
Abstract
Ecological processes in the deep sea are poorly understood due to the logistical constraints of sampling thousands of metres below the ocean’s surface and remote from most land masses. Under such circumstances, genetic data provides unparalleled insight into biological and ecological relationships. We use microsatellite DNA to compare the population structure, reproductive mode and dispersal capacity in two deep sea corals from seamounts in the Southern Ocean. The solitary coral Desmophyllum dianthus has widespread dispersal consistent with its global distribution and resilience to disturbance. In contrast, for the matrix-forming colonial coral Solenosmilia variabilis asexual reproduction is important and the dispersal of sexually produced larvae is negligible, resulting in isolated populations. Interestingly, despite the recognised impacts of fishing on seamount communities, genetic diversity on fished and unfished seamounts was similar for both species, suggesting that evolutionary resilience remains despite reductions in biomass. Our results provide empirical evidence that a group of seamounts can function either as isolated islands or stepping stones for dispersal for different taxa. Furthermore different strategies will be required to protect the two sympatric corals and consequently the recently declared marine reserves in this region may function as a network for D. dianthus, but not for S. variabilis.
Similar content being viewed by others

Introduction
Despite recent developments in ocean observing and exploration, the deep sea remains one of the last scientific frontiers. The marine communities that exist at depths in excess of 1000 m are some of the least understood ecosystems on the planet, but they face considerable threats from fishing, mining and climate change and there is an urgency to document their diversity and understand threatening processes to provide a foundation for effective management and conservation of deep sea resources1,2,3.
Evidence of anthropogenic-induced declines in deep sea populations is growing. Fishing, particularly on seamounts, has resulted in depletion of many fish stocks, as well as having indirect effects on benthic communities including ecosystem engineers such as sponges and corals that are very slow to recover4,5,6. Population declines can result in reduced genetic diversity and resilience, hence ensuring connectivity among populations is integral to recovery of exploited populations and future conservation of these vulnerable marine ecosystems7. Accordingly, research on connectivity, particularly in deep sea ecosystems, must be prioritised for future management needs1.
For many marine species, connectivity is maintained through the planktonic dispersal of microscopic larvae or juveniles, and an assumption of a direct link between larval duration and dispersal distance has formed the basis for predicting connectivity in poorly studied ecosystems such as the deep sea8,9. Increasingly, however, studies are revealing such paradigms are not widely upheld in marine systems, and that many factors will collectively influence realised dispersal10,11. These include hydrodynamic features such as oceanic fronts, upwellings, and the stratification of water masses, all of which can constrain the dispersal of long-lived larvae12,13,14,15. In contrast, rafting can result in long-range dispersal for species that have direct-developing larvae with no dispersal phase16 and larval behaviours can facilitate dispersal in currents17,18. Even anthropogenic factors such as pollution and coastal runoff can affect dispersal and population genetic structure19 and isolation-by-distance, although intuitively satisfying as a model for larval dispersal, is only rarely found in marine populations with planktonic larvae e.g. ref. 20.
The mismatch between expected and observed dispersal of marine invertebrates has highlighted the need to incorporate environmental parameters with biological, ecological and genetic data to parametrise models that better explain population or metapopulation structure and predict dispersal in the ocean; hence a growing focus on “seascape genetics” in studies of connectivity in the sea21,22,23,24,25. Even so, there exist limitations to understanding dispersal at depths >1000 m due to a paucity of data on deep ocean currents and the biological and environmental conditions that might affect dispersal including the reproductive mode and timing of most deep sea species26,27. Moreover, despite a burgeoning literature on deep sea genetic connectivity, studies are geographically disparate, taxonomically diverse, plagued by low sample sizes, and poor in spatial and temporal replication1,28 making it hard to draw general conclusions29.
Notably, most deep sea genetic studies have focussed on large-scale patterns of dispersal across thousands of kilometres between geographic features such as oceanic ridges or at continental scales e.g. refs 15, 30, 31, 32, 33, 34, and there have been few studies of small-scale (m to km) patterns of connectivity in the deep sea. In large part this is due to the logistical difficulties of sampling at depth, resulting in fewer samples per site, and confounded by low resolution of genetic markers used28. Increasingly, however, shallow water studies are showing that patchiness in population genetic structure usually exists at small scales, and that for many species, dispersal may be limited to just a few hundred metres or kilometres35,36,37,38. Thus to fully understand ecological relationships in deep sea communities it will be important to study processes occurring both at small, as well as large, spatial scales. Of the few deep-sea studies that have examined small spatial scales, there is evidence that patterns similar to those seen in shallow water may exist. For example, Shank & Halanych39 were able to distinguish among populations and cohorts of the vent tubeworm Riftia pachyptila, and reported genetic structure across distances as little as 400 m. Baco & Shank40 found genetic differentiation among some seamount populations of the gorgonian Corallium lauuense in Hawaii. Similarly, genetic studies of the deep sea coral Lophelia pertusa have revealed evidence of limited dispersal and genetic differentiation among some populations at scales of tens of kilometres41.
Here we combine population genetic data based on microsatellite genotypes of two sympatric and common deep sea coral species - the solitary coral Desmophyllum dianthus and the matrix-forming, colonial coral Solenosmilia variabilis - to predict dispersal within and among seamount populations in the Southern Ocean. These two species of deep sea coral belong to the scleractinian family Caryophyllidae and are abundant and widely distributed on seamounts in the Southern Ocean. The solitary coral, Desmophyllum dianthus, has been recorded to depths of almost 2400 m42 and occurs throughout the world’s oceans. Genetic studies of ITS, 16S, and the mitochondrial control region have shown that populations of D. dianthus on widely separated ridge systems are isolated (1000s of km30) and there is evidence that oceanographic features such as interfaces between vertically stratified water masses within the Southern Ocean are barriers to vertical dispersal in this species15. Unfortunately little is known about the early life history of D. dianthus, but it is presumed to be a seasonal gonochoric spawner like most other species within the family43,44 and is therefore likely to have a relatively short (e.g. days-weeks) planktonic dispersal phase.
The branching coral Solensomilia variabilis has been likened to the Southern Ocean equivalent of Lophelia pertusa both structurally and ecologically. It forms extensive beds on shelf and seamount structures and is an important habitat forming species. In contrast to D. dianthus, DNA sequence data of the 16 S and mitochondrial control regions showed no evidence of subdivision among populations of S. variabilis from southern Australia and New Zealand30 which may reflect long-distance dispersal of larvae, or this may be an artefact of the conserved nature of mtDNA in anthozoans45. Like most deep sea corals, we also know little about reproductive mode and timing in S. variabilis, although mature gametes have been observed in New Zealand specimens in April44, confirming that it is gonochoric and most likely broadcast spawns in late April or May (Autumn) in the Southern Ocean, although reproduction in S. variabilis in the South Atlantic may be continuous46. Solenosmilia variabilis likely undergoes asexual reproduction based on observations of intratentacular budding44.
Here we have sampled both species across the same spatial scales, from oceanic ridges to multiple sites on a single seamount (Fig. 1) in order to make direct comparisons of the scale of dispersal and better understand ecological and evolutionary processes in these deep seamount ecosystems. In addition, recognising the increasing exploitation of deep sea ecosystems, we were able to compare genetic diversity and structure between fished and unfished seamounts to determine if there was evidence that fishing activities had reduced resilience of seamount coral communities.

