
Abstract
Age-related increases in prevalent dementia over the next 30–40 years risk collapsing medical resources or radically altering the way we treat patients. Better prevention of dementia therefore needs to be one of our highest medical priorities. We propose a perspective on the pathological basis of dementia based on a cerebrovascular-Alzheimer disease spectrum that provides a more powerful explanatory framework when considering the impact of possible public health interventions. With this in mind, a synthesis of evidence from basic, clinical and epidemiological studies indeed suggests that the enhanced treatment of hypertension could be effective for the primary prevention of dementia of either Alzheimer or vascular etiology. In particular, we focus on candidate preventative mechanisms, including reduced cerebrovascular disease, disruption of hypoxia-dependent amyloidogenesis and the potential neuroprotective properties of calcium channel blockers. Following the successful translation of large, long-term and resource-intense trials in cardiology into improved vascular health outcomes in many countries, new multinational prevention trials with dementia-related primary outcomes are now urgently required.
Similar content being viewed by others

Introduction
Hypertension and dementia affect an extraordinary number of individuals. Population-based figures suggest that one in three adults has hypertension as defined by a blood pressure ⩾140/90 mm Hg,1 rising to 60–70% in those over 60 years of age.2 At the same time, about 34 million individuals are affected by dementia worldwide, and by 2050 this is expected to swell to 100 million.3 Together, these two conditions already account for the bulk of disease burden in modern society, and given the continuation of the status quo, risk the viability of modern health-care systems. For example, in Australia, health-care spending on dementia alone will rise to $84 billion by 2060,4 more than any other health condition and equivalent to the total current health budget, clearly an unsustainable scenario.
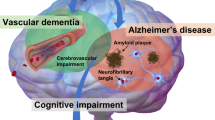
With this in mind, for the past two decades, evidence has accumulated for and against the idea that hypertension and dementia may be causally linked. To date there remains no consensus, reflected by contradictory conclusions in recent systematic and expert reviews.5, 6, 7, 8 In our opinion, at the heart of the debate has been a false dichotomy over presumed etiology—either Alzheimer disease (AD) or Vascular dementia (VaD or alternatively any one of the many related syndromes9). Yet, as will be reviewed below, the pathogenesis of late-onset dementia in the wider community is likely to originate from an interaction of cerebrovascular disease (CVD) and AD processes and in recent years has driven a reconceptualization of mixed dementia as a more accurate description.10, 11
When trying to understand why antihypertensive (AHT) treatment may help prevent dementia, it is also helpful to consider that distinct mechanisms may apply at extreme ends of an AD–CVD spectrum. At the CVD extreme, linkages are seemingly straightforward: Hypertension raises risk for cardiac disease, CVD and stroke, increasingly powerful antecedents of cognitive dysfunction. AHTs may thereby protect against dementia by averting the development of CVD and stroke. At the other extreme, there is a significant literature of complex interactions between hypertension, CVD and AD. Details of the molecular and pathophysiological links between cerebral hypoxia, ischemic injury, aberrant beta-amyloid processing and neuronal dysfunction are becoming clearer. Finally, a third possible mechanism may be specific to the calcium channel blocker (CCB) class of AHTs, which potentially exhibit a neuroprotective mode of action independent of either CVD, AD or their antipressor action. Here we briefly review the clinical and biological evidence base linking hypertensive (HT), AHT treatment and dementia prevention in particular focusing on these three candidate mechanisms.
The CVD–AD dementia spectrum
Even in the most specialized centers, clinically diagnosed Alzheimer dementia patients will invariably be at risk for CVD by virtue of one or more cardiovascular risk factors or will exhibit CVD on fluid-attenuated inversion recovery-weighted magnetic resonance imaging. A degree of CVD is extremely common after the age of 5012 and refers to a collection of distinct disease processes, including large-vessel (macrovascular) and small-vessel (microvascular) disease. Aggregate data from a number of longitudinal studies found one or more different types of CVD in 68% of their sample.13 Yet surprisingly, no imaging study has to date compared the frequency of CVD (using magnetic resonance imaging) with that of AD (using molecular Pittsburgh compound B (PiB) imaging, University of Pittsburgh, PA, USA) in individuals with probable AD dementia. Best estimates from longitudinal, community-based postmortem brain series indicate that AD and CVD often co-occur. The MRC CFAS (Medical Research Council Cognitive Function and Ageing Study) found 61% of demented individuals had AD pathology and 54% CVD.14 A mixed pathological basis for all-cause dementia was also the most common finding in the Rush Memory and Aging Project.15 In those clinically diagnosed with probable AD dementia, postmortem analysis similarly reveals high levels of dual AD and CVD pathology—in fact, coincident disease is more common than pure AD.16 The converse also appears to follow: 30–50% of those with clinical VaD express histological evidence of AD pathology at postmortem,17, 18 corroborated in vivo by a recent study of subcortical VaD patients, which found that 31% were PiB positive.19 These studies are likely to have underestimated the true level of mixed pathology because the standard neuropathological analyses are relatively insensitive to microvascular disease.
In the community, the ‘default’ etiology for dementia may therefore best approximate a mix of AD and CVD along a spectrum. This simple idea has several implications. In the clinic, an optimal dementia-treatment strategy may require management of the AD process, for example via cholinesterase inhibitors, as well as the CVD process, by aggressive reduction of cardiac risk factors and secondary stroke prevention.20 In the context of public health, it elevates elimination of cardiac risk factors as the pre-eminent strategy for dementia prevention,21 for there remain no proven methods for prospectively retarding the AD process (see Figure 1). As we will argue, elimination of HT is arguably the most effective tool at our disposal for primary dementia prevention.

Conceptual diagram depicting the most common type of dementia being of mixed pathology—a combination of AD and CVD. Reducing vascular risk factors not only will have an impact on the CVD end of the spectrum but also will reduce dementia risk in general.
