
Abstract
Genetic variation, both single-nucleotide variations and copy number variations (CNV), contribute to changes in gene expression. In some cases these variations are meaningfully correlated with disease states. We hypothesized that in a genetically heterogeneous disorder such as sporadic Alzheimer’s disease (AD), utilizing gene expression as a quantitative trait and CNVs as a genetic marker map within the same individuals in the context of case–control status may increase the power to detect relevant loci. Using this approach an 8-kb deletion was identified that contains a PAX6-binding site on chr2q33.3 upstream of CREB1 encoding the cAMP responsive element-binding protein1 transcription factor. The association of the CNV to AD was confirmed by a case–control association study consisting of the Texas Alzheimer Research and Care Consortium and NIA-LOAD Family Study data sets.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder affecting ∼5.4 million individuals in the United States and is the most common cause of dementia in North America and Europe.1, 2, 3 Genetic factors have an important role in the pathogenesis of AD.4, 5 Rare Mendelian forms of AD have confirmed and elucidated pathways involved in amyloid accumulation, but only account for a small percentage of AD.6 GWAS studies have identified 10 susceptibility loci for the more common, late-onset form of AD7 using single-nucleotide polymorphisms (SNPs) as a genetic marker map. The non-APOE loci account for 35% of the population attributable fractions, suggesting that there is still missing heritability.7 Published copy number variations (CNV) studies in AD8, 9, 10, 11 have focused on large events (>100 kb) to achieve high confidence calls and performed the tests of association on these events.
Genetic variation, both single-nucleotide variations and CNV, contribute to changes in gene expression. In some cases these variations are meaningfully correlated with disease states.12 As GWAS studies are performed with increasing sample sizes7, 13, 14, 15 it is becoming clear that in disorders with marked genetic heterogeneity where the marker specific risk is low in case–control sets, it is difficult to identify the true positives from the false positives and to replicate the results.16, 17 In addition, case–control design in AD suffers from additional confounders, such as misclassification bias due to age-dependent penetrance. The approach of using gene expression data from pathologically ascertained cases and controls eliminates the misclassification bias and gene expression serves as a refined phenotype in a heterogeneous disorder, both increasing the power to detect association signals. We hypothesized that in a genetically heterogeneous disorder such as sporadic AD, utilizing gene expression as a quantitative trait (eQTL) and CNVs as a genetic marker map within the same individuals may increase the power to detect relevant loci, and by incorporating case–control status, these loci will be candidates for case–control association studies.
Materials and methods
Subject cohorts
The discovery cohort consisted of 22 pathological confirmed AD (mean age at death 80, range 61–93) temporal lobes and 15 control temporal lobes (mean age at death 65, range 41–93). Human frozen temporal lobe tissue was obtained from the Alzheimer’s Disease and Memory Disorders Center tissue collection and the New York Brain Bank. Neuropathological diagnosis was assigned by board-certified neuropathologists. AD or control neuropathology was confirmed by plaque and tangle assessment and Braak staging. The brain cohort was exempt from IRB approval as the specimens were deidentified.
The case–control association study consisted of the Texas Alzheimer Research and Care Consortium (TARCC) and the NIA-LOAD Family Study (LOAD) probands http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000168.v1.p1.) The TARCC data set including 381 Caucasian subjects with probable AD and 191 neuropsychologically tested normal controls.18 Probable AD was assigned by NINCDS-ADRDA criteria after consensus review.19 Controls were recruited at each participating site by the same inclusion criteria, including age over 55 years, male and female, unrelated to AD subjects, CDR global score 0, normal performance on activities of daily living, and all information was obtained from surrogate historian. After enrollment all control subjects underwent neuropsychological testing including assessment of global cognitive functioning (MMSE and CDR), attention (Digit Span and Trails A), executive function (Trails B and Clock Drawing), memory (WMS Logical Memory I and WMS Logical Memory II), language (Boston Naming and FAS Verbal Fluency), Premorbid IQ (AMNART), Visuospatial Memory (WMS-Visual Reproduction I and II), Psychiatric (Geriatric Depression Scale; Neuropsychiatric Inventory-Questionnaire) and Functional assessment (Lawton-Brody ADL: PSMS, IADL). Control subjects showing impairment were excluded from the control cohort after consensus review.
The Institutional Review Board of BCM and all participating sites approved the study. Informed consent was obtained from all subjects before inclusion. Genomic DNA was isolated from whole blood by the Puregene DNA isolation kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions.