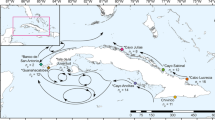
Map of four sampling regions (a) for the two deep sea coral species Desmophyllum dianthus and Solenosmilia variabilis in the Southern Ocean and details of individual seamounts (b) sampled within the Tasmanian Seamounts complex, noting five replicate sites were sampled on each of Mini Matt and Hill U seamounts. Maps were generated using Manifold GIS (V8.29) www.manifold.net based on the publically available Australia Bathymetry and Topography Grid, June 2009 obtained from Geoscience Australia (www.ga.gov.au).
Results
Genetic diversity in deep sea corals
Genetic diversity in D. dianthus from nine seamounts in the Southern Ocean was high. Across the nine microsatellite loci genotyped in this species, we recorded between 13 and 31 alleles, with an average of 21.25 alleles per locus overall, and an average of 8.3 alleles per locus within each of the 16 sampling sites (Table S4). Of the 326 D. dianthus genotyped, there were 324 unique genotypes. Only two genotypes occurred more than once in the sample; two corals from Macquarie Ridge Seamount 5 Station 50 shared the same 8-locus genotype and two individuals from Hill Z56 in the Tasmanian Seamounts shared the same 8-locus genotype. The probability that each of these repeat genotypes arose more than once in the sample through sexual reproduction was incredibly low (<5.75 × 10−11) and so it is likely these are the result of asexual reproduction.
Heterozygosity observed in D. dianthus populations ranged from 0.438 to 0.778 and was lower than expected in all populations (Table S4). Of the eighty single-locus x site tests for Hardy-Weinberg equilibrium in D. dianthus (for those sites where n > 12), more than half (52%) of these were significant, with all representing heterozygote deficits. This pattern did not change after data were adjusted for the presence of null alleles.
Genetic diversity in S. variabilis was lower than that observed in D. dianthus. We recorded between 3 and 34 alleles per locus across the nine seamounts (average = 15.2), and an average of 5.4 alleles per locus at each of the 18 sites (Table S5). Observed heterozygosity ranged from 0.335 to 0.529 and was lower in all populations than expected (Table S5). There were 85 instances of departures from HWE in 134 tests, and 71 of these represented heterozygote deficits. These were in part related to clonality within the populations. Of the 589 samples genotyped, there were only 327 unique genotypes of which 83 were represented by more than one sample. Some of these were quite common (represented by 10–15 individuals) and specimens that shared genotypes were usually collected from the same site. Four clonal genotypes were recorded from two different sites; with two of these being different sites on the same seamount. The probability that repeat genotypes arose through sexual reproduction was low (<1.2 × 10−4) hence they most likely represent the product of asexual reproduction. Estimates of genotypic richness in S. variabilis averaged 0.56 across all sites, but ranged from 0.22 to 0.88 across the nine seamounts (Table 1) indicating varying levels of asexual reproduction at different sites. Clonal genotypes were subsequently removed from the S. variabilis data set and estimates of HWE recalculated. This decreased the number of departures from HWE to just 32 of 134 tests (28 of which were heterozygote deficits), but most of these were associated with Locus SvD213 which had large heterozygote deficits at 14 of the 18 sites sampled and was likely to have null alleles. Excluding SvD213, and with only 18 of the remaining 116 locus x site tests being significant, we considered the sexual populations of S. variabilis at each site to be in Hardy Weinberg Equilibrium.
Despite the known impacts of deep sea fishing on Tasmanian seamounts47,48, there was no evidence that fishing activity had reduced genetic diversity in the coral populations. Comparisons of allelic richness and expected heterozygosity revealed no significant differences in diversity between fished and unfished seamounts either for D. dianthus or S. variabilis (Table 2). Furthermore private alleles were evident in most populations of both species. For D. dianthus multiple private alleles were detected at all but three of the 16 sampling sites and with an average of 2.8 (±0.65SE) private alleles/site. For S. variabilis, there were private alleles recorded at 16 of the 17 sites studied, with an average of 2.2 (±0.39SE) private alleles/site.
Spatial genetic structure within species
There was evidence of multiple genetic groups within each of the two deep sea coral species. For D. dianthus STRUCTURE analysis suggested K = 2 (based on ΔK) (Figs 2a and S1a) or K = 3 (based on L(K)) (Figs 2b and S1b). For K = 3 there was a clear link between genetic structure and depth (Fig. 2), which agrees with findings based on DNA sequence data15 that depth is an important structuring force in this coral species. Principal coordinates analysis also revealed clear groupings of sites based on depth rather than geographic location (Fig. 3) and provided further support for the existence of three depth-related groups based on microsatellite data. Additional STRUCTURE runs on the mid-depth cluster and mid+deep cluster did not reveal any evidence of sub-structure linked to geographic location (i.e. seamount).

Genetic groups within Desmophyllum dianthus, based on output from STRUCTURE analysis and grouped by collection depth.

Principal coordinates analysis showing three clear groups associated with collection depth within the Desmophyllum dianthus samples.
For S. variabilis, the number of populations within the data set was also K = 2 (based on ΔK) (Figs 4a and S2a) or K = 3 (based on L(K)) (Figs 4b and S2b). However, in contrast to D. dianthus, this structure exists within a single depth stratum (mid-depth 1000–1400 m). Interestingly, while some sites appear to be a mix of the 2 (or 3) genetic groups (e.g. HillU Sites 13 & 14, Little Mongrel) other sites are dominated by a single type (e.g. Hill U Sites 12 & 15, Tas 1200, Mini Matt Site 34) (Fig. 4a,b), although there was no clear relationship evident between genetics and location based on principal coordinates analysis (results not shown).

Genetic groups within Solenosmilia variabilis based on output from STRUCTURE analysis and grouped by seamount sampling location.
For D. dianthus it was not possible to test for oceanographic-scale genetic structure as replication was confounded by the depth stratification. However for S. variabilis there was significant genetic differentiation between the Macquarie Ridge and all other sites (pairwise FST ranged from 0.044–0.162, p < 0.001 across all comparisons). This is quite different to the findings based on DNA sequence data (16 S and control region) which revealed a single common haplotype in S. variabilis populations spanning Western Australia, Tasmania, Macquarie Ridge and New Zealand30, reinforcing that microsatellite markers are better suited to revealing population structure than mtDNA sequences for corals45.
Gene flow among seamounts within a single depth stratum (1000–1400 m)
Within the 1000–1400 m depth stratum on Tasmanian Seamounts, the two closely related deep sea corals have very different population genetic structure and gene flow. For D. dianthus there are only very low levels of genetic subdivision, with two of three test statistics suggesting gene flow occurs among the seamounts (FST = 0.009, p < 0.001; G’ST = 0.036, p = 0.128; DEST = 0.03, p = 0.129 considering all sites; FST = 0.006, p < 0.001; G’ST = 0.024, p = 0.061; DEST = 0.021, p = 0.062 across sites where n > 12). Pairwise comparisons showed significant differences between sites in only one comparison, between Hill Z15 and Dory Hill and again this was significant based on FST, but not DEST (Table 3). Estimates of gene flow for D. dianthus were moderate, with Nem = 43.6 (based on FST), Nem = 5.6 (based on private alleles) and Nem = 19.6 based on coalescent analyses.
For S. variabilis there is significant genetic subdivision among all the Tasmanian seamounts (FST = 0.111, p < 0.001; G’ST = 0.167, p < 0.001; DEST = 0.103, p < 0.001) indicating populations are not panmictic. Most pairwise comparisons among seamounts showed significant genetic differentiation indicative of limited gene flow, with the exception of four of the 153 tests (between L Mongrel and Mini Matt Stn 34, Dory Hill and Tasman 1200, Dory Hill and Mini Matt Stn 33 and Hill Z56 and Mini Matt Stn 33; Table 4). Estimates of gene flow were considerably lower in S. variabilis than in D. dianthus, with Nem = 3.752 (based on FST), Nem = 2.25 (based on private alleles) and Nem = 0.94 based on coalescent analyses.