Pathophysiology of hypertension in late life
Hypertension in later life is characterized by a disproportionate elevation of systolic pressure, attributable to reduced distension of the large arteries.22 In the extreme form, this is expressed as isolated systolic hypertension, accompanied by normal (or even low) diastolic pressure. Beyond 50 years of age, biophysical properties of the central arteries change, with collagenosis, loss of elastic laminae and reduced elasticity and elastic recoil in arterial walls.23 Consequently, in older people, the arterial pressure wave is less damped during ventricular systole, pressure rising more with left ventricular ejection and falling more during diastole (because of the lost elastic recoil). Activation of the sympathetic nervous system, an important pathophysiological mechanism of HT in younger patients, is not a feature in elderly patients.24 Further, HT in later life is not typically renin-dependent, with renin activity commonly subnormal.25
Unsurprisingly, HT also produces a number of vascular changes in the brain. Most characteristically, increased pressure in the cerebral vessels produces vascular remodeling, an adaptive but ultimately pathological process that leads to elevated resistance.26 Pathology includes development of lipohyalinosis, wall thickening, luminal narrowing and smooth muscle hypertrophy.27 These degenerative vasostructural changes can disrupt blood flow, eventually leading to focal ischemia, perivascular edema, blood-brain barrier disruption and, in severe cases, cortical deafferentation.28
Effect of hypertension on brain structure and function
HT is closely associated with development of white matter hyperintensities (WMHs) on magnetic resonance imaging.29, 30 WMHs image multiple neuropathological processes, including microvascular disease, inflammation and blood-brain barrier breakdown.31, 32, 33 As the severity of WMHs increases, cognitive function deteriorates. In non-demented adults, WMH severity predicts decreased performance on a number of cognitive domains, particularly information processing speed, attention and executive tasks.34, 35 Furthermore, WMHs are associated with subjective memory complaints,36 predict the development of severe cognitive decline37 and independently predict conversion from mild cognitive impairment to clinically diagnosed AD dementia over 3 years.38
In midlife, hypertension-related microaneurysms, microhemorrhages and soft exudates can be detected in vivo using retinal scans and are associated with subtle cognitive impairment.39 Hypertension also increases the frequency of ‘microbleeds’ on specialized T2-weighted imaging, small 1–2 mm3 areas of hemosiderin deposition, likely due to small hemorrhages as well as leakage of blood through damaged blood vessels.40 Furthermore, Arterial spin labeling magnetic resonance imaging has revealed that HT is linked to widespread cerebral blood-flow deficits, including hypoperfusion of the hippocampus and several cortical areas.41 This may be relevant to the pathogenesis of AD, because the hippocampus is uniquely vulnerable to degeneration early in the disease.42, 43 In Figure 2, we show an example of a cognitively impaired HT individual with severe WMHs indicative of CVD, as well as severe atrophy cortical and hippocampal atrophy, suggestive of an AD process. In general, WMHs are correlated with reduced hippocampal volumes,44 but the temporal sequencing of these two pathologies is not clear.

Fluid-attenuated inversion recovery-weighted magnetic resonance imaging of an elderly cognitively impaired individual with severe white matter disease (hyperintense lesions around ventricles, arrow) as well as severe hippocampal atrophy (highlighted boxed area).
Overall, there appears to be a close link between HT and microvascular disease in the brain, the latter particularly relevant to cognitive dysfunction in later life. One possible mechanism by which AHT treatment may help to minimize the occurrence of cognitive dysfunction is therefore by reducing the incidence of microvascular disease. The PROGRESS (Perindopril Protection Against Recurrent Stroke Study) (see Table 1) has indeed shown that AHT leads to a significant reduction in WMH volume,45 matched by a significant reduction in dementia incidence in those individuals who were cognitively intact at baseline.46 There is even stronger evidence between CVD and AD when examining macrovascular disease in the form of stroke. HT doubles the likelihood of stroke47 and AHT treatment significantly reduces the chances of stroke by 30–40%.48, 49 Given that a history of stroke is a criterion in many classic definitions of VaD, there is a straightforward and valid argument that elimination or better management of HT will lower VaD burden through the prevention of CVD.
Molecular links between CVD and AD
Cerebral ischemia, hypoperfusion and hypoxia appear to be linked with the development of AD pathology. Several journals have dedicated entire special issues to this topic (see editions of Journal of Alzheimer's Disease50 and Annals of the New York Academy of Sciences51)—the breadth of this work is beyond the scope of this review. Rather, here we highlight a few key themes and single out a possible root cause in the form of shared regulatory control of gene-expression networks.
Early clinical data highlighted a synergistic risk between stroke and AD dementia. A stroke increases the likelihood of AD dementia by a factor of three52 and the two diseases potentiate one another, such that the likelihood of dementia for a given level of Alzheimer pathology is eight times higher in the presence of infarction.17 The relationship also appears to be bidirectional, for hippocampal tissue from individuals who died from infarction exhibits increased beta-amyloid (Aβ) pathology as well as greater expression of the APOE protein.53
Ischemia upregulates and deregulates the entire amyloidogenic cascade. Animal models of ischemic injury have found increased expression of not only Aβ protein54, 55 but also its molecular antecedent, amyloid precursor protein (APP),54, 55, 56, 57 and the pathological APP cleavage enzyme, beta-secretase.58, 59 In humans, a detailed spatial analysis of postmortem tissue from individuals with AD dementia found that senile plaques were more likely situated near microhemorrhages and broken capillaries than could be expected by chance,60 hence exposing neural populations to localized hypoxia. A study of the effects of chronic environmental hypoxia in the APP23 transgenic AD mouse found that beta-amyloid cleavage enzyme levels are increased by 151%, Aβ40 generation by 358%, Aβ42 by 185% and numbers of neuritic plaques 1.5 times; moreover, memory performance was impaired over and above that seen in normoxic APP23 mice.58 A causal relationship between CVD and AD is hence quite plausible. However, a key missing link for the field remains the identification of the specific mechanisms by which hypoxia and ischemic injury impinges upon pathological amyloidogenesis.61
Origins for these links may well lie at the molecular level. Laser dissection of hippocampal neurons obtained from individuals with pathologically confirmed AD revealed >1600 gene-expression differences in comparison with control tissue.62 Furthermore, these researchers used a series of novel systems biology analyses to identify underlying patterns within this rich data set. First, AD neuron-specific gene expression changes grouped into several distinct clusters, the primary cluster unique for demonstrating the most coherent changes, focused around altered protein synthesis. Interestingly, functional annotation of deregulated genes within the primary cluster found a series of AD-related gene expression abnormalities (as expected), as well as abnormalities related to cardiovascular disease. For example, greater expression of apolipoprotein E (ApoE) was found in AD neurons and agrees with ApoE4 as the only corroborated genetic risk factor for sporadic AD dementia.63 Less well known is that APOE4 also increases the risk for cardiovascular disease by 42%.64 Moreover, abnormal APOE expression in the brain correlated with abnormal expression of cardiovascular disease-related genes such as kinase deficient protein (WNK1), timpmetallopeptidase inhibitor 1 (TIMP1) and cystathionine-beta-synthase (CBS), as well as several coronary artery disease-related genes. Accordingly, APOE was identified as a hub gene within the primary cluster, maximally connected with other gene-expression changes. Many of the 17 other hub genes have also been implicated in both neurodegenerative and vascular disease processes: microtubule-associated protein 4 (a microtubule stabilization protein important to cell growth and motility in both neurons and cardiocytes), serpin peptidase inhibitor clade A, member 3 (SERPINA3, an enzyme regulator implicated in inflammatory processes relevant to myocardial infarction65 as well as amyloid fibril formation in AD66) and paraoxonase 2 (PON2, a reactive oxygen species scavenger implicated in both atherosclerosis67 and AD68). Evidently, at the neuronal level, gene-expression changes linked to both AD and vascular disease regularly co-occur.