Isolation of DNA and RNA from brain tissue, expression profiling and genotyping
DNA was prepared with QIAamp DNA mini kit (Qiagen) according to the manufacturer’s instructions. For RNA preparation, brain tissue was homogenized with a tissue homogenizer in Trizol (Invitrogen, Madison, WI, USA) and purified with Rneasy mini kit (Qiagen). DNA and RNA QC criteria for proceeding to array experiments were 260/280 nm 1.6–1.8 for DNA and 1.9–2.0 for RNA, 260/230 nm >2.0 for DNA and >1.5 for RNA by Nanodrop 1000 (Wilmington, DE, USA). RNA quality was further assessed by calculating RIN (RNA integrity number) with Agilent 2100 (Santa Clara, CA, USA) Bioanalyzer (Microarray core facility, Baylor College Medicine). Sample was entered to expression array experiment if RIN >4 (range 4.3–6.9).
Microarray expression profiling was performed on the Illumina Human-6 V3 BeadChip (Illumina, San Diego, CA, USA) (Supplementary information). Genotyping was performed on the Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA, USA) according to the manufacturer’s instructions (Supplementary information). Long-range PCR was designed for the deletion genotyping (Supplementary information). APOE genotyping was performed according to manufacturer‘s instruction with real-time PCR using custom TaqMan probes (Applied Biosystems, Carlsbad, CA, USA) (Supplementary information).
Analysis of array data
Genome-wide CNV inference for the initial analysis
CNV states for each subject were determined by segmentation of the Genome-wide Human SNP Array 6.0 (Affymetrix) data with the Hidden Markov Model algorithm in the Genotyping Console 3.0 software (Affymetrix, Santa Clara, CA, USA) package using detection threshold of 1 kb.
Expression data
Normalized expression array values were obtained by processing Illumina BeadStudio summary data using the VST transform method as implemented in the lumi R package,20, 21 which performs variance stabilization on the low level data to achieve normal distribution. We validated the normal distribution for the significant probes by visualizing residual plots. We also required Illumina BeadStudio probewise detection P-values <0.05 to be considered in the expression analysis. To ensure cross array comparability after the detection filter quantile normalization was applied to the VST summaries across all subjects.
Definition of CNV expression
The UCSC browser track was utilized for mapping every probe to the human genome Build 36; 500 Kb padding was added on each side of the expression probe (1 Mb total window) and any CNV calls overlapping the 1 Mb window was associated with that probe. The 500 kb was selected based on previously published data22 suggesting that within this distance the likelihood of an effect of a SNP on gene regulation is over 99%. All CNV calls were tabulated as integer values 0 (homozygous loss, extremely rare), 1 (single-copy loss) 2 (diploid) 3 (gain) and 4 (multi-gain) within 500 kb upstream and downstream from each transcript for each person. To avoid distortions for rare multi-copy gains or losses, the encoding of one for both homozygous and hemizygous loss, two for normal CNV state and three for gain CNV state was used.
Statistical analysis of the discovery cohort incorporating expression, disease state and CNV information
The following filters were applied to reduce the data set to meaningful comparisons for the hypothesis: (i) expressed in brain (bead level data detection P<0.05), (ii) differentially expressed between cases and controls (t-test, P<0.05) and (iii) CNV events in at least two individuals that are within 1 Mb of the expression probe (frequency >0.05) to avoid spurious associations caused by single events at the extremes of the expression spectrum.
The null hypothesis is that the difference in expression between cases and controls does not correlate with cis-CNV state. Analysis of covariance (ANCOVA) was performed for each expression probe where a linear effect for CNV together with a shift term for case–control status was estimated by least squares. The fraction of variation (partial R^2) attributable to CNV state was assessed by taking the ratio between the regression sum of squares for the CNV information to the regression sum of squares for the full model. The F-statistic and a corresponding P-value were determined by dividing the mean square regression for CNV by the mean square error of the full ANCOVA. This analysis can be performed by selecting corresponding entries from the ANCOVA table from the appropriate statistical software.