There was no evidence of isolation by distance in either species across the Tasmanian Seamounts (D. dianthus: Mantel’s R2 = 0.025, p = 0.321; S. variabilis Mantel’s R2 = 0.014, p = 0.106). For D. dianthus, values of Nem based on coalescent estimates of migration were generally large and ranged from 0.09 to 156. Of the 50 estimates of migration between all pairs of sites, 38 resulted in Nem > 1, and 20 of these with Nem > 10 indicating sufficient gene flow for ecological connectivity among most seamounts49. Seventy three percent of pairwise estimates of Nem among seamounts were significantly greater than 1 (and only 3% significantly <1; Table S6) emphasising the considerable gene flow among populations of D. dianthus. Furthermore, in 15 of the 25 seamount pair comparisons there was significant bi-directionality in migration (Fig. 5, Table S6) and overall ≈70% of migration in D. dianthus occurred in a westerly direction. This is consistent with estimates of particle dispersal at ~1000 m in the Tasmanian Seamount region which predicts a westward flow in the months when D. dianthus is most likely to be spawning (Fig. 6). However, tests of four different models of gene flow showed strong support only for a full model of dispersal (p > 0.9) above a westward, eastward or panmictic pattern of dispersal, suggesting either that reproduction and dispersal occurs outside of the period of predominately westward flow or other mechanisms are influencing gene flow in this species.

Arrows indicate where there is statistically significant directional gene flow between pairs of seamounts, with the arrow indicating the direction of the highest gene flow. The width of lines is directly proportional to the magnitude of gene flow, which ranges from Nem = 6.96 (Hill Z9 to Hill U) to Nem = 151.5 (Hill U to Dory Hill). Dashed lines show other instances where Nem >10 (i.e. sufficient to be considered ecologically connected). Red arrows indicate westerly flow, green arrows indicate easterly flow. The figure was created using Microsoft PowerPoint based on migration rates calculated in LAMARC.

The dispersal probabilities were generated using “Connie2.0” CSIRO Connectivity Interface http://www.csiro.au/Connie2/ and are based on the sum across years 1993–2007. Dispersal was modelled at 995 m depth (the black cross marks release point), with a dispersal period of 25 days, and across the likely reproductive period for D. dianthus and S. variabilis in the Southern Ocean (January to May44). The figure was created in R version 3.1.289 using the packages raster90, rasterVis91 and sp92.
For S. variabilis, pairwise estimates of Nem based on coalescent estimates of migration among seamounts were low and ranged from 0.001 to 10.76 indicating insufficient gene flow between any seamounts for ecological connectivity. In fact 49 of the 56 estimates were <1 indicating that seamount populations of S. variabilis are ecologically isolated and only 5% of estimates showed Nem significantly greater than one (Table S7). For S. variabilis nine of the 28 pairwise estimates of Nem were significantly different to each other, although since most of these involved Nem < 1, are likely meaningless in the context of directional gene flow. Notably the highest estimated gene flow occurred into Hill Z15 from Hill Z56 (Nem = 10.75; eastward flow) compared with Nem of 0.49 in the opposite direction and into MiniMatt from Hill Z56 (Nem = 9.35; westward flow) compared with Nem of 2.84 in the opposite direction.
Estimates of effective population size also differed between the two coral species, being much smaller in S. variabilis (mean Θ = 1.31; 95% CI 1.19–1.47) than in D. dianthus (mean Θ = 9.81; 95% CI 7.61–10.01). Contemporary estimates of effective population size revealed similar patterns, with populations of S. variabilis relatively small but more variable across sites than Θ (mean Ne = 11.69; 95% CI 6.65–68.7) and populations of D. dianthus very large (mean Ne = infinite; although this likely reflects limitations of calculating Ne for populations where Ne is >1000, rather than an accurate estimate of effective population size50).
Gene flow within seamounts
The genetic structure of coral populations among sites within the same depth stratum on a seamount was very different for D. dianthus and S. variabilis. There was small-scale structure in S. variabilis both at Hill U and Mini Matt, with pairwise comparisons (based on FST and Jost’s D) significantly different to zero in most comparisons (Table 4) suggesting limited gene flow among sites even separated by as little as a few hundred metres on the same seamount. Different genetic populations among the sites at Hill U are also apparent from the STRUCTRUE analysis (Fig. 4a,b). The structuring in S. variabilis is the opposite of what we found at mid-depth for D. dianthus, although unfortunately sample sizes were less robust at the within-seamount scale in this species and consequently the results are less clear. On Hill U, there was no significant genetic differentiation between Stations 13 and 14 which are separated by approximately 700 m based on G’ST (0.005, p = 0.217) and Jost’s D (0.021, p = 0.229) although FST was significant (0.011, p < 0.05). Similarly on the Macquarie Ridge, the two shallow sites sampled on the top of Seamount 5 (~3 km apart) were not significantly differentiated from each other based on G’ST (0.003, p = 0.229) and Jost’s D (0.021, p = 0.242) but were based on FST (=0.016, p < 0.05) and the same pattern was evident for the three deep sites sampled on the Cascade Plateau and that are separated by as little as 300 m. However the consensus between G’ST and Jost’s D is compelling evidence51 that there is no genetic differentiation among D. dianthus populations at the within-seamount scale, and that contemporary gene flow occurs among these sites. Coalescent estimates of migration among sites are, however, low (Table S6) and in line with significant FST results found in comparisons among sites both within and among seamounts for D. dianthus which likely reflects historical structure among populations.
Within seamounts there was little evidence of directional migration among sites within the same depth stratum. For D. dianthus, coalescent estimates of m and Nem indicated similar levels of exchange in either direction among sites on Hill U, among sites on Cascade Plateau and between sites on Seamount 5 on the Macquarie Ridge (Table S8), the only exception being that there was almost ten times more migration from Station 15 to Station 14 on Hill U than in the opposite direction (net north-westerly flow). Furthermore, most estimates of Nem were significantly greater than one (Table S8). Overall these results suggest D. dianthus populations on seamounts are well mixed, at least within depth zones.
For S. variabilis, there was also limited evidence of directional migration among sites within seamounts, particularly at Mini Matt, noting that all estimates of m and Nem are very low, with many being significantly less than one indicating populations are effectively isolated (Table S9). However on Hill U there was some indication that Stations 12 and 13 were a source of migrants to Stations 14, 15 and 16, although this resulted in little effective gene flow among sites as Nem was <10 in all instances (Table S9).
Discussion
Seamounts have variously been considered as isolated islands of endemicity or stepping stones for dispersal across ocean divides52; here we provide genetic evidence that both hypotheses are valid. Data from hypervariable microsatellite DNA markers have shown the solitary coral Desmophyllum dianthus has relatively large effective population sizes and considerable gene flow indicating the seamounts are acting as stepping stones for connectivity among sites in the Tasmanian Seamount Region at depths of 1000–1400 m. In contrast, the matrix-forming Solenosmilia variabilis across the same set of seamounts and depth range has small effective population size, populations are genetically differentiated, asexual reproduction appears to be an important aspect of its life history, and there is limited gene flow and larval dispersal even on relatively small spatial scales among sites on a single seamount. The starkly contrasting nature of these coral populations provides considerable insight into the variety of ecological processes operating in the deep sea.
