in BALB/cJ mice do not require adult hippocampal neurogenesis or the serotonin 1A receptor. Neuropsychopharmacology 2008; 33: 406–417.
Hanson ND, Owens MJ, Nemeroff CB . Depression, antidepressants, and neurogenesis: a critical reappraisal. Neuropsychopharmacology 2011; 36: 2589–2602.
Siesser WB, Sachs BD, Ramsey AJ, Sotnikova TD, Beaulieu JM, Zhang X et al. Chronic SSRI treatment exacerbates serotonin deficiency in humanized Tph2 mutant mice. ACS Chem Neurosci 2013; 4: 84–88.
Mjellem N, Lund A, Hole K . Reduction of NMDA-induced behaviour after acute and chronic administration of desipramine in mice. Neuropharmacology 1993; 32: 591–595.
Sachs BD, Rodriguiz RM, Siesser WB, Kenan A, Royer EL, Jacobsen JP et al. The effects of brain serotonin deficiency on behavioural disinhibition and anxiety-like behaviour following mild early life stress. Int J Neuropsychopharmacology 2013; 14: 1–14.
Encinas JM, Vaahtokari A, Enikolopov G . Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci USA 2006; 103: 8233–8238.
Dzirasa K, Kumar S, Sachs BD, Caron MG, Nicolelis MA . Cortical-amygdalar circuit dysfunction in a genetic mouse model of serotonin deficiency. J Neurosci 2013; 33: 4505–4513.
Nibuya M, Morinobu S, Duman RS . Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 1995; 15: 7539–7547.
Nibuya M, Nestler EJ, Duman RS . Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 1996; 16: 2365–2372.
O'Leary OF, Bechtholt AJ, Crowley JJ, Hill TE, Page ME, Lucki I . Depletion of serotonin and catecholamines block the acute behavioral response to different classes of antidepressant drugs in the mouse tail suspension test. Psychopharmacology (Berl) 2007; 192: 357–371.
Cesana R, Ceci A, Ciprandi C, Borsini F . Mesulergine antagonism towards the fluoxetine anti-immobility effect in the forced swimming test in mice. J Pharm Pharmacol 1993; 45: 473–475.
Page ME, Detke MJ, Dalvi A, Kirby LG, Lucki I . Serotonergic mediation of the effects of fluoxetine, but not desipramine, in the rat forced swimming test. Psychopharmacology (Berl) 1999; 147: 162–167.
Gavioli EC, Vaughan CW, Marzola G, Guerrini R, Mitchell VA, Zucchini S et al. Antidepressant-like effects of the nociceptin/orphanin FQ receptor antagonist UFP-101: new evidence from rats and mice. Naunyn Schmiedebergs Arch Pharmacol 2004; 369: 547–553.
Jacobsen JP, Nielsen EO, Hummel R, Redrobe JP, Mirza N, Weikop P . Insensitivity of NMRI mice to selective serotonin reuptake inhibitors in the tail suspension test can be reversed by co-treatment with 5-hydroxytryptophan. Psychopharmacology 2008; 199: 137–150.
Blakely RD . Overview: a rare opportunity or just one less reason to be depressed. Neuron 2005; 48: 701–702, author reply 705-6.
Narboux-Neme N, Sagne C, Doly S, Diaz SL, Martin CB, Angenard G et al. Severe serotonin depletion after conditional deletion of the vesicular monoamine transporter 2 gene in serotonin neurons: neural and behavioral consequences. Neuropsychopharmacology 2011; 3: 2538–2550.
Mosienko V, Bert B, Beis D, Matthes S, Fink H, Bader M et al. Exaggerated aggression and decreased anxiety in mice deficient in brain serotonin. Transl Psychiatry 2012; 2: e122.
Bechtholt AJ, Hill TE, Lucki I . Anxiolytic effect of serotonin depletion in the novelty-induced hypophagia test. Psychopharmacology 2007; 190: 531–540.
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 2009 14 8: 764–773, 739.
Petrik D, Lagace DC, Eisch AJ . The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology 2012; 62: 21–34.
Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E . Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci 1997; 17: 2492–2498.
Gould E, Tanapat P, McEwen BS, Flugge G, Fuchs E . Proliferation of granule cell precursors in the dentate gyrus of adult monkeys is diminished by stress. Proc Natl Acad Sci USA 1998; 95: 3168–3171.