Case–control association study
Subsequently to test the hypothesis whether this association could be detected in a larger case–control study we performed a metaanalysis of the TARCC cohort and the publicly available NCRAD-LOAD (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000168.v1.p1) Caucasian probands (N=2166). The TARCC cohort was genotyped on the Affymetrix 6.0 array. The genotype calls were generated by the GoldenHelix software. The NCRAD-LOAD data were analyzed by the Beadstudio software for log2ratio calculation and the genotype calls were based on the log2ratio distributions by inspection of the kernel density estimate of log2ratios and identification of cutpoints that distinguished the three genotype classes. Fisher’s exact test was calculated for the TARCC, LOAD and the metaanalysis. In addition, we developed a factorial model using logistic regression with the logitlink function, which allowed the addition of sex and age as covariates. Odds ratio was calculated for the deletion carrier status in the metaanalysis by collapsing the homozygous and heterozygous deletion carriers.
Results
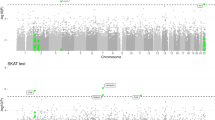
Thrity-seven expression arrays passed QC criteria and 31 genotyping arrays were adequate for copy number analysis. Consideration was limited to (i) CNVs where at least two subjects had other than two copies of the region (locus-specific frequency >0.05), (ii) genes where expression level could be distinguished from background with P-value of <0.05 and that were within 500 kb upstream and downstream from each CNV. These interval parameters detect 90% of interactions extrapolated from SNP data.23 Combining the CNV variation and gene expression filters resulted in 2455 probes corresponding to 2063 genes to be considered in our eQTL and CNV integrated analysis. As the aim is to identify genes that have an association with disease state, the analysis was applied to genes (probes) that are differentially expressed between AD cases and controls (P<0.05). Five hundred seventy-four probes corresponding to 531 genes were differentially expressed between cases and controls. These genes were tested for whether expression was correlated with CNV genotype status. Regression results (F-statistic P-values) for the component of variance explained by CNV in the ANCOVA are depicted in the Manhattan plot (Figure 1). Six probes had P-values of <0.001. These probes also had over 50% of the variance in expression explained by the ANCOVA attributable to CNV. (Table 1) The top hit ARL17P1 corresponds to CNVR 7114.8 and CNVR 7114.10 (http://www.sanger.ac.uk/cgi-bin/humgen/cnv/42mio/downloadBigDB.cgi),24 and the subsequent three probes coding CREB1 and FAM119A correspond to CNVR 1123.1. These latter three probes have similar P-values and identical slopes and thus appear to represent a triplicate experiment. The two additional genes identified on chromosome 1 are SDF4 and NBPF10. SDF4 does not have a known corresponding CNV and NBPF10 corresponds to CNVR 93.13 and CNVR 94.3. The distribution of the expression data is represented with box plots for the three probes on chromosome 2 in Figure 2. Multiple testing corrections for this analysis are confounded by the potential of linked probes to have correlated results because they share the associated CNV resulting in overcorrection. An alternative approach is to calculate the probability of a run of 3 in a row in 574 trials where 6/574 are positives, which is 0.0006. The assumption was made that the modest number of linked probes represents replicate experiments examining the association with CNV state instead of independent experiments; any result identified in the eQTL study was anticipated to be confirmed by tests in a larger case–control cohort. Gene expression levels were validated by TaqMan assay for CREB1 (Supplementary Figure 1). The differential expression of CREB1 between cases and controls was replicated in the data set by Myers et al.23 (#10) (GSE15222); the AD cases had higher expression levels compared with the normal controls (t-test, P=1.36 × 10−18) (Supplementary Figure 2).

The Manhattan plot depicts the −log10p for the F-statistic corresponding to the portion of variation of expression explained by copy number variation (CNV) state in the analysis of covariance (ANCOVA) in the reduced data set after the filters (N=574). The chromosomes are shaded alternately and each expression probe is represented by a dot. Probes with P<0.001 were considered for a case–control association study (red).

The box plots depict the expression levels in Alzheimer’s disease (AD) and controls by copy number state (1 or deletion and 2 or diploid) for the three probes corresponding to the two adjacent genes (CREB1 and FAM199A) on chromosome 2. These probes represent a triplicate experiment as they are associated with the same copy number variation (CNV; CNVR 1123.1) and have similar expression patterns. The association of CNVR 1123.1 with AD was confirmed in the case–control association study.
