
Abstract
Subtilases belong to a superfamily of serine proteases which are ubiquitous in fungi and are suspected to have developed distinct functional properties to help fungi adapt to different ecological niches. In this study, we conducted a large-scale phylogenomic survey of subtilase protease genes in 83 whole genome sequenced fungal species in order to identify the evolutionary patterns and subsequent functional divergences of different subtilase families among the main lineages of the fungal kingdom. Our comparative genomic analyses of the subtilase superfamily indicated that extensive gene duplications, losses and functional diversifications have occurred in fungi, and that the four families of subtilase enzymes in fungi, including proteinase K-like, Pyrolisin, kexin and S53, have distinct evolutionary histories which may have facilitated the adaptation of fungi to a broad array of life strategies. Our study provides new insights into the evolution of the subtilase superfamily in fungi and expands our understanding of the evolution of fungi with different lifestyles.
Similar content being viewed by others

Introduction
To survive, fungi depend on their ability to harvest nutrients from living or dead organic materials. The ecological diversification of fungi is profoundly affected by the array of enzymes they secrete to help them obtain such nutrients. Thus, the differences in their secreted enzymes can have a significant impact on their abilities to colonize plants and animals1. Among these enzymes, a superfamily of serine proteases named subtilases, ubiquitous in fungi, is suspected of having distinct functional properties which allowed fungi adapt to different ecological niches. For many saprophytes, subtilases are the principal broad-spectrum proteases which play a role in nutrition acquisition, such as digesting proteins to release peptides and amino acids for fungal cell absorption and growth2,3. In many pathogenic fungi, subtilases are believed to play important roles in host invasion3,4. For instance, subtilases derived from several entomopathogous and nematophagous fungi have been characterized as having the ability to disrupt the physiological integrity of insect and/or nematode cuticles during penetration and colonization. Thus, they are also called cuticle-degrading proteases5,6,7,8,9.
According to the MEROPS peptidase classification10, subtilases are divided into two clans: S8 (subtilisins) and S53 (sedolisins). Clan S8 utilizes a catalytic triad of three amino acids, Aspartate, Histidine, and Serine (DHS catalytic triad), to catalytically hydrolyze peptide bonds, while the catalytic triad of the S53 clan is Serine, Glutamate and Aspartate (SED catalytic triad). The S8 proteases are further classified into S8A and S8B subclans11. At present, subclan S8A has been divided into several distinct families such as proteinase K-like proteases, pyrolisin proteases and oxidatively stable proteases (OSP), whereas S8B subclan is composed of those proteases belonging to the kexin subfamily1,3,12.
Proteinase K-like protease, which was first identified in the ascomycete Tritirachium album, is a large subfamily of endopeptidases found in fungi2. Proteases within this subfamily may play important roles during the evolution of pathogenicity. The cuticle-degrading proteases mentioned above belong to the proteinase K-like subfamily5,6,7,8,9. Pyrolisin, also named class I subtilisin, was first characterized from the hyperthermophilic archaeon Pyrococcus furiosus. It is the most thermostable protease identified to date and retains half of its activity after boiling for several hours. A characteristic feature of Pyrolisin is the fact that these enzymes are substrates of their own proteolytic activity and degrade themselves in a process termed autoproteolysis13. However, although Pyrolisin has long been known in fungi, their ecological function is still uncertain3. Kexin, which was first identified in the yeast Saccharomyces cerevisiae, can process the yeast precursors of alpha-mating factor and killer toxin, as well as play a significant role in post-translational modification in eukaryotes14. OSP proteases were identified in the endophytic fungus Epichloë festucae and other Hypocreales fungi1. S53 family proteases probably contribute to the extracellular digestion of food proteins. Recently, based on the differences in the closest amino acids from the DHS catalytic triad, six new subtilase groups (named new 1 to new 6) were identified by Muszewska et al.15. Previous studies suggested that most of the subtilases in fungi were extracellular secreted proteases, which were thought to play a nutritive role16. However, there are several intracellular proteases localized to the vacuole4. The vacuolar proteases appear to play a specialized role in the breakdown of autophagic bodies in the vacuole during autophagy17. Moreover, most members of the subtilase superfamily are inhibited by general serine peptidase inhibitors such as diisopropyl fluorophosphate (DFP) and phenylmethane sulfonylfluoride(PMSF), but kexin is resistant to PMSF and requires high concentrations of DFP, which initially led to its misidentification as a cysteine peptidase18.