The significance of AD–CVD gene coexpression is not entirely clear, and requires replication, but one possible explanation is that coexpressed genes may also be coregulated.62 Exploratory analysis looked for commonalities in the upstream promoter region of those genes identified within the primary cluster and found 26 transcription factors with binding sites that matched the most common cis-regulatory motifs. Some of these included transcriptional factors that target dozens of different AD-related and CVD-related genes, including Arnt-Ahr (crucial to activating hypoxia-related genes) and Hand1-TCF3 (a member of the basic helix-loop-helix family that has a very wide impact on gene regulation). It is hence possible that CVD and AD co-occur because abnormal protein processes in both diseases share a common transcriptional regulatory network, and so compromise the neurovascular unit. Further research of this nature is highly prized as it may reveal entirely new vascular pharmacological targets for combating dementia across the CVD–AD spectrum.
Hypertension and cardiac risk factors are also dementia risk factors
Long-term population-based cohort studies link hypertension to increased risk for AD dementia69 as well as VaD.70 Two cohorts, each with >15 years follow-up, have shown that hypertension in middle age increases the risk of AD dementia in late life by between 100 and 380%.21, 71 In one study, midlife hypertension increased AD dementia risk independent of other risk factors such as elevated cholesterol and APOEɛ4.72 Intriguingly, studies from Japan have failed to replicate a relationship between midlife HT and AD dementia, whereas confirming links with VaD, leaving open the question of possible cohort differences.73, 74 In later life, hypertension may also be associated with increased risk for dementia, but findings have not been as consistent.69, 75 A possible explanation is that extended follow-up may be essential, because blood pressure falls in the years prior to dementia onset, to the extent that it is often similar or even lower than in non-demented individuals.76
Beyond hypertension, each known cardiac risk factor has also been linked to AD dementia (and VaD), including smoking,77 diabetes78 and midlife obesity.79 Hence, there is an inherent challenge when trying to parse the relative contribution of hypertension from these other metabolic disorders. On the other hand, there is emerging evidence for dose-dependency across cardiovascular risk factors. Luchsinger et al.80 directly compared long-term AD dementia risk in those with and without vascular risk factors, including hypertension, diabetes mellitus, positive smoking history and heart disease. Figure 3 shows the results of a multivariate Hazard model. Compared with those with no risk factors, any single cardiovascular factor significantly increased risk for AD dementia by 70%, two risk factors by 150% and three or four risk factors by an alarming 240%. Accumulative risk for dementia across vascular risk factors has also been reported in an independent cohort with 20-year follow-up data.81 Epidemiological studies are therefore beginning to suggest that hypertension and the other cardiovascular risk factors not only manifest a common risk for dementia but also possess additive independent risks.

A 5-year hazard risk for incident dementia as a function of number of cardiovascular risk factors (adapted from text of Luchsinger et al.80).
Does AHT treatment prevent dementia?
Six large, longitudinal, double-blind, randomized and placebo-controlled trials of AHT medication have investigated effects on dementia incidence and cognitive decline (see Table 1). Importantly, none of these trials was designed for the purpose of dementia prevention, but rather for assessing primary cardiac or stroke outcomes. Hence, baseline and follow-up evaluation of cognition—the key variable of interest in dementia trials—were very limited. The degree of pre-existing cardiac disease for trial entry and the nature and combination of specific AHTs have also varied. A quantitative meta-analytic integration of these results is arguably premature and may explain conflicting outcomes from systematic reviews.6, 7, 8 Nevertheless, results from these trials have been very informative.
The SYST-EUR (Systolic Hypertension in Europe) trial produced the most emphatic results and is unique among the AHT trials for testing CCBs as first-line therapy. After an average follow-up of 2 years, the trial was stopped prematurely because of overwhelming evidence of benefit in the active group: stroke rates had dropped by 42% and cardiac endpoints by 26%.82 At this time incident, dementia was also analysed:48 overall rates were lower than expected with only 32 new cases, mirroring low cardiac and stroke events generally. Despite early cessation of the trial, active treatment reduced the incident dementia rate from 7.7 cases per 1000 patient-years(py) to 3.8 cases per 1000 py, a 50% relative reduction (P=0.05 intention-to-treat analysis, P=0.03 per-protocol analysis). Interestingly, all original placebo subjects were then offered the active-therapy regime for a further 2 years of open-label follow-up.83 At the 4-year stage from randomization, twice as many dementia cases were observed, and moreover, the protective effects of initial CCB AHT treatment appeared to increase rather than diminish: in the active group, 3.3 cases per 1000 py were observable and in the original placebo group, 7.4 cases per 1000 py, a 55% relative decrease (P<0.001). Furthermore, this protective effect was found for both clinical AD dementia and VaD independently. The SYST-EUR trial is therefore the only multicentre, double-blind, placebo-controlled and longitudinal randomized controlled trial (RCT) to provide positive evidence—based on any medical treatment—for effective AD dementia prevention. As summarized in Table 1, no RCT has subsequently replicated the CCB –first-line approach used in the SYST-EUR trial. There are theoretical and experimental reasons to suggest that the strong positive effects in this trial may have been related to both antihypertension-related mechanisms as well as neuroprotective effects unique to CCBs (see next section).
The MRC trial of beta blockers or diuretics found an altogether negative result on cognitive performance in a study administered through general practitioners.84 Interestingly, this study produced an average systolic blood pressure reduction twice as great as the SYST-EUR trial (that is, 16 vs 8 mm Hg), with no effect detected on two simple measures of cognition. Choice and method of cognitive assessment may therefore be as critical as choice of first-line AHT.