The case–control cohort (1230 AD subjects and 936 normal controls) consisted of the TARCC data set (381 cases and 191 controls) and the LOAD Caucasian probands (N=848) and Caucasian unrelated controls (N=745). First we studied the TARCC data set as it had adequate coverage of the candidate regions to perform segmentation. The cohort characteristics are summarized in Supplementary Table 1. In the chromosome 2 region the contiguous significant P-values of the overlapping sliding windows delineate the position of the deletion (CNVR 1123.1) detected in the discovery cohort. The association between copy number and case–control status for the sliding windows across the 1 Mb regions are depicted in Table 2 for the chromosome 2 region. The genomic locations of the associated CNVRs are depicted by the bolded probe identifiers in each table. The other regions which were not considered significant after correction for multiple comparisons are summarized in Supplementary tables 2–4. As the LOAD genotyping platform offered a limited number of probes (7), the segmentation algorithms had limited utility.
Second, in order to further validate the association of CNVR 1123.1 the association was tested after generating CNV calls (CN 0, 1 and 2 quantitating homozygous deletion, heterozygous deletion and normal diploid state) in both the TARCC and LOAD cohorts. The 8 kb deletion was covered with 23 probes on the Affymetrix array and 7 probes on the Illumina array. Genotype calls were based on the log2ratio distributions by inspection of the kernel density estimate of log2ratios and identification of cutpoints that distinguished the three genotype classes. Long-range PCR confirmed the chip based genotyping of CNVR 1123.1 for 277 TARCC samples in which DNA was available for this study. The genotyping accuracy of the Affymetrix array is 96%. Typical genotyping results are depicted in Figure 3. The frequencies of the homozygous and heterozygous deletion samples did not defer between the TARCC and LOAD cohorts genotyped on the Affymetrix and Illumina platforms, respectively (Supplementary Table 5). The PCR validation and similar allele frequencies suggest that the genotyping is accurate. The Fisher’s exact test resulted in a P-value of 0.008 in the combined set and the Fisher’s exact test was significant in both the TARCC (for both the Affymetrix and the PCR (Supplementary Table 7) based genotype calls) and LOAD cohorts when analyzed separately (Supplementary Table 6). The odds ratio for the deletion carrier status (collapsed homozygous or heterozygous deletion) was calculated for the metaanalysis (OR 1.23 (1.02–1.49)). Furthermore, we applied a factorial model in the replication set (1230 AD subjects and 936 normal controls) to study the effects of age and gender as covariates. The association remained significant after adding age and gender into the model (P=0.03). APOE data was available for the TARCC set; the regression model remained significant for CNV effect (Table 3).

Genotyping, genomic context and breakpoint analysis of CNVR 1123.1. (a) Genome-Wide Human SNP Array 6.0 is presented as plot of normalized probewise log2ratio data in the three panels for diploid, heterozygous and homozygous deletion carriers (CN=2,1,0). (b) Electrophoresis of long-range PCR products of the same samples. The heterozygous deletion carrier generates two PCR products from the normal and the deletion alleles, respectively. (c) The genomic context of the deletion (black) is outlined in relation to the two genes CREB1 (light purple) and FAM119A (dark purple) in cis. The deletion encompasses a conserved PAX6-binding site (orange arrow). The breakpoint for the deletion was sequenced in five individuals and detected the same inserted sequence at the breakpoint. The inserted sequence is present 400 kb downstream from the deletion suggesting a replicative mechanism for rearrangement.
Third, we studied whether this association is expected to be detected in GWAS studies. SNP association with case–control status was explored using the genotypes of 24 SNPs in the region using the TARCC cohort (Supplementary table 8). The strongest linkage disequilibrium with CNVR 1123.1 was for rs2113383 and rs7561038 with r2 value of 0.39 and 0.22, respectively (Supplementary figure 3). These two SNPs although did not survive multiple testing correction, had the strongest level of association with case–control status (corrected P=0.26 and 0.055, respectively). To delineate the potential mechanism for the deletion event the breakpoint of five homozygous deletion samples was sequenced. Interestingly the breakpoint sequences were identical (Figure 3). In the breakpoint sequence an inserted sequence was identified 400 kb downstream from the deletion (Figure 3).