In previous studies, a large variation in the number of subtilase genes was observed among fungal species1,3,4, implying that many changes have likely occurred in subtilases during fungal evolution. It is generally accepted that gene duplication and loss were the main sources of the gene copy number changes among species19, and were a significant source of functional innovation20,21,22. In view of this, although the biological functions of most fungal subtilases are not yet described, gene duplication and sequence diversification have likely resulted in functional diversifications of fungal subtilases beyond simple nutrient utilization. Further, these functional differences among subtilases likely enabled fungi to develop new life strategies and adapt to a broad array of habitats as saprophytes and pathogens. Therefore, investigation of the evolution of the subtilase superfamily may help us understand the specialization and adaptation of fungi. In recent years, the increasing availability of completely sequenced fungal genomes provided a good opportunity for us to characterize the subtilases superfamily in different fungal taxa and to infer their evolutionary patterns. In this study, by analyzing the distribution of subtilases genes among 83 fungal species, we investigated the evolutionary patterns of the main subtilase families, and also examined the possible gene duplication and loss which may have contributed to the functional divergence of these subtilase genes.
Data and Methods
Identification of subtilase genes
Based on the phylogeny of the fungal kingdom which constructed by James et al.23 and Spatafora et al.24, we selected representatives of the whole genome sequenced fungi that are distributed among all the main fungal lineages, including the main phyla of Ascomycota, Basidiomycota, Mucormycotina and the very ancestral Microsporidia for this study. A total of 83 fungal genome sequences and the corresponding annotated protein databases are retrieved from the Fungal Genome Initiative at the BROAD Institute (http://www.broad.mit.edu/annotation/fungi/fgi), Fungal Genome Research (http://fungalgenomes.org/), and the NCBI database.
To find putative homologs of subtilases in the 83 fungal genome sequences employed in this study, the HMM profile Peptidase_S8 (PF00082; http://pfam.xfam.org/family/PF00082#curationBlock), which includes both S8 and S53 domains, was downloaded and used as a query in the search for homologous proteins using the program HMMSEARCH from the HMMER package (http://hmmer.wustl.edu/). Hits were considered significant when they matched the Pfam HMM profile with E values <10−5.
CLANS, a Java-based software program which visualizes pair-wise sequence similarities25, was used to elucidate the relationships between and within subfamilies of the S8 peptides. Also, the amino acid sequences of each subgroup were aligned using MUSCLE Version 3.726, and those sequences with ambiguously aligned regions around the three active catalytic residues (Asp-His-Ser triad in S8 and Ser-Glu-Asp triad in S53) were eliminated in the subsequent analyses.
The conserved functional domain structures of the identified subtilases were predicted using the MEME/MAST motif discovery tool (http://meme.sdsc.edu)27 and InterProScan 5.0 (http://www.ebi.ac.uk/interpro/) with default parameter settings28.
Phylogenetic analysis
Before phylogenetic analyses, MUSCLE v3.7 was used to generate protein alignment with default settings26. The ambiguous areas of alignment were located and removed by using the program Gblocks 0.91b29,30 with default parameters. The gap selection criterion “with half” was used here. Finally, alignments with 184 amino acid sequences were identified for the proteinase K group in Sordariomycetes fungi, and 282, 465, and 363 amino acid sequences were respectively identified for the pyrolisin, kexin and S53 families among the 83 included fungal genomes.
In this study, the aligned amino acid sequences of each gene family/subfamily were performed using two different tree construction methods: Neighbor-joining (NJ) analysis and Maximum likelihood (ML) analysis. For NJ analysis, the data was analyzed using MEGA 631 using bootstrap analysis (BS) with 1,000 replicates. For ML analysis, the best-fit models of protein evolution for each subfamily were first estimated using the program ProtTest32, then the recommended models and parameters were used for ML analysis with PHYML 3.033. In the ML analysis, the reliability of the tree topology was evaluated using bootstrap support with 100 repeats34.