The remaining three large RCTs have produced mixed results. The SHEP trial (Systolic Hypertension in the Elderly Program) found a nonsignificant trend for a 16% reduction in dementia incidence in the treatment group compared with controls.85 However, as has been noted in a subsequent reanalyses,86 a higher drop-out rate in the placebo group may have biased results towards the null hypothesis, particularly in the context of a disease with progressive cognitive decline.
The PROGRESS trial targeted individuals with a recent cerebrovascular history rather than hypertension per se using a specific ACEi.87 When analysis was restricted to those without cognitive symptoms at baseline (84% of the original sample), the results for prevention were in favor of a 31% reduction in incident dementia (P=0.02). However, when combined data were used, including those with baseline impairment, the result was lost (nonsignificant 12% risk reduction). As PROGRESS was primarily designed for evaluating the secondary prevention of stroke, dementia associated with recurrent stroke was also investigated. Results were positive in this respect, with findings of a 34% reduction in risk (P=0.03).
The HOPE (Heart Outcomes Prevention Evaluation) trial was also designed for the secondary prevention of stroke.88 Analysis of functional outcomes was restricted to those incident cases of stroke in the four-and-a-half-year follow-up phase. There were 156 new strokes in the treatment group and 226 cases in the placebo group, a significant 32% relative risk reduction. Functional outcomes—including global cognition, motor weakness, speech and swallowing—were also better in the treatment group: 1.1% of individuals with recurrent stroke in the ramipril group had no functional impairment in activities of daily living compared with 1.7% in the placebo group, a significant 39% relative decrease.
The most recent HYVET (Hypertension in the Very Elderly) trial was designed to assess fatal and non-fatal strokes in response to indapamide (±ACEi perindopril) in individuals ⩾80 years.49 Like the SYST-EUR trial, the Hypertension in the Very Elderly was stopped prematurely after an average of 2 years of follow-up because of overwhelming evidence of positive vascular effects: the treatment group had a 30% reduction in strokes, 39% reduction in mortality from stroke and 21% reduction in all-cause mortality. A substudy examining dementia and cognitive outcomes (HYVET-COG) found a nonsignificant decrease in dementia incidence of 14%, equivalent across both VaD and AD dementia.6 The authors noted that their study was under-powered to detect an effect size of this magnitude, and also carried out a meta-analysis of the field including results from their own trial. This most recent meta-analysis found a significant protective effect of AHTs on dementia incidence, with risk reduced by 13%.
Overall, there is imperfect but nevertheless suggestive evidence that AHT treatment may have a useful role in helping prevent dementia and age-related cognitive impairment. To date, only a single RCT has specifically found a protective effect for incident AD dementia, and so further trials of this kind are urgently required, designed intentionally to assess incident dementia. Because a number of trials have been required to stop prematurely because of overwhelming positive effects on myocardial and cerebral infarction, use of placebo arms are no longer justifiable. Rather, dose-finding or head-to-head drug comparisons could be employed. But which AHT to use? Below, we briefly outline the benefits for considering CCBs as first-line treatment.
Candidate CNS mechanisms
Neuroprotective properties of CCBs
Several cohort studies have noted lower rates of dementia (including AD dementia) in individuals treated for hypertension compared with those never exposed to AHTs.69, 89 Furthermore, a more recent study has established that each year of AHT use is linked with an 8% reduction in prospective dementia risk in individuals <75 years of age.90 The question therefore arises whether AHTs may have an intrinsic neuroprotective mode of action?
Experimental studies suggest CCBs may possess neuroprotective properties. For example, cultured neurons are protected in vitro from the effects of hypoxia when pretreated with CCBs.91, 92 Animal models of ischemia also exhibit CCB-dependent neuroprotection in vivo,93, 94 with particularly interesting findings in spontaneously HT rats.95 These animals exhibit widespread neuronal loss, including in hippocampus and cortical regions. Treatment with a blood pressure-lowering dose of nicardipine (3 mg kg−1) rescued hypertension-related neuronal damage.96 Interestingly, a non-hypotensive dose (0.1 mg kg−1) was also sufficient to rescue neuronal numbers in the frontal lobe and protects neuronal loss in the hippocampus. In contrast, the loop diuretic hydrazaline was ineffectual, despite equivalent antipressor action. Potential neuroprotective effects of CCBs may well be independent of their effect on blood pressure, and rather reflect their effect on calcium metabolism.
Dysfunction of intracellular calcium metabolism is widely implicated in brain ageing, ischemia and AD.97, 98 Interaction of the classic AD-related molecule, beta-amyloid (Aβ), with the neuronal plasma membrane leads to elevated intracellular calcium and consequent excitotoxicity.99, 100 The potentially more lethal oligomeric Aβ species also disrupt the cell membrane to produce calcium-mediated toxicity in vitro.101 Calcium metabolism may thereby undergo a ‘double hit’ in the context of hypertension because consequent neuronal injury also leads to calcium pump failure.102, 103
CCBs could thereby exert a neuroprotective effect on vulnerable neuronal populations by normalizing calcium metabolism, in addition to attenuating the development of CVD through their antipressor action. A dual mode of action is also supported by the findings of the SYST-EUR study—unique for using CCBs as first-line AHT therapy—which of all the RCTs found the strongest preventative effects.83 A dual mode of action is hence an interesting hypothesis, but will require more research. In particular, new studies are required using CCBs as first-line therapy or inclusion as a comparison arm in head-to-head trials. To specifically test a potential neuroprotective mechanism, a subtherapeutic trial of CCBs in normotensive individuals using neuroimaging outcomes would be of high interest.
AHTs as AD modifiers
Provocatively, results from one study suggest that several different AHTs may be effective in attenuating the development of Aβ pathology.104 In this study, 55 different AHT medications were first screened in a high-throughput system to analyze their effect on in vitro production of Aβ1−40 and Aβ1−41. Seven different AHT drugs, from across the class spectrum, were found to significantly reduce Aβ production by cultured embryonic neurons. Because the main neurotoxic Aβ species may include soluble oligomers,105 these seven compounds were further screened using an oligomerization assay. Only valsartan, an angiotensin receptor blocker, significantly prevented the polymerization of monomeric Aβ1-41 into Aβ oligomers. Next, 6-month-old transgenic Tg2576 AD mice were treated for 5 months with valsartan at subtherapeutic doses. Accordingly, blood pressure in these animals was unchanged. However, in mice killed at 11 months of age, oligomeric Aβ was reduced by 2–3 times and insoluble Aβ plaques by 50–75%. Furthermore, animals were benefited from improved spatial working memory. Although the mechanism of this action is unknown, an increase in insulin-degrading enzyme protein in the brains of treated animals suggests a possible amyloid breakdown and clearance effect. These interesting data, however, have not been replicated in a subsequent study that also tested valsartan,106 possibly because a different transgenic mouse model was studied (3xTGAD), and were killed at a younger age (6 months), prior to the age of development of significant AD pathology.