Links between ocean currents, distribution patterns and larval dispersal in deep sea corals
The distinct genetic structures and contrasting connectivity in D. dianthus and S. variabilis are remarkably consistent with the species’ ranges. D. dianthus is common across all oceans53,54. The cosmopolitan distribution of many marine species has been used to infer widespread dispersal55 and this is supported in D. dianthus by genetic studies that reveal shared nuclear (ITS and 28S) and mitochondrial (16S and CO1) haplotypes in specimens from Australia to the Mediterranean (Addamo et al. 2012). Our microsatellite genetic data provides further evidence that larval dispersal and gene flow can occur in this species across hundreds of kilometres. Indeed patterns of migration appear to be influenced, at least in part, by the prevailing westerly currents across the Tasmanian Seamounts suggesting passive larval dispersal facilitated by stepping-stones such as intermediate seamounts may play a role in the distribution of this widespread and common species.
Solenosmilia variabilis is common in the southern hemisphere, forming extensive reef-like structures particularly around southern Australia and New Zealand47,56. It has been reported from the North Pacific and North Atlantic but is rare in these locations (www.gbif.org). A much reduced dispersal capacity, as inferred from genetic differentiation within and among Tasmanian seamounts, may explain the restricted distribution and localised abundance of S. variabilis. For the confamilial and matrix-forming Lophelia pertusa, planulae are active swimmers and can survive for at least eight weeks in the water column, although they will commence settlement behaviour after 3–5 weeks57. These behaviours suggest potential for long-distance dispersal, although genetic structure among reefs indicates L. pertusa populations are not panmictic and that dispersal may be affected by local hydrodynamic and geographic features41. Arguably, S. variabils and L. pertusa are the southern and northern hemisphere ecological equivalents of each other, and it is interesting that genetic structure indicative of restricted larval dispersal is apparent in both these matrix-forming species that have apparently constrained distributions.
Unfortunately too little is known about reproduction and the early life history of D. dianthus and S. variabilis to know if the approximately three times greater level of genetic differentiation seen in S. variabilis compared with D. dianthus (based on G’st and Jost’s D) links directly to larval duration, other aspects of their life history, or environment. For example, spawning in winter compared with summer may result in very different dispersal potential based on the magnitude of currents at 995 m in the Tasmanian Seamounts (Figure S3). Furthermore strong support of a full model of gene flow reflecting different levels of dispersal among all sites, in contrast to the more simplistic model of westerly dispersal in D. dianthus suggests either a different reproductive period than assumed here (January to May), or that other factors such as larval behaviour are influencing dispersal patterns. Of note also is that the hydrodynamic model used here (Figs 6 and S3) only considers processes at large scales (~10 km grid). At smaller within-seamount scales, other processes associated with boundary currents, eddies and larval behaviour will affect the dispersal of larvae, and this is evident in the variation in migration estimates among sites even within seamounts (Tables S4 and S5). Clearly more information on the timing and nature of reproduction in will be required to be able to refine oceanographic predictions and fully understand the links between currents, larval behaviour and dispersal for these deep sea coral species.
One final consideration is that the contrasting genetic structure in D. dianthus and S. variabilis could be linked to different reproductive modes. We know that S. variabilis is gonochoric and probably a broadcast spawner; but nothing is known of reproductive mode in D. dianthus. Conceivably, D. dianthus may brood larvae that are well provisioned and better suited to long-distance dispersal58. Recruitment success in deep sea gorgonians has been linked to differences between brooding and spawning life histories59, although notably the broadcast spawning species was more successful than the brooding species, possibly linked to larval supply rather than dispersal. However in shallow water corals, there is little evidence that brooders will be more widely dispersed or more successful than spawners60,61 or that there will be a predictable link between life history and dispersal62, hence reproductive mode (brooding vs. spawning) is an unsatisfactory explanation for the differences in gene flow between D. dianthus and S. variabilis.
The importance of asexual reproduction in maintaining deep sea coral populations
We found evidence that asexual reproduction is important for localised recruitment in S. variabilis. This finding suggests even closer affinity with its northern hemisphere ecological equivalent L. pertusa which also recruits through a combination of sexual and asexual reproduction41,63,64. On some reefs, clonal reproduction can account for more than 50% of recruitment in L. pertusa41 and in S. variabilis we found as many as 76% of samples were clonal, although clearly there is variation in the importance of asexual reproduction in both species (Table 1 and refs 41, 63, 65). One concern in this study was our power to detect clonality due to the indirect method of sampling using benthic sleds which will invariably result in some fragmentation of colonies. However, with this in mind, we targeted our genetic collections at each site so as to sample progressively through each sled sample to avoid colony fragments (assuming fragments would be grouped together as a colony was crushed in the net) and in an attempt to obtain accurate estimates of genetic diversity across the sampling site. Future targeted ROV collections would be required to precisely estimate the extent and nature of clonality in S. variabilis i.e. through natural fragmentation, colony extension or the production of asexual larvae. However, given S. variabilis has been reported to reproduce asexually44, forms extensive mono-specific stands on seamounts47,56 and that each of our sleds likely covered tens to hundreds of metres of ground, we expect that asexual reproduction is an important life history strategy in this species. In contrast, sexual reproduction is the main source of recruitment in D. dianthus, further emphasising the variation of life histories even in closely related deep sea corals.
Effects of deep-sea fishing on seamount coral populations
Although there may be evidence of reduced biomass of corals on seamounts that have been fished4,5,48 we found no evidence that corals on fished seamounts had lower genetic diversity than those on unfished seamounts. The loss of genetic diversity may be buffered in species with long generation times66, and this may explain why genetic diversity in these long-lived coral species on the Tasmanian seamounts does not appear to have been reduced to a level likely to affect their evolutionary resilience.
Our genetic data, which gives insight into life history processes in these little-understood corals, may help explain the observed effects and limited recovery of some deep sea corals following fishing. For example, S. variabilis populations on Tasmanian seamounts showed almost no signs of recovery 5–10 years after trawling ceased48. This slow recovery matches with our discovery of the importance of asexual reproduction for local recruitment in this species, as well as the limited capacity for larval dispersal and importance of local recruitment. In combination, these life history characters would certainly hinder recovery of populations where adult biomass was severely reduced.
Solitary corals are among the species that seem least affected by fishing5, and may also represent early colonisers on seamounts that have been impacted by fishing. Again our genetic data, which suggests a greater capacity for larval dispersal in the solitary D. dianthus (compared with S. variabilis), is consistent with their reported relative capacity to recover and/or recolonise following disturbance, and supports the suggestion that solitary corals such as D. dianthus will be less vulnerable to fishing effects than colonial species5. Importantly, it is worth noting that although the capacity for dispersal and gene flow is higher in D. dianthus than in S. variabilis, migration rates are still low to moderate in both species, and arguably lower than expected if populations were panmictic. Although only “one migrant per generation” is considered sufficient gene flow to prevent evolutionary divergence among populations, from a conservation and management perspective, Nem of >10 migrants per generation may be a more realistic measure of connectivity49,67, which is apparent at most scales for D. dianthus, but not for S. variabilis. In this context, S. variabilis is likely to be more vulnerable to anthropogenic impacts than D. dianthus.
Implications for high seas management and conservation
Collectively, the results from this study provide critical input to the future management of deep sea corals. Based on genetic data it is clear that these two closely related coral species have very different population structures and capacity for dispersal. In this context, conservation measures such as Marine Protected Areas will need to consider the different scales across which dispersal processes are operating in order to achieve adequate protection or ensure the provision of networks of connectivity for all taxa. For example, the recently declared Commonwealth Marine Reserves in SE Australia will most likely be operating as networks for D. dianthus, but not for S. variabilis. Similarly, the size and timing of management actions such as fishing closures would have different consequences for recovery in each species. Given the importance placed on conservation of ecosystem engineers in managing marine ecosystems more broadly68,69 then clearly any measures that capture the breadth of life histories and connectivity for these two corals will undoubtedly have consequences for the persistence of a broad suite of seamount species.