Discussion
Transcribed sequences were identified with evidence of differential expression in the temporal lobe of post mortem patients with and without AD in which a significant portion of the variation in gene expression was attributable to CNV (Figure 1 and Table 1). Five genes were both differentially expressed between cases and controls and had >50% of the variance explained attributable to the cis-CNV state. Three of the six probes are adjacent to CNVR 1123.1 and fall into a linked region in AD on chromosome 2. Two of these probes are replicates of CREB1 and the third probe is FAM119A, a gene adjacent to CREB1. The sign and size of the regression coefficients and P-values are similar suggesting that the association is observed in triplicate. CREB1 has been implicated in memory by multiple lines of evidence.25, 26 The associated deletion, CNVR 1123.1 has been reported and validated in endeavors to catalog CNVs genome-wide (http://www.sanger.ac.uk/cgi-bin/humgen/cnv/42mio/downloadBigDB.cgi). Genotyping was confirmed by long-range PCR.
Subsequently, the associated CNVs were followed-up in a case–control association study. The case–control association study confirmed that harboring the CNVR 1123.1 deletion increases the risk of AD with an odds ratio of 1.23 (1.02–1.49). The TARCC data set was used (i) to perform a SNP association study with SNPs in the same haplotype block with CNVR 1123.1, and (ii) to evaluate linkage disequilibrium between the SNPs and the deletion in the haplotype block. None of the SNPs tag the deletion perfectly (r2 max 0.39) and while rs2113383 and rs7561038 showed an association with the disease phenotype it was weaker than that of the deletion and did not survive multiple testing correction.
In order to elucidate the mechanism of rearrangement, the breakpoints of five homozygous deletion samples were sequenced. Interestingly the breakpoint sequences were identical. The breakpoint sequence suggests a replication-dependent mechanism27 as the inserted sequence was identified 400 kb downstream from the deletion making recurrent events less likely. The linkage disequilibrium map and the identical breakpoints suggest that the deletion occurred on the ancestral haplotype. As a result the deletion association is a haplotype association where the haplotype is tagged by a single marker (CNVR 1123.1). This haplotype structure likely increased the statistical power and allowed the detection of the association signal. GWAS studies using single SNPs may have not been able to detect this association signal as they did not incorporate the haplotype information.
Variation CNVR 1123.1 400 Kb upstream of CREB1 encompasses a conserved PAX6-binding site,28 which could explain the altered gene expression. PAX6 has been implicated in the development of the eye and nervous system,29, 30 and effects of PAX6 mutations were associated with eye developmental anomalies31and developmental anomalies of the brain.32 The effect of loss of this specific PAX6-binding site has not been studied, and the regulation of CREB1 and FAM119A by PAX6 has not yet been described to our knowledge. Further experimental work will be needed to delineate the relationship between PAX6 and CREB1 or FAM119A. Interestingly, other deletion CNVs in noncoding regions have recently been linked to disease, even common disease.33 A deletion polymorphism upstream of IRGM was found to alter IRGM expression and explained the known association signal in Crohn’s disease.34
The expression of FAM119A adjacent to CREB1 was also significant in the analysis of CNV-gene expression interaction accounting for the disease state; therefore, its potential role cannot be excluded. FAM119A is a predicted methyltransferase with unknown function. CREB1 belongs to the CREB family of leucine-zipper transcription factors that have a central role in cell signal transduction. In a molecular triangulation analysis in AD with APOE, APP, PSEN1 and 2, CREB1 has been predicted as one of the top 10 network nodes.35 Disruption of CREB1 in a mouse model showed neurodegeneration in the hippocampus.36 Expression of a constitutively active form of CREB1 in mice interfered with retrieval of spatial information.37 CREB1 is regulated by alternative splicing and alternative promoters in Aplysia.38, 39
This study presents an approach to detect a phenotypically relevant association in a genetically heterogeneous disorder using gene expression as an eQTL and genetic markers (CNVs in this case) in the same individuals. One limitation of the study is the use of post mortem human brain tissue in the discovery cohort. Interestingly, three probes corresponding to two genes showed very consistent coefficients and P-values; this suggests that degradation was not a major problem at least for these two genes. In addition, the case–control association study used only CNV genotypes derived from blood, with independent methods and statistical approaches, and confirmed the association.
The increased power of the discovery cohort to detect the association is attributable to multiple factors: (i) elimination of misclassification bias by using pathologically confirmed cases and controls, (ii) refined phenotype by using gene expression as eQTL and (iii) deriving genotype and expression data from the same individual allowing for direct correlation. GWAS studies with increasing sample sizes7, 13, 14, 15 suggest that disorders with marked genetic heterogeneity where the marker specific risk is low in case–control sets, may have limitations to detect and replicate association signals.16, 17 This approach may complement the robust GWAS studies to identify biologically relevant loci in this devastating disease with the ultimate goal to find intervention.