Surprisingly, a recent autopsy series has found evidence for possible disease-modifying effects in humans.107 This group compared AD pathology at postmortem in demented individuals with those who had untreated hypertension, treated hypertension or normal blood pressure prior to death. Results are difficult to interpret because dementia severity was different between groups, but nevertheless it was surprising that the treated HT group had significantly less neuropathology than even the normotensive group. There is hence an intriguing possibility that AHT medication may exert an AD-modifying role in humans as it may in rodent models, an idea well suited to further testing with PiB imaging methods.
A hypothetical disease and prevention pathway
Arguably the most controversial idea in the area is that CVD and AD are not spontaneously parallel pathologies but rather that CVD promotes and precedes development of AD. In Figure 4, we propose a hypothetical disease pathway. It begins with hypertension in midlife, itself an endpoint in an idiopathic and multifactorial disease process. Hypertension drives two major brain changes. First, cerebral angiopathy, which at the macroscopic scale resembles that of coronary artery disease and in the most severe form can lead to stroke, while at the microscopic scale, arteriolar and capillary disease in subcortical areas is already highly prevalent by the age of 50 years.12 Second, these structural changes lead to dynamic vascular changes, the most relevant being a significant reduction in cerebral blood flow in the hippocampus.41 Capillary wall damage may be most severe in the hippocampus, for it is a part of the brain with the greatest vessel tortuosity.60 Microhemorrhages appear here first,108 leading to localized microbleeds, hypoxia and neuronal and synaptic damage—the latter is the most faithful biological correlate of clinical symptoms109, 110 and so may coincide with the onset of amnestic problems that are the first clinical sign of incipient dementia. In parallel and perhaps temporally offset, hypoxia potentiates all parts of the amyloidogenic cascade,58 further exacerbated by ischemic excitotoxic and inflammatory byproducts. Beta-amyloid is itself vasconstrictive,111 and so propagates a vicious cycle. As alluded to, these two pathological processes may be joined at a fundamental molecular level, with vulnerable hippocampal neurons exhibiting both AD-related and CVD-related gene expression abnormalities. By the time the individual comes to postmortem, there will be a convergence of AD and CVD pathology in the brain.

The hypothetical disease pathway for development of mixed dementia. AHT medication may have three hypothetical preventative actions: direct antipressor effects, neuroprotection and disease modification.
Following the logic of this schema, AHT medication may have three main benefits that together help prevent and delay dementia onset. First, hypertension is addressed at outset, reducing the degree of vascular remodeling and angiopathy in the brain, and so reversing impediment to cerebral blood flow. This in turn leads to decreased development of microbleeds, microischemia, infarction, and ultimately, reduced synaptic loss. Second, CCBs may be neuroprotective by stabilizing calcium metabolism and also by blocking Aβ-dependent vasoconstriction.111 Third, some AHTs may also act as possible disease modifiers, and so directly decrease Alzheimer pathology.
Conclusions
Control of hypertension is one of the adult medicine's finest preventative treatments, reducing the incidence of stroke and heart attack by between 30 and 40%. For this reason alone, AHTs may have a role in the better prevention of dementia, especially at the CVD end of the spectrum. Many pieces of evidence suggest that AHTs may also have a positive impact against dementia at the AD end of the spectrum. First, AD and CVD are most likely to be fundamentally connected, with evidence of a shared gene regulatory network, frequent coexpression on postmortem analysis, and as well the accumulation of dementia risk across vascular risk factors. Second, clinical trials indicate that AHTs can help combat incident dementia, one multinational RCT of CCBs specifically finding a preventative effect on AD dementia. Because the etiology for dementia in the community is most often a mix of AD and CVD pathology, the relevance of AHT treatment to dementia prevention is in general likely to be greater than previously acknowledged. At a mechanistic level, there are several plausible biological pathways that may be involved, none so far definitive. Primary prevention of stroke and microvascular disease is the logistical first candidate. In addition, CCBs may benefit from a neuroprotective mode of action that is unrelated to their effect on blood pressure. More speculatively, AHTs may interfere with amyloid pathogenesis by reducing hypoxia-dependent enzymatic cleavage of amyloid precursor protein or with greater amyloid breakdown and clearance. AHTs may therefore help combat against dementia by CVD- and AD-related mechanisms, as well as through neuroprotection.
Overall, the multiple lines of evidence reviewed here suggest that better management of hypertension could be effective for the prevention of dementia. Given the forecasts of a global dementia epidemic to accompany the graying of modern nations, better hypertension control may well turn out to also be one of the most important preventative tools of psychiatry and neurology. Several lacunae exist in our current state of knowledge, including a molecular basis for how CVD may lead to AD and a better distinguishing signal from noise in longitudinal epidemiological studies. Perhaps the most glaring deficit is the absence of large-scale RCTs orientated towards dementia prevention outcomes—these are urgently needed. In this regard, the field could learn from a history of long-term, resource-intensive, transnational and multicentre prevention trials in cardiology, and indeed may profit by a closer collaboration between heart and brain specialists.
References
Briganti EM, Shaw JE, Chadban SJ, Zimmet PZ, Welborn TA, McNeil JJ et al. Untreated hypertension among Australian adults: the 1999–2000 Australian Diabetes, Obesity and Lifestyle Study (AusDiab). Med J Aust 2003; 179: 135–139.
Lloyd-Jones D, Evans J, Levy D . Hypertension in Adults across the age spectrum: current outcomes and control in the community. JAMA 2005; 294: 466–472.
Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM . Forecasting the global burden of Alzheimer's disease. Alzheimers Dement 2007; 3: 186–191.
Keeping Dementia Front of Mind: Incidence and Prevalence 2009-2050. Report by Access Economics Pty Ltd for Alzheimer's Australia, 2009.