From a conservation perspective, the magnitude of difference in effective population size between the two coral species must also be considered. Maintaining large effective size of populations is considered paramount in conservation, as small populations will be susceptible to the effects of drift and concomitant loss of genetic diversity and adaptive capacity70. Here we have shown that S. variabilis has relatively small effective population sizes compared with D. dianthus both in evolutionary and ecological timescales, hence S. variabilis populations may be particularly vulnerable to the effects of genetic drift, and this may be exacerbated if populations are further reduced e.g. from fishing or climate change. Furthermore, the low apparent migration rate and importance of asexual reproduction in S. variabilis are also life history traits that emphasise the vulnerability of this species71 relative to the sympatric D. dianthus.
This study provides another example of the value of genetic data to underpin informed management and conservation measures, but this is particularly so in ecosystems such as the deep sea which is remote and where it is logistically difficult to obtain ecological data in alternate ways.
Methods
Study species and sample collection
Specimens of both species were collected by benthic sled from seamount and shelf locations at depths ranging from 489–2395 m during three research voyages in the Southern Ocean: The South East MPA Survey south of Tasmania on the RV Southern Surveyor in March/April 2007 (Voyage SS200702), the Macquarie Ridge on the RV Tangaroa in 2008 (Voyage TAN0803) and the Tasmanian Seamounts on the Thomas T. Thompson in 2008 (Voyage TT200801; samples collected by sled, and also using the Jason ROV). Sleds typically covered a few hundred metres of ground at each site. All samples were preserved whole in 95% ethanol or ultra-frozen for genetic analysis. In total 10 different seamounts and one slope location were sampled for D. dianthus and eight seamounts and one slope location for S. variabilis (Fig. 1) spanning a range of spatial scales from ~100 m to >2000 km. For both species, seamounts were classified as either “unfished” (i.e. those that were located in the Tasman Seamounts Reserve that had been closed to fishing since 1997) or “fished” (i.e. fishing activities were still permitted at the time of sampling).
Microsatellite genotyping
The development of microsatellite loci for the two deep sea coral species is described in the Supplementary Material. DNA was extracted from a total of 326 D. dianthus individuals from 16 sites (Table S1) using the Qiagen DNeasy Blood and Tissue DNA Extraction Kit. PCR amplification of eight microsatellite loci was done as two separate multiplexes because allele sizes overlapped in many loci; Panel 1 - DdB4, DdB9, DdC102, DdD109 and Panel 2 - DdB114, DdB118, DdC6, DdC107. The forward primer for each locus was 5′ labelled with either 6FAM, VIC, NED or PET fluorescent dyes (Table S1). Multiplex amplifications were performed in 25 μL volumes comprising 12.5 μL of 2× Qiagen Multiplex PCR Master Mix, 0.1 μM of each primer, 2.5 μL of 5× Q-solution and 10 ng DNA. Thermal cycling comprised 15 min at 95 °C, followed by 10 cycles of 94 °C for 30 sec, 59 °C annealing for 90 sec and 72 °C extension for 60 sec, then 25 cycles of 94 °C for 30 sec, 57 °C annealing for 90 sec and 72 °C extension for 60 sec with a final 30 min extension at 72 °C.
For S. variabilis, DNA was extracted from a total of 589 samples from 18 sites using the Qiagen DNeasy Blood and Tissue DNA Extraction Kit. Three PCR reactions were performed to amplify the nine loci (Table S2); two multiplexes (Panel 1- SvB106, SvC107, SvD5, SvB1; Panel 2 - SvB116, SvD103, SvB123, SvD213) and Locus SvC4 amplified in a separate reaction. Amplifications were in 25 μL volumes comprising 12.5 μL of 2× Qiagen Multiplex PCR Master Mix, 0.1 μM of all primers excepting B116-F, B116-R, D213-F and D213-R which were added to a concentration of 0.2 μM, 2.5 μL of 5× Q-solution and 10ng DNA. Thermal cycling comprised 15 min at 95 °C, followed by 10 cycles of 94 °C for 30 sec, 57 °C annealing for 90 sec and 72 °C extension for 60 sec, then 25 cycles of 94 °C for 30 sec, 55 °C annealing for 90 sec and 72 °C extension for 60 sec with a final 30 min extension at 72 °C.
For both species, PCR products were diluted 10–20 fold into a 20 μL volume containing 9.8 μL HiDi Formamide and 0.2 μL GeneScan 500 LIZ (ABI) internal size standard, prior to electrophoresis on an ABI 3730. Alleles were analysed and scored based on fragment size using the GeneMapper V4 software (Applied Biosystems Inc.) and binned using Tandem V 1.0272.
Genetic data analysis
The final data sets for each species were checked for evidence of linkage disequilibrium using Genepop 4.273,74 and because no evidence of linkage was detected in either species, all loci were retained for further analysis. We assessed levels of intra-specific genetic variation by calculating allelic and genetic diversity in Genalex 6.575, and compared the observed levels of heterozygosity within each sampling location against those expected under conditions of Hardy Weinberg Equilibrium (HWE) using Exact Tests in Genepop 4.2, and with p-values estimated using the Markov chain method based on 10,000 iterations. Significance levels were adjusted using a Bonferroni correction to account for multiple tests. Wright’s inbreeding coefficient (calculated as Weir and Cockerham’s FIS in Genepop 4.2) was used to examine the size and nature of any departures from random mating. We calculated allelic richness using FSTAT76 for both species, and used ANOVA to compare allelic richness and expected heterozygosity between sites on fished and unfished seamounts to test the hypothesis that mean diversity differs between fished and unfished seamounts. We also calculated genotypic richness to assess the contribution of asexual reproduction to population structure as R = (G − 1)/(N − 1)77.
We tested for the presence of null alleles using Micro-checker 2.2.378. Null alleles were evident in some populations at all loci for D. dianthus except locus DdC107 and at two loci (SvB116 and SvD213) in S. variabilis. We therefore adjusted each data set to account for null alleles using Micro-checker, based on the Oosterhout correction algorithm. Null alleles at each locus were labelled as the largest allele + one repeat. Because these adjustments are based on assumptions of HWE, which may not be appropriate for coral populations as they often reproduce asexually and can be inbred, the population genetic analyses were done on the raw data as well as the data adjusted for the presence of null alleles, for comparison.
We used STRUCTURE 2.3.379 to infer the number of distinct genetic groups within the data set for both species. For each potential value of K, 20 replicate runs (including a 50,000 run burn- in and 1,000,000 MCMC replicates, using the admixture model and correlated allele frequencies) were done for the likely range of K for each species (K = 1–16 for D. dianthus and K = 1–17 for S. variabilis). Structure Harvester80 was used to infer the true number of genetic groups in the data set. In addition we used principal coordinates analysis using Genalex 6.5 to visualise clustering of sites related to location (for both species) and depth (for D. dianthus).