Alonso R, Griebel G, Pavone G, Stemmelin J, Le Fur G, Soubrie P . Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry 2004; 9: 278–286, 224.
Pham K, Nacher J, Hof PR, McEwen BS . Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci 2003; 17: 879–886.
Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA . Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011; 476: 458–461.
Revest JM, Dupret D, Koehl M, Funk-Reiter C, Grosjean N, Piazza PV et al. Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol Psychiatry 2009; 14: 959–967.
Kempermann G, Kronenberg G . Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003; 54: 499–503.
Jacobs BL . Adult brain neurogenesis and depression. Brain Behav Immun 2002; 16: 602–609.
Huang GJ, Bannerman D, Flint J . Chronic fluoxetine treatment alters behavior, but not adult hippocampal neurogenesis, in BALB/cJ mice. Mol Psychiatry 2008; 13: 119–121.
Kitamura T, Saitoh Y, Takashima N, Murayama A, Niibori Y, Ageta H et al. Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 2009; 139: 814–827.
Hanson ND, Owens MJ, Boss-Williams KA, Weiss JM, Nemeroff CB . Several stressors fail to reduce adult hippocampal neurogenesis. Psychoneuroendocrinology 2011; 36: 1520–1529.
Sahay A, Scobie KN, Hill AS, O'Carroll CM, Kheirbek MA, Burghardt NS et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 2011; 472: 466–470.
Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry 2008; 64: 293–301.
Wu X, Castren E . Co-treatment with diazepam prevents the effects of fluoxetine on the proliferation and survival of hippocampal dentate granule cells. Biol Psychiatry 2009; 66: 5–8.
Klempin F, Babu H, Tonelli Dde P, Alarcon E, Fabel K, Kempermann G . Oppositional effects of serotonin receptors 5-HT1a, 2, and 2c in the regulation of adult hippocampal neurogenesis. Front Mol Neurosci 2010; 3.
Sairanen M, Lucas G, Ernfors P, Castren M, Castren E . Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci 2005; 25: 1089–1094.
Egeland M, Warner-Schmidt J, Greengard P, Svenningsson P . Neurogenic effects of fluoxetine are attenuated in p11 (S100A10) knockout mice. Biol Psychiatry 2010; 67: 1048–1056.
Choi SH, Li Y, Parada LF, Sisodia SS . Regulation of hippocampal progenitor cell survival, proliferation and dendritic development by BDNF. Mol Neurodegener 2009; 4: 52.
Acknowledgements
This work was supported in part by grants from the National Institutes of Health (MH79201 and MH60451) to MGC. Support from the Lennon Family Foundation to MGC for the initial part of this work is also greatly appreciated. WBS was the recipient of an NRSA postdoctoral fellowship (F32-MH-083404) and BDS was the recipient of a Minority Supplement award from the National Institutes of Health (MH79201-03S1) and is currently the recipient of an NRSA postdoctoral fellowship (F32-MH093092). JPRJ is the grateful recipient of an individual grant from The Lundbeck Foundation of Denmark. We thank Meghan Rudder for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Sachs, B., Jacobsen, J., Thomas, T. et al. The effects of congenital brain serotonin deficiency on responses to chronic fluoxetine. Transl Psychiatry 3, e291 (2013). https://doi.org/10.1038/tp.2013.65
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2013.65
Keywords
This article is cited by
-
Organic cation transporter 2 contributes to SSRI antidepressant efficacy by controlling tryptophan availability in the brain
Translational Psychiatry (2023)
-
Brain serotonin deficiency and fluoxetine lead to sex-specific effects on binge-like food consumption in mice
Psychopharmacology (2022)
-
Antidepressant Like Effect of Ascorbic Acid in Mice: Possible Involvement of NO-sGC-cGMP Signaling
Neurochemical Research (2022)
-
Possible involvement of NO-sGC-cGMP signaling in the antidepressant like effect of pyridoxine in mice
Metabolic Brain Disease (2022)
-
Effects of chronic fluoxetine treatment on anxiety- and depressive-like behaviors in adolescent rodents – systematic review and meta-analysis
Pharmacological Reports (2022)