Daviglus ML, Plassman BL, Pirzada A, Bell CC, Bowen PE, Burke JR et al. Risk Factors and Preventive Interventions for Alzheimer Disease: State of the Science. Arch Neurol 2011; 68: 1185–1190.
Peters R, Beckett N, Forette F, Tuomilehto J, Clarke R, Ritchie C et al. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG): a double-blind, placebo controlled trial. Lancet Neurol 2008; 7: 683–689.
Feigin V, Ratnasabapathy Y, Anderson C . Does blood pressure lowering treatment prevents dementia or cognitive decline in patients with cardiovascular and cerebrovascular disease? J Neurol Sci 2005; 229-230: 151–155.
McGuinness B, Todd S, Passmore P, Bullock R . The effects of blood pressure lowering on development of cognitive impairment and dementia in patients without apparent prior cerebrovascular disease. Cochrane Database Syst Rev 2006; CD004034.
Looi JC, Sachdev PS . Differentiation of vascular dementia from AD on neuropsychological tests. Neurology 1999; 53: 670–678.
Korczyn AD . Mixed dementia–the most common cause of dementia. Ann N Y Acad Sci 2002; 977: 129–134.
Langa KM, Foster NL, Larson EB . Mixed dementia: emerging concepts and therapeutic implications. JAMA 2004; 292: 2901–2908.
Dickson D . Structural Changes in the Aged Brain. In: Timiras P, Bittar E (eds). Advances in Cell Aging and Gerontology. Jai Press: Greenwich, 1997, pp 51–76.
Strozyk D, Dickson DW, Lipton RB, Katz M, Derby CA, Lee S et al. Contribution of vascular pathology to the clinical expression of dementia. Neurobiol Aging 2010; 31: 1710–1720.
Neuropathology Group Medical Research Council Cognitive Function and Aging Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 2001; 357: 169–175.
Schneider JA, Arvanitakis Z, Bang W, Bennett DA . Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007; 69: 2197–2204.
Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA . The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009; 66: 200–208.
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR . Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277: 813–817.
Victoroff J, Mack WJ, Lyness SA, Chui HC . Multicenter clinicopathological correlation in dementia. Am J Psychiatry 1995; 152: 1476–1484.
Lee JH, Kim SH, Kim GH, Seo SW, Park HK, Oh SJ et al. Identification of pure subcortical vascular dementia using 11C-Pittsburgh compound B. Neurology 2011; 77: 18–25.
Deschaintre Y, Richard F, Leys D, Pasquier F . Treatment of vascular risk factors is associated with slower decline in Alzheimer disease. Neurology 2009; 73: 674–680.
Launer LJ, Hughes T, Yu B, Masaki K, Petrovitch H, Ross GW et al. Lowering midlife levels of systolic blood pressure as a public health strategy to reduce late-life dementia: perspective from the Honolulu Heart Program/Honolulu Asia Aging Study. Hypertension 2010; 55: 1352–1359.
Safar M . Hemodynamic concepts of hypertension: from the Eindkessel model to large artery compliance and wave reflections. In: Birkenhager W, Robertson J, Zanchetti A (eds). Handbook of Hypertension. Hypertension in the Twentieth Century: Concepts and Achievements, vol. 22, Elsevier: Amsterdam, 2004, pp 173–191.
Glagov S, Vito R, Giddens DP, Zarins CK . Micro-architecture and composition of artery walls: relationship to location, diameter and the distribution of mechanical stress. J Hypertens Suppl 1992; 10: S101–S104.
Esler M, Lambert G, Jennings G . Regional norepinephrine turnover in human hypertension. Clin Exp Hypertens A 1989; 11 (Suppl 1): 75–89.
Padfield PL, Brown JJ, Lever AF, Schalekamp MA, Beevers DG, Davies DL et al. Is low-renin hypertension a stage in the development of essential hypertension or a diagnostic entity? Lancet 1975; 1: 548–550.
Manolio TA, Olson J, Longstreth WT . Hypertension and cognitive function: pathophysiologic effects of hypertension on the brain. Curr Hypertens Rep 2003; 5: 255–261.
Mulvany MJ . Small artery remodeling in hypertension. Curr Hypertens Rep 2002; 4: 49–55.
Semplicini A, Maresca A, Sartori M, Calo L, Pessina AC . Hypertension and cerebrovascular diseases: a specific role of vascular protection for the prevention of dementia. J Cardiovasc Pharmacol 2001; 38 (Suppl 2): S79–S82.
Dufouil C, de Kersaint-Gilly A, Besancon V, Levy C, Auffray E, Brunnereau L et al. Longitudinal study of blood pressure and white matter hyperintensities: the EVA MRI Cohort. Neurology 2001; 56: 921–926.
Longstreth Jr WT, Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke 1996; 27: 1274–1282.
Pantoni L, Garcia JH . The significance of cerebral white matter abnormalities 100 years after Binswanger's report. A review. Stroke 1995; 26: 1293–1301.
Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R et al. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006; 37: 1391–1398.
Englund E . Neuropathology of white matter lesions in vascular cognitive impairment. Cerebrovasc Dis 2002; 13 (Suppl 2): 11–15.
de Groot JC, de Leeuw FE, Oudkerk M, van Gijn J, Hofman A, Jolles J et al. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol 2000; 47: 145–151.
Au R, Massaro JM, Wolf PA, Young ME, Beiser A, Seshadri S et al. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch Neurol 2006; 63: 246–250.
Stewart R, Dufouil C, Godin O, Ritchie K, Maillard P, Delcroix N et al. Neuroimaging correlates of subjective memory deficits in a community population. Neurology 2008; 70: 1601–1607.
Dufouil C, Godin O, Chalmers J, Coskun O, MacMahon S, Tzourio-Mazoyer N et al. Severe cerebral white matter hyperintensities predict severe cognitive decline in patients with cerebrovascular disease history. Stroke 2009; 40: 2219–2221.
van Straaten EC, Harvey D, Scheltens P, Barkhof F, Petersen RC, Thal LJ et al. Periventricular white matter hyperintensities increase the likelihood of progression from amnestic mild cognitive impairment to dementia. J Neurol 2008; 255: 1302–1308.
Wong TY, Klein R, Sharrett AR, Nieto FJ, Boland LL, Couper DJ et al. Retinal microvascular abnormalities and cognitive impairment in middle-aged persons: the Atherosclerosis Risk in Communities Study. Stroke 2002; 33: 1487–1492.