Analysis of connectivity among seamounts was focussed on a single depth stratum in the Tasmanian Seamounts (mid-depth range 1000–1400 m) as this sub-set of the data represented the best level of replication for both species (Table S3). Here we were able to test for differentiation at two spatial scales both for D. dianthus and S. variabilis; among sites on the same seamount (<1 km) and among seamounts (5–106 km) using AMOVA and by calculating a range of pairwise genetic differentiation measures including FST, G’ST and DEST (using Genalex 6.5) to provide insight on evolutionary vs. contemporary relationships81. Significance levels in all tests were based on 10,000 permutations of the data. Only data from sites where n > 12 were used for statistical analysis (Table S3). For connectivity analyses we condensed clonal genotypes to a single representative individual at each site so as to specifically assess gene flow mediated via sexual propagules.
To assess gene flow in both species, we calculated Nem using private alleles82 in Genepop 4.2 as well as from estimates of FST using the formula Nem = (1/FST) − 1 (Wright 1931) as each gives a different temporal perspective i.e. private alleles reflect relatively recent gene flow and FST methods represent gene flow through longer time spans83. Isolation by distance was examined by plotting pairwise FST against geographic distance among all sites within the mid-depth range in the Tasmanian Seamounts. Tests for a significant relationship between genetic and geographic distance were done using a Mantel Test in Genalex 6.5. In addition we examined variation in the directionality of dispersal among mid-depth seamount sites in the Tasmanian Seamounts using LAMARC 2.1.984. Because there was considerable variation in sample size among sites. and to facilitate analysis of a large, multi-locus data set, we reduced sample size (by random selection) so that replication was no greater than 20 individuals at all sites. Calculation of migration among all sites within the Tasmanian Seamounts was based on a Bayesian approach. Starting values for Θ and M were based on the average of three preliminary runs using default settings and uniform prior distributions. Final runs consisted of 1 final chain, 1,000,000 steps and a burn-in of 10,000 steps, and were run with two simultaneous searches with adaptive heating and temperatures set to 1 and 1.1. Following the final run, we plotted the curve files to confirm that a single optimum value was estimated for each parameter. We calculated Nem from the LAMARC estimates of migration by multiplying effective population size (Θ) by migration (M) among all sites. Additionally we calculated contemporary effective population size using the linkage disequilibrium method of Waples and Do (2008) using NeEstimator V285.
To better understand if patterns of dispersal across the Tasmanian Seamounts at mid-depth were linked to the directionality of oceanographic currents, we tested four alternate models of gene flow among the seamounts: (1) Panmixis, (2) Westward, (3) Eastward, and (4) a full model that considered gene flow among all pairs of sites. This analysis was conducted using Migrate-N v3.6.1186 according to the method described by Beerli & Palczewski87. Runs for each model used Bayesian Inference, a long chain of 10,000,000 steps with a 10,000 step burnin and were run with four simultaneous chains using static heating and swapping at every 10th interval. Bayes factors and the probability of each model were calculated according to Beerli88. This analysis was done only for D. dianthus as S. variabilis populations were considered effectively isolated (see results).
For D. dianthus it was also possible to assess migration within seamounts for deep (Cascade Plateau, >2000 m) and shallow (Maquarie Ridge, <500 m) sites. These analyses were conducted as described above for the mid-depth sites in the Tasmanian Seamounts.
Additional Information
How to cite this article: Miller, K. J. and Gunasekera, R. M. A comparison of genetic connectivity in two deep sea corals to examine whether seamounts are isolated islands or stepping stones for dispersal. Sci. Rep. 7, 46103; doi: 10.1038/srep46103 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Clark, M. R., Schlacher, T. A., Rowden, A. A., Stocks, K. I. & Consalvey, M. Science Priorities for Seamounts: Research Links to Conservation and Management. PLOS One 7, e29232 (2012).
Van Dover, C. L. et al. Designating networks of chemosynthetic ecosystem reserves in the deep sea. Marine Policy 36, 378–381 (2012).
Danavaro, R., Snelgrove, P. V. R. & Tyler, P. Challenging the paradigms of deep-sea ecology. Trends in Ecology & Evolution 29, 464–475 (2014).
Clark, M. R. & Koslow, J. A. Impacts of fisheries on seamounts. (Blackwell, 2007).
Williams, A. et al. Seamount megabenthic assemblages fail to recover from trawling impacts. Marine Ecology 31, 183–199 (2010).
Clark, M. R. et al. The Ecology of Seamounts: Structure, Function, and Human Impacts. Annual Review of Marine Science 2, 253–278 (2010).
Cowen, R. K., Gawarkiewicz, G., Pineda, J., Thorrold, S. R. & Werner, F. E. Population Connectivity in Marine Systems. An Overview. Oceanography 20, 14–21 (2007).
Bohonak, A. J. Dispersal, gene flow, and population structure. Q. Rev. Biol. 74, 21–43 (1999).
Bradbury, I. R. & Snelgrove, P. V. R. Contrasting larval transport in demersal fish and benthic invertebrates: the role of behaviou and advective processes in determining spatial pattern. Can. J. Fish. Aquat. Sci. 58, 811–823 (2001).
Lester, S. E., Ruttenberg, B. I., Gaines, S. D. & Kinlan, B. P. The relationship between dispersal ability and geographic range size. Ecology Letters 10, 745–758, doi: 10.1111/j.1461-0248.2007.01070.x (2007).
Weersing, K. & Toonen, R. J. Population genetics, larval dispersal, and connectivity in marine systems. Mar. Ecol. Prog. Ser. 393, 1–12, doi: 10.3354/meps08287 (2009).
Hunter, R. L. & Halanych, K. M. Evaluating connectivity in the brooding brittle star Astrotoma agassizii across the Drake Passage in the Southern Ocean. Journal of Heredity 99, 137–148 (2008).
Hunter, R. L. & Halanych, K. M. Phylogeography of Antarctic planktotrophic brittle star Ophionotus victoriae reveals genetic structure inconsistent with early life history. Mar. Biol. 157, 1693–1704 (2010).
Morgan, S. G., Fisher, J. L., Miller, S. H., McAfee, S. T. & Largier, J. L. Nearshore larval retention in a region of strong upwelling and recruitment limitation. Ecology 90, 3489–3502 (2009).
Miller, K. J., Rowden, A. A., Williams, A. & Haussermann, V. Out of their depth? Isolated deep populations of the cosmopolitan coral Desmophyllum dianthus may be highly vulnerable to environmental change. PLOS One 6, e19004 (2011).
Leese, F., Agrawal, S. & Held, C. Long-distance island hopping without dispersal stages: transportation across major zoogeographic barriers in a Souther Ocean isopod. Naturwissenschaften 97, 583–594 (2010).
Arellano, S. M., Van Gaest, A. L., Johnson, S. B., Vrijenhoek, R. C. & Young, C. M. Larvae from deep-sea methane seeps disperse in surface waters. Proc. R. Soc. B-Biol. Sci. 281, doi: 10.1098/rspb.2013.3276 (2014).
Martinez-Quintana, A., Bramanti, L., Viladrich, N., Rossi, S. & Guizien, K. Quantification of larval traits driving connectivity: the case of Corallium rubrum (L. 1758). Mar. Biol. 162, 309–318 (2015).
Puritz, J. B. & Toonen, R. J. Coastal Pollution limits pelagic larval dispersal. Nat. Commun. 2, 226 (2011).
Andras, J. P., Rypien, K. L. & Harvell, C. D. Range-wide population genetic strucutre of the Caribbean sea fan coral. Gorgonia ventalina. Mol. Ecol. 22, 56–73 (2013).
Galindo, H. M., Olson, D. B. & Palumbi, S. R. Seascape genetics: A coupled oceanographic-genetic model predicts population structure of Caribbean corals. Curr. Biol. 16, 1622–1626, doi: 10.1016/j.cub.2006.06.052 (2006).