References
Ebly EM, Parhad IM, Hogan DB, Fung TS . Prevalence and types of dementia in the very old: results from the Canadian Study of Health and Aging. Neurology 1994; 44: 1593–1600.
Kukull WA, Higdon R, Bowen JD, McCormick WC, Teri L, Schellenberg GD et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol 2002; 59: 1737–1746.
Rocca WA, Hofman A, Brayne C, Breteler MM, Clarke M, Copeland JR et al. Frequency and distribution of Alzheimer’s disease in Europe: a collaborative study of 1980-1990 prevalence findings. The EURODEM-Prevalence Research Group. Ann Neurol 1991; 30: 381–390.
Daw EW, Heath SC, Wijsman EM . Multipoint oligogenic analysis of age-at-onset data with applications to Alzheimer disease pedigrees. Am J Hum Genet 1999; 64: 839–851.
Daw EW, Payami H, Nemens EJ, Nochlin D, Bird TD, Schellenberg GD et al. The number of trait loci in late-onset Alzheimer disease. Am J Hum Genet 2000; 66: 196–204.
Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 1999; 65: 664–670.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 2011; 43: 436–441.
Heinzen EL, Need AC, Hayden KM, Chiba-Falek O, Roses AD, Strittmatter WJ et al. Genome-wide scan of copy number variation in late-onset Alzheimer’s disease. J Alzheimers Dis 2010; 19: 69–77.
Brouwers N, Cauwenberghe CV, Engelborghs S, Lambert JC, Bettens K, Bastard NL et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol psychiatry 2012; 17: 223–233.
Swaminathan S, Shen L, Kim S, Inlow M, West JD, Faber KM et al. Analysis of copy number variation in Alzheimer’s disease: the NIA-LOAD/NCRAD Family Study. Curr Alzheimer Res 2012; 9: 801–814.
Ghani M, Pinto D, Lee JH, Grinberg Y, Sato C, Moreno D et al. Genome-wide survey of large rare copy number variants in Alzheimer’s disease among Caribbean hispanics. G3 (Bethesda) 2012; 2: 71–78.
Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ . Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 2010; 6: e1000888.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 2009; 41: 1088–1093.
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010; 303: 1832–1840.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 2009; 41: 1094–1099.
Ku CS, Loy EY, Pawitan Y, Chia KS . The pursuit of genome-wide association studies: where are we now? J Hum Genet 2010; 55: 195–206.
Florez JC . Clinical review: the genetics of type 2 diabetes: a realistic appraisal in 2008. J Clin Endocrinol Metab 2008; 93: 4633–4642.
O’Bryant SE, Waring SC, Cullum CM, Hall J, Lacritz L, Massman PJ et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer’s research consortium study. Arch Neurol 2008; 65: 1091–1095.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM . Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984; 34: 939–944.
Du P, Kibbe WA, Lin SM . Lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24: 1547–1548.
Lin SM, Du P, Huber W, Kibbe WA . Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res 2008; 36: e11.
Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007; 315: 848–853.
Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L et al. A survey of genetic human cortical gene expression. Nat Genet 2007; 39: 1494–1499.
Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C et al. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet 2010; 42: 385–391.
Barco A, Pittenger C, ER Kandel . CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets 2003; 7: 101–114.
Han JH, Kushner SA, Yiu AP, Hsiang HL, Buch T, Waisman A et al. Selective erasure of a fear memory. Science 2009; 323: 1492–1496.
Hastings PJ, Ira G, Lupski JR . A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 2009; 5: e1000327.
Epstein J, Cai J, Glaser T, Jepeal L, Maas R . Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J Biol Chem 1994; 269: 8355–8361.
Sansom SN, Griffiths DS, Faedo A, Kleinjan DJ, Ruan Y, Smith J et al. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet 2009; 5: e1000511.
Riesenberg AN, Le TT, Willardsen MI, Blackburn DC, Vetter ML, Brown NL . Pax6 regulation of Math5 during mouse retinal neurogenesis. Genesis 2009; 47: 175–187.
Hewitt AW, Kearns LS, Jamieson RV, Williamson KA, van Heyningen V, Mackey DA . PAX6 mutations may be associated with high myopia. Ophthalmic Genet 2007; 28: 179–182.