Kato H, Izumiyama M, Izumiyama K, Takahashi A, Itoyama Y . Silent cerebral microbleeds on T2*-weighted MRI: correlation with stroke subtype, stroke recurrence, and leukoaraiosis. Stroke 2002; 33: 1536–1540.
Dai W, Lopez OL, Carmichael OT, Becker JT, Kuller LH, Gach HM . Abnormal regional cerebral blood flow in cognitively normal elderly subjects with hypertension. Stroke 2008; 39: 349–354.
Braak H, Braak E . Staging of Alzheimer-related cortical destruction. Int Psychogeriatr 1997; 9 (Suppl 1): 257–261.
Jack Jr CR, Petersen RC, Xu Y, O'Brien PC, Smith GE, Ivnik RJ et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology 2000; 55: 484–489.
de Leeuw FE, Barkhof F, Scheltens P . White matter lesions and hippocampal atrophy in Alzheimer's disease. Neurology 2004; 62: 310–312.
Dufouil C, Chalmers J, Coskun O, Besancon V, Bousser MG, Guillon P et al. Effects of blood pressure lowering on cerebral white matter hyperintensities in patients with stroke: the PROGRESS (Perindopril Protection Against Recurrent Stroke Study) Magnetic Resonance Imaging Substudy. Circulation 2005; 112: 1644–1650.
Tzourio C, Anderson C, Chapman N, Woodward M, Neal B, MacMahon S et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med 2003; 163: 1069–1075.
O'Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 2010; 376: 112–123.
Forette F, Seux ML, Staessen JA, Thijs L, Birkenhager WH, Babarskiene MR et al. Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet 1998; 352: 1347–1351.
Beckett NS, Peters R, Fletcher AE, Staessen JA, Liu L, Dumitrascu D et al. Treatment of hypertension in patients 80 years of age or older. N Engl J Med 2008; 358: 1887–1898.
Special Issue ‘Basics of Alzheimer's Disease Prevention’. J Alzheimer's Dis 2010; 20 (guest editor: Jack de la Torre).
Special Issue ‘Vascular Factors in Alzheimer's Disease’. Ann NY Acad Sci 2000; 903.
Kalaria R, Ince P . Vascular factors in Alzheimer's disease. Ann NY Acad Sci 2000; 903.
Qi JP, Wu H, Yang Y, Wang DD, Chen YX, Gu YH et al. Cerebral ischemia and Alzheimer's disease: the expression of amyloid-beta and apolipoprotein E in human hippocampus. J Alzheimers Dis 2007; 12: 335–341.
Nihashi T, Inao S, Kajita Y, Kawai T, Sugimoto T, Niwa M et al. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir (Wien) 2001; 143: 287–295.
Popa-Wagner A, Schroder E, Walker LC, Kessler C . Beta-amyloid precursor protein and ss-amyloid peptide immunoreactivity in the rat brain after middle cerebral artery occlusion: effect of age. Stroke 1998; 29: 2196–2202.
Hall ED, Oostveen JA, Dunn E, Carter DB . Increased amyloid protein precursor and apolipoprotein E immunoreactivity in the selectively vulnerable hippocampus following transient forebrain ischemia in gerbils. Exp Neurol 1995; 135: 17–27.
Li L, Zhang X, Yang D, Luo G, Chen S, Le W . Hypoxia increases Abeta generation by altering beta- and gamma-cleavage of APP. Neurobiol Aging 2009; 30: 1091–1098.
Sun X, He G, Qing H, Zhou W, Dobie F, Cai F et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci USA 2006; 103: 18727–18732.
Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW . Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res 2004; 1009: 1–8.
Cullen K, Kocsi Z, Stone J . Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiol Aging 2006; 27: 1786–1796.
Ogunshola OO, Antoniou X . Contribution of hypoxia to Alzheimer's disease: is HIF-1alpha a mediator of neurodegeneration? Cell Mol Life Sci 2009; 66: 3555–3563.
Ray M, Ruan J, Zhang W . Variations in the transcriptome of Alzheimer's disease reveal molecular networks involved in cardiovascular diseases. Genome Biol 2008; 9: R148.
Myers RH, Schaefer EJ, Wilson PW, D'Agostino R, Ordovas JM, Espino A et al. Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology 1996; 46: 673–677.
Wilson PW, Schaefer EJ, Larson MG, Ordovas JM . Apolipoprotein E alleles and risk of coronary disease. A meta-analysis. Arterioscler Thromb Vasc Biol 1996; 16: 1250–1255.
Licastro F, Chiapelli M, Caldarera CM, Caruso C, Lio D, Corder EH . Acute myocardial infarction and proinflammatory gene variants. Ann N Y Acad Sci 2007; 1119: 227–242.
Kalsheker NA . Alpha 1-antichymotrypsin. Int J Biochem Cell Biol 1996; 28: 961–964.
Ringelstein E, Koschorke S, Holling A . Computed Tomographic Patterns of Proven Embolic Brain Infarctions. Ann Neurol 1989; 26: 759–765.
Shi T, Iverson GM, Qi JC, Cockerill KA, Linnik MD, Konecny P et al. Beta 2-Glycoprotein I binds factor XI and inhibits its activation by thrombin and factor XIIa: loss of inhibition by clipped beta 2-glycoprotein I. Proc Natl Acad Sci USA 2004; 101: 3939–3944.
Qiu C, Winblad B, Fratiglioni L . The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol 2005; 4: 487–499.
Sharp SI, Aarsland D, Day S, Sonnesyn H, Ballard C . Hypertension is a potential risk factor for vascular dementia: systematic review. Int J Geriatr Psychiatry 2011; 26: 661–669.
Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K et al. Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ 2001; 322: 1447–1451.
Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K et al. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med 2002; 137: 149–155.
Ninomiya T, Ohara T, Hirakawa Y, Yoshida D, Doi Y, Hata J et al. Midlife and late-life blood pressure and dementia in Japanese elderly: the hisayama study. Hypertension 2011; 58: 22–28.
Yamada M, Kasagi F, Sasaki H, Masunari N, Mimori Y, Suzuki G . Association between dementia and midlife risk factors: the Radiation Effects Research Foundation Adult Health Study. J Am Geriatr Soc 2003; 51: 410–414.
Power MC, Weuve J, Gagne JJ, McQueen MB, Viswanathan A, Blacker D . The Association Between Blood Pressure and Incident Alzheimer Disease: A Systematic Review and Meta-analysis. Epidemiology 2011; 22: 646–659.
Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L et al. 15-year longitudinal study of blood pressure and dementia. Lancet 1996; 347: 1141–1145.
Anstey K, von Sanden C, Salim A, O'Kearney R . Smoking as a risk factor for dementia and cognitive decline: a meta-analysis of prospective studies. Am J Epidemiol 2007; 166: 367–378.
Jan Biessels G, Staekenborg S, Brunner E, Brayne C, Scheltens P . Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006; 5: 64–74.
Anstey KJ, Cherbuin N, Budge M, Young J . Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev 2011; 12: e426–e437.
Luchsinger J, Reitz C, Honig L, Tang M, Shea S, Mayeax R . Aggregation of vascular risk factors and risk of incident Alzheimer's disease. Neurology 2005; 65: 545–551.
Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J . Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population-based study. Lancet Neurol 2006; 5: 735–741.
Staessen JA, Thijs L, Bieniaszewski L, O'Brien ET, Palatini P, Davidson C et al. Ambulatory monitoring uncorrected for placebo overestimates long-term antihypertensive action. Systolic Hypertension in Europe (SYST-EUR) Trial Investigators. Hypertension 1996; 27 (Pt 1): 414–420.
Forette F, Seux ML, Staessen JA, Thijs L, Babarskiene MR, Babeanu S et al. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med 2002; 162: 2046–2052.
Prince MJ, Bird AS, Blizard RA, Mann AH . Is the cognitive function of older patients affected by antihypertensive treatment? Results from 54 months of the Medical Research Council's trial of hypertension in older adults. BMJ 1996; 312: 801–805.
Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA 1991; 265: 3255–3264.
Di Bari M, Pahor M, Franse LV, Shorr RI, Wan JY, Ferrucci L et al. Dementia and disability outcomes in large hypertension trials: lessons learned from the systolic hypertension in the elderly program (SHEP) trial. Am J Epidemiol 2001; 153: 72–78.
van Gijn J . The PROGRESS Trial: preventing strokes by lowering blood pressure in patients with cerebral ischemia. Emerging therapies: critique of an important advance. Stroke 2002; 33: 319–320.
Arnold JM, Yusuf S, Young J, Mathew J, Johnstone D, Avezum A et al. Prevention of Heart Failure in Patients in the Heart Outcomes Prevention Evaluation (HOPE) Study. Circulation 2003; 107: 1284–1290.
Khachaturian ZS . When will dementia become a curable disease and Alzheimer's a forgotten word? Alzheimers Dement 2006; 2: 1.
Haag MD, Hofman A, Koudstaal PJ, Breteler MM, Stricker BH . Duration of antihypertensive drug use and risk of dementia: A prospective cohort study. Neurology 2009; 72: 1727–1734.
Yamada H, Chen YN, Aihara M, Araie M . Neuroprotective effect of calcium channel blocker against retinal ganglion cell damage under hypoxia. Brain Res 2006; 1071: 75–80.
Wildburger NC, Lin-Ye A, Baird MA, Lei D, Bao J . Neuroprotective effects of blockers for T-type calcium channels. Mol Neurodegener 2009; 4: 44.
Korenkov AI, Pahnke J, Frei K, Warzok R, Schroeder HW, Frick R et al. Treatment with nimodipine or mannitol reduces programmed cell death and infarct size following focal cerebral ischemia. Neurosurg Rev 2000; 23: 145–150.
Mossakowski MJ, Gadamski R . Effect of a calcium channel blocker on the development of ischemic neuron damage in sector CA1 of the horn of Ammon in Mongolian gerbils]. Neuropatol Pol 1987; 25: 439–450.
Amenta F, Tayebati SK, Tomassoni D . Spontaneously hypertensive rat neuroanatomy: applications to pharmacological research. Ital J Anat Embryol 2010; 115: 13–17.
Sabbatini M, Mignini F, Venarucci D, Vega JA, Amenta F . Effect of nicardipine treatment on the expression of neurofilament 200 KDa immunoreactivity in the brain of spontaneously hypertensive rats. Clin Exp Hypertens 2001; 23: 127–141.
Bezprozvanny I, Mattson MP . Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci 2008; 31: 454–463.
Khachaturian Z . The five-five, ten-ten plan for Alzheimer's disease. Neurobiol Aging 1992; 13: 197–198; discussion 199.
Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE . beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 1992; 12: 376–389.
Arispe N, Rojas E, Pollard HB . Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci USA 1993; 90: 567–571.
Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG . Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 2005; 280: 17294–17300.
Grotta JC, Picone CM, Dedman JR, Rhoades HM, Strong RA, Earls RM et al. Neuronal protection correlates with prevention of calcium-calmodulin binding in rats. Stroke 1990; 21 (11 Suppl): III28–III31.
Randall RD, Thayer SA . Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci 1992; 12: 1882–1895.
Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X et al. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest 2007; 117: 3393–3402.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 2008; 14: 837–842.
Ferrington L, Miners JS, Palmer LE, Bond SM, Povey JE, Kelly PA et al. Angiotensin II-inhibiting drugs have no effect on intraneuronal Abeta or oligomeric Abeta levels in a triple transgenic mouse model of Alzheimer's disease. Am J Transl Res 2011; 3: 197–208.
Hoffman LB, Schmeidler J, Lesser GT, Beeri MS, Purohit DP, Grossman HT et al. Less Alzheimer disease neuropathology in medicated hypertensive than nonhypertensive persons. Neurology 2009; 72: 1720–1726.
Cullen K, Stone J . Vascular relationships of heam-rich deposits in the aging cerebral cortex. J Cereb Blood Flow Metab 2005; 25: 1656–1667.
Scheff SW, Price DA . Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging 2003; 24: 1029–1046.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991; 30: 572–580.
Paris D, Town T, Mori T, Parker TA, Humphrey J, Mullan M . Soluble beta-amyloid peptides mediate vasoactivity via activation of a pro-inflammatory pathway. Neurobiol Aging 2000; 21: 183–197.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Valenzuela, M., Esler, M., Ritchie, K. et al. Antihypertensives for combating dementia? A perspective on candidate molecular mechanisms and population-based prevention. Transl Psychiatry 2, e107 (2012). https://doi.org/10.1038/tp.2012.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.28
Keywords
This article is cited by
-
Therapeutically relevant structural and functional mechanisms triggered by physical and cognitive exercise
Molecular Psychiatry (2016)
-
Chronic arterial hypertension impedes glioma growth: a multiparametric MRI study in the rat
Hypertension Research (2015)