Selkoe, K. A., Henzler, C. M. & Gaines, S. D. Seascape genetics and the spatial ecology of marine populations. Fish and Fisheries 9, 363–377, doi: 10.1111/j.1467-2979.2008.00300.x (2008).
Selkoe, K. A. et al. Taking the chaos out of genetic patchiness: seascape genetics reveals ecological and oceanographic drivers of genetic patterns in three temperate reef species. Mol. Ecol. 19, 3708–3726, doi: 10.1111/j.1365-294X.2010.04658.x (2010).
White, C. et al. Ocean currents help explain population genetic structure. Proc. R. Soc. B-Biol. Sci. 277, 1685–1694, doi: 10.1098/rspb.2009.2214 (2010).
Alberto, F. et al. Isolation by oceanographic distance explains genetic structure for Macrocystis pyrifera in the Santa Barbara Channel. Mol. Ecol. 20, 2543–2554, doi: 10.1111/j.1365-294X.2011.05117.x (2011).
Waller, R. G. & Tyler, P. A. Reproductive patterns in two deep-water solitary corals from the north-east Atlantic-Flabellum alabastrum and F. angulare (Cnidaria: Anthozoa: Scleractinia). Journal of the Marine Biological Association of the United Kingdom 91, 669–675, doi: 10.1017/s0025315410000822 (2011).
Brooke, S. & Jarnegren, J. Reproductive periodicity of the scleractinian coral Lophelia pertusa from the Trondheim Fjord, Norway. Mar. Biol. 160, 139–153, doi: 10.1007/s00227-012-2071-x (2013).
Audzijonyte, A. & Vrijenhoek, R. C. When gaps really are gaps: statistical phylogeography of hydrothermal vent invertebrates. Evolution 64–8, 2369–2384 (2010).
Vrijenhoek, R. C. Genetic diversity and connectivity of deep-sea hydrothermal vent metapopulations. Mol. Ecol. 19, 4391–4411, doi: 10.1111/j.1365-294X.2010.04789.x (2010).
Miller, K., Williams, A., Rowden, A. A., Knowles, C. & Dunshea, G. Conflicting estimates of connectivity among deep-sea coral populations. Marine Ecology 31, 144–157 (2010).
France, S. C., Hessler, R. R. & Vrijenhoek, R. C. Genetic differentiation between spatially-disjunct populations of the deep-sea, hydrothermal vent-endemic amphipod {IVentiella sulfuris}. Mar. Biol. 114, 551–559 (1992).
Herrera, S., Shank, T. M. & Sanchez, J. A. Spatial and temporal patterns of genetic variation in the widespread antitropical deep-sea coral Paragorgia arborea . Mol. Ecol. 21, 6053–6067 (2012).
Bors, E. K., Rowden, A. A., Maas, E. W., Clark, M. R. & Shank, T. M. Patterns of Deep-Sea Genetic Connectivity in the New Zealand Region: Implications for Management of Benthic Ecosystems. PLOS One 7, e49474 (2012).
Clague, G. E., Jones, W. J., Paduan, J. B., Clague, D. A. & Vrijenhoek, R. C. Phylogeography of Acesta clams from submarine seamounts and escarpments along the western margin of North America. Marine Ecology 33, 75–87 (2012).
Miller, K. J., Maynard, B. T. & Mundy, C. N. Genetic diversity and gene flow in collapsed and healthy abalone fisheries. Mol. Ecol. 18, 200–211, doi: 10.1111/j.1365-294X.2008.04019.x (2009).
Vollmer, S. V. & Palumbi, S. R. Restricted gene flow in the Caribbean staghorn coral Acropora cervicornis: implications for the recovery of endangered reefs. Journal of Heredity 98, 40–50 (2007).
Baird, H. P., Miller, K. J. & Stark, J. S. Genetic Population Structure in the Antarctic Benthos: Insights from the Widespread Amphipod, Orchomenella franklini. PLOS One 7, e34363 (2012).
Miller, K. J., Mundy, C. N. & Mayfield, S. Molecular genetics to inform spatial management in benthic invertebrate fisheries: a case study using Australian Greenlip Abalone. Mol. Ecol. 23, 4958–4975 (2014).
Shank, T. M. & Halanych, K. M. Toward a mechanistic understanding of larval dispersal: insights from genomic fingerprinting of the deep-sea hydrothermal vent tubeworm Riftia pachyptila. Marine Ecology 28, 25–35, doi: 10.1111/j.1439-0485.2007.00146.x (2007).
Baco, A. R. & Shank, T. M. In Cold-Water Corals and Ecosystems Erlangen Earth Conference Series (eds Freiwald, A. & Roberts, J. M. ) 663–678 (Springer-Verlag Berlin, 2005).
Dahl, M. P., Pereyra, R. T., Lundalv, T. & Andre, C. Fine-scale spatial genetic structure and clonal distribution of the cold-water coral Lophelia pertusa. Coral Reefs 31, 1135–1148, doi: 10.1007/s00338-012-0937-5 (2012).
Thresher, R. et al. Strong Depth-Related Zonation of Megabenthos on a Rocky Continental Margin (700–4000 m) off Southern Tasmania, Australia. PLoS ONE 9, e85872, doi: 10.1371/journal.pone.0085872 (2014).
Harrison, P. L. & Wallace, C. C. In Coral Reef Ecosystems (ed. Z., Dubinsky ) 133–207 (Elsevier, 1990).
Burgess, S. N. & Babcock, R. C. In Cold-water Corals and Ecosystems. (eds Freiwald, A. & Roberts, J. M. ) 701–713 (Springer-verlag 2015).
Shearer, T. L., Van Oppen, M. J. H., Romano, S. L. & Worheide, G. Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol. Ecol. 11, 2475–2487 (2002).
Pires, D. O., Silva, J. C. & Bastos, N. D. Reproduction of deep-sea reef-building corals from the southwestern Atlantic. Deep Sea Research Part II: Topical Studies in Oceanography 99, 51–63, doi: 10.1016/j.dsr2.2013.07.008 (2014).
Koslow, J. A. et al. Seamount benthic macrofauna off southern Tasmania: community structure and impacts of trawling. Mar. Ecol. Prog. Ser. 213, 111–125 (2001).
Althaus, F. et al. Impacts of bottom trawling on deep-coral ecosystems of seamounts are long-lasting. Marine Ecology-Progress Series 397, 279–294 (2009).
Mills, L. S. & Allendorf, F. W. The One-Migrant-per-Generation Rule in Conservation and Management. Cons. Biol. 10, 1509–1518 (1996).
Waples, R. S. & Do, C. LDNE: a program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources 8, 753–756 (2008).
Leng, L. & Zhang, D. Measuring population differentiation using Gst or D? A simulation study with microsatellite DNA markers under a finite island model and nonequilibrium conditions. Mol. Ecol. 20, 2494–2509 (2011).
McClain, C. R. Seamounts: identity crisis or split personality? Journal of Biogeography 34, 2001–2008 (2007).
Cairns, S. D. Scleractinia of the temperate North Pacific. Smithsonian Contributions to Zoology 557, 150 (1994).
Cairns, S. D. The Marine Fauna of New Zealand: Scleractinia (Cnidaria: Anthozoa) (1995).
Teixeira, S. et al. High connectivity across the fragmented chemosynthetic ecosystems of the deep Atlantic Equatorial Belt: efficient dispersal mechanisms or questionable endemism. Mol. Ecol. 22, 4663–4680 (2013).