Numayama-Tsuruta K, Arai Y, Takahashi M, Sasaki-Hoshino M, Funatsu N, Nakamura S et al. Downstream genes of Pax6 revealed by comprehensive transcriptome profiling in the developing rat hindbrain. BMC Dev Biol 2010; 10: 6.
Benko S, Fantes JA, Amiel J, Kleinjan DJ, Thomas S, Ramsay J et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet 2009; 41: 359–364.
McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet 2008; 40: 1107–1112.
Krauthammer M, Kaufmann CA, Gilliam TC, Rzhetsky A . Molecular triangulation: bridging linkage and molecular-network information for identifying candidate genes in Alzheimer’s disease. Proc Natl Acad Sci USA 2004; 101: 15148–15153.
Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 2002; 31: 47–54.
Viosca J, Malleret G, Bourtchouladze R, Benito E, Vronskava S, Kandel ER et al. Chronic enhancement of CREB activity in the hippocampus interferes with the retrieval of spatial information. Learn Mem 2009; 16: 198–209.
Mohamed HA, Yao W, Fioravante D, Smolen PD, Byrne JH . cAMP-response elements in Aplysia creb1, creb2, and Ap-uch promoters: implications for feedback loops modulating long term memory. J Biol Chem 2005; 280: 27035–27043.
Bartsch D, Casadio A, Karl KA, Serodio P, Kandel ER . CREB1 encodes a nuclear activator, a repressor, and a cytoplasmic modulator that form a regulatory unit critical for long-term facilitation. Cell 1998; 95: 211–223.
Acknowledgements
This work was supported by the Cynthia and George Mitchell fund at Baylor College of Medicine. We thank Dr John W Belmont for valuable comments in the manuscript and the Texas Alzheimer Research Consortium (TARC) for the Affymetrix data. Members of TARC (listed in Alphabetical Order): Adams P, Alvarez L, Barber R, Darby E, Diaz-Arrastia R, Dugas DV, Fairchild T, Hittle A, Kneebl J, Mains D, O’Bryant SE, Reisch JS, Rosenberg R, Svetlik D, Tindall M, Waring SC, Williams B and Zhang Y.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Suzanne Z Powell was a speaker at the AAN meeting in Honolulu, and donated her honorarium to the AAN Foundation. She was the ground rounds speaker in July 2011 at the University of Indiana, Department of Pathology and also in March 2012 at the University of Pennsylvania, Department of Pathology and serves on the Editorial Boards of the Archives of Pathology and Laboratory Medicine and Modern Pathology. James R Lupski is a consultant for Athena Diagnostics, holds stock ownership of 23 and Me and Ion Torrent Systems, and is a co-inventor on multiple United States and European patents for DNA diagnostics. The Baylor College of Medicine and Department of Molecular and Human Genetics derive revenue from molecular genetics testing clinical services provided by the Medical Genetics Laboratories; MGL https://www.bcm.edu/geneticlabs. Dr Doody provided consultations to AC Immune, Allon, Astellas, Avanir, Biote, Cardeus, Chiesi, Elan, ExonHit, Genzyme, GlaxoSmithKline, Hoffman LaRoche, Medivation, Merck, Nutricia, Otsuka, Pfizer, Prana, QR Pharma, Sanofi-Aventis, Shire, Sonexa, Takeda, Targacept, Transition and Zinfandel regarding experimental approaches to Alzheimer’s disease and has stock options in QR Pharma, Sonexa and Transition. She receives funding from the NIH Alzheimer’s Disease Cooperative Study from a contract they have with Lilly. She receives funding from NIH AGO 10483 and AGO 224904, and from the Texas Alzheimer’s Research and Care Consortium, and serves as PI on clinical trials funded by Genentech, Janssen with Pfizer, and Pfizer. She serves on the editorial boards of Alzheimer’s Research and Therapy and Dementia and Geriatric Cognitive Disorders. Yanchun Li, Chad A Shaw, Irene Sheffer, Norbert Sule, Brian Dawson, Syed NY Zaidi, Kristine L Bucasas, Kirk C Wilhelmsen and Kinga Szigeti declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, Y., Shaw, C., Sheffer, I. et al. Integrated copy number and gene expression analysis detects a CREB1 association with Alzheimer’s disease. Transl Psychiatry 2, e192 (2012). https://doi.org/10.1038/tp.2012.119
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.119

{kind=link}
{kind=link}
{kind=link}