Tracey, D. M., Rowden, A. A., Mackay, K. A. & Compton, T. Habitat-forming cold-water corals show affinity for seamounts in the New Zealand region. Mar. Ecol. Prog. Ser. 430, 1–22 (2011).
Larsson, A. I. et al. Embryogenesis and Larval Biology of the Cold-Water Coral Lophelia pertusa . PLoS ONE 9, e102222, doi: 10.1371/journal.pone.0102222 (2014).
Richmond, R. H. Energetics, competency, and long-distance dispersal of planulae larvae of the coral {IPocillopora damicornis}. Mar. Biol. 93, 527–533 (1987).
Lacharite, M. & Metaxas, A. Early Life History of Deep-Water Gorgonian Corals May Limit Their Abundance. PLOS One 8, e65394 (2013).
Glynn, P. W. & Colley, S. B. Survival of Brooding and Broadcasting Reef Corals Following Large Scale Disturbances: Is There Any Hope for Broadcasting Species During Gloabl Warming? Proccedings of the 11th International Coral Reef Symposium, 361–365 (2008).
Ayre, D. J. & Hughes, T. P. Genotypic diversity and gene flow in brooding and spawning corals along the Great Barrier Reef, Australia. Evolution 54, 1590–1605 (2000).
Miller, K. J. & Ayre, D. J. Population structure is not a simple function of reproductive mode and larval type: insights from tropical corals. Journal of Animal Ecology 77, 713–724 (2008).
Le Goff-Vitry, M. C., Pybus, O. G. & Rogers, A. D. Genetic structure of the deep-sea coral Lophelia pertusa in the northeast Atlantic revealed by microsatellites and internal transcribed spacer sequences. Mol. Ecol. 13, 537–549 (2004).
Morrison, C. L. et al. Genetic discontinuity among regional populations of Lophelia pertusa in the North Atlantic Ocean. Conserv. Genet. 12, 713–729, doi: 10.1007/s10592-010-0178-5 (2011).
Becheler, R. et al. Low incidence of clonality in cold water corals revealed through the novel use of a standardized protocol adapted to deep sea sampling. Deep Sea Research Part II: Topical Studies in Oceanography, doi: 10.1016/j.dsr2.2015.11.013 (2015).
Lippé, C., Dumont, P. & Bernatchez, L. High genetic diversity and no inbreeding in the endangered copper redhorse, Moxostoma hubbsi (Catostomidae, Pisces): the positive sides of a long generation time. Mol. Ecol. 15, 1769–1780 (2006).
Lowe, W. H. & Allendorf, F. W. What can genetics tell us about population connectivity? Mol. Ecol. 19, 3038–3051 (2010).
Underwood, J. N., Wilson, S. K., Ludgerus, L. & Evans, R. D. Intergrating connectivity science and spatial conservation management of coral reefs in north-west Australia. Journal for Nature Conservation 21, 163–172 (2013).
Palumbi, S. R. et al. Managing for ocean biodiversity to sustain marine ecosystem services. Frontiers in Ecology and the Environment 7, 204–211 (2009).
Frankham, R., Ballou, J. D. & Briscoe, D. A. Introduction to conservation genetics. Second edition., 618 pp (Cambridge University Press, 2010).
Roberts, C. M. & Hawkins, J. P. Extinction risk in the sea. TREE 14, 241–246 (1999).
Matschiner, M. & Salzburger, W. TANDEM: integrating automated allele binning into genetics and genomics workflows. Bioinformatics 25, 1982–1983 (2009).
Raymond, M. & Rousset, F. Genepop (version 1.2): Population genetics software for exact tests and ecumenicism. J. Heredity 86, 248–249 (1995).
Rousset, F. Genepop’007: a complete reimplementation of the Genepop software for Windows and Linux. Molecular Ecology Resources 8, 103–106 (2008).
Peakall, R. & Smouse, P. E. GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6, 288–295 (2005).
Goudet, J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3) Available from www.unil.ch/izea/softwares/fstat.html (2001).
Dorken, M. E. & Eckert, C. G. Severely reduced sexual reproduction in northern populations of a clonal plant, Decodonverticillatus (Lythraceae). J. Ecol 89, 339–350, doi: 10.1046/j.1365-2745.2001.00558.x (2001).
Van Oosterhout, C., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. Micro-Checker: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes 4, 5353–5538 (2004).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Earl, D. A. & vonHoldt, B. M. Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conservation Genetics Resources 4, 359–361 (2012).
Whitlock, M. C. G’ST and D do not replace FST. Mol. Ecol. 20, 1083–1091 (2011).
Barton, N. H. & Slatkin, M. A quasi-equilibrium theory of the distribution of rare alleles in a subdivided population. Heredity 56, 401–415 (1986).
Yamamichi, M. & Innan, H. Estimating the migration rate from genetic variation data. Heredity 108, 362–363 (2012).
Kuhner, M. K. LAMARC 2.0: maximum liklihood and Bayesian estimation of population parameters. Bioinformatics 22, 768–770 (2006).
Do, C. et al. NeEstimator V2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Molecular Ecology Resources 14, 209–214 (2014).
Beerli, P. How to use migrate or why are markov chain monte carlo programs difficult to use? Cons. Biol. 17, 42–79 (2009).
Beerli, P. & Palczeweski, M. Unified Framework to Evaluate Panmixia and Migration Direction Among Multiple Sampling Locations. Genetics 185, 313–326 (2010).
Beerli, P. Tutorial: Comparison of gene flow models using Bayes Factors. Available at http://popgen.sc.fsu.edu (2012).
R. Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/ (2014).
Hijmans, R. J. Raster: Geographic Data Analysis and Modeling. R package version 2.5-8. https://CRAN.R-project.org/package=raster (2016).
Lamigueiro, O. P. & Hijmans, R. rasterVis: visulalisation methods for raster data V0.10 (2011).
Pebesma, E. J. & Bivand, R. S. Classes and methods for spatial data in R. R News 5 (2), https://cran.r-project.org/doc/Rnews/ (2005).
Acknowledgements
This study was funded through the Australian Government Commonwealth Environmental Research Fund (CERF) Marine Biodiversity Hub and the Australian Pacific Science Foundation. The authors thank Alan Williams, Ashley Rowden, Di Tracey and Mireille Consalvey for facilitating field collections of corals and Phillip England for facilitating microsatellite development and laboratory work. Alyce Hancock assisted with DNA extractions for S. variabilis with genotyping completed by the Australian Genome Research Foundation.
Author information
Authors and Affiliations
Contributions
K.J.M. developed the project, participated in field collections, and analysed the data. R.M.G. developed microsatellite markers and protocols for both coral species and completed laboratory genotyping for D. dianthus. Both authors contributed to the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Miller, K., Gunasekera, R. A comparison of genetic connectivity in two deep sea corals to examine whether seamounts are isolated islands or stepping stones for dispersal. Sci Rep 7, 46103 (2017). https://doi.org/10.1038/srep46103
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46103
This article is cited by
-
Opportunistic observations of reproductive mode, larval settlement and development of a New Zealand deep-sea stony coral, Goniocorella dumosa
Marine Biology (2024)
-
Tropical seamounts as stepping-stones for coral reef fishes: range extensions and new regional distributions from mesophotic ecosystems in the Coral Sea, Australia
Marine Biodiversity (2024)
-
A review of current knowledge on reproductive and larval processes of deep-sea corals
Marine Biology (2023)
-
Population genetic structure of the deep-sea precious red coral Hemicorallium laauense along the Hawaiian Ridge
Marine Biology (2023)
-
The shrunk genetic diversity of coral populations in North-Central Patagonia calls for management and conservation plans for marine resources
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