Results
Number of subtilase genes from different fungi
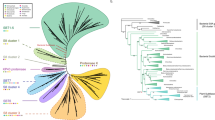
We identified the near-complete repertories of subtilase genes from 83 fungal species with different life-styles. After using HMM profile Peptidase_S8 as query model for Hmmsearch, a total of 993 subtilase genes were identified. To elucidate the relationships among the 993 subtilase genes, a clustering analysis which relied on the sequence similarities of proteins was conducted using the CLANS program25. As seen in Fig. 1, cluster analysis of the 993 subtilase genes resulted in nine distinct clades. The domain architectures of each clade were analyzed in our study (Supplementary Fig. S1). Although all the S8 family subtilases contain the common DHS catalytic triad, the closest amino acid sequences around the three catalytic amino acids were not the same. For example, the closest amino acid sequences from the DHS catalytic triad of the sequences grouped together in the proteinase K-like clade are Y/I/LDT/SG, HGT/SH/AV/CA/SG/A, and GT/SS, respectively. Also, beside the canonical DHS catalytic triads, the proteinase K-like subtilases contain an N-terminal subtilisin propeptide Inhibitor_I9 (InterPro acc. no.: IPR010259), which is thought to act as an intramolecular chaperone to assist protein folding as well as inhibit enzyme activity35. For the Pyrolisin proteases, the sequences around the catalytic triads are I/V/LDT/SGD/N/W, HGT/SH/F/A, and GT/SSM/F/YA/S, respectively, and they usually contain a Fn3-like domain (InterPro acc. no.: IPR010435), which maybe functionally related to adhesin/invasin36. For kexins, the catalytic triads are V/I/LDDGL/YD, HGTRCAG/AE/QI/VA/S/GA, and HGGTSAAA/GP, respectively. Moreover, kexins usually have a pro-protein C-terminal convertase domain (P_protein; InterPro acc. no.: IPR002884) and a Galactose binding-like domain (InterPro acc. no.: IPR008979). For OSP proteases, beside the canonical peptidase S8 domain, most of them also contain a Galactose binding-like domain (InterPro acc. no.: IPR008979). Also, few of them contain a Ankyrin repeat domain(InterPro acc. no.: IPR002110) which is one of the most common protein-protein interaction motifs in nature. For S53 proteases, the canonical SED catalytic triads are EA/S/GXLD, S/A/GSGD, and GTSA/CS/AA/S/TP, respectively(Supplementary Fig. S1). Generally, combined with the results based on CLAN and their motif feature of each clade, these nine distinct clades were divided into different subfamilies/families: S53 proteases were clustered into a single compact clade, while the genes of the S8 peptides were divided into eight separate clades: proteinase K proteases, pyrolisin proteases, kexin, OSP, and four newly identified groups (new 1 to new 4).

The name of each group is marked. Four new groups are identified in our study with new 1, new 3 and new 4 groups being the same as the classification of Muszewska et al., and new 2 group in our study corresponding to the new 6 group proposed by Muszewska et al.15.
Because the DHS and SED catalytic triads are the key active centers of S8 and S53 proteases, respectively, any mutation within or around the region encoding the catalytic triads may result in the inactivation of a protein. Thus, those genes with ambiguously aligned regions around the three active catalytic residues were considered to be pseudogenes and removed from subsequent analyses. Finally, a total of 904 putative functional subtilase genes (Supplementary data S2), including 429 proteinase K-like subtilases, 26 OSPs, 136 pyrolisins, 84 kexins, 126 S53 proteases, 53 new 1 group proteases, 9 new 2 group proteases, 33 new 3 group proteases and 8 new 4 group proteases were used in subsequent analyses. The identified number of subtilases for the 83 fungal species are presented in Table 1 (see Supplementary Table S3). These variations imply that gene duplication and loss may have played significant roles during the evolution of the fungal subtilases.
Among the 83 analyzed fungal genomes, remarkable variations in the number of subtilase genes were observed among different fungal species (Table 1 and Supplementary Table S3). The entomopathogous fungus Metarhizium robertsii had the largest number of subtilase genes (48), whereas the Saccharomycotina fungus Saccharomyces kluyveri contained only 1 subtilase gene. Among the families in the S8 clan, the numbers of proteinase K-like subfamily genes in fungal species were the most variable, ranging from 0 to 19 (Table 1 and Supplementary Table S3). This proteinase K-like subfamily of genes was common to all the species examined in this study, suggesting its functional importance. In our study, Eurotiomycetes and Sordariomycetes fungi showed a large variation in the number of proteinase K-like subfamily genes, and the fungi which contained fewer than 10 subtilase genes were mainly saprophytic fungi, such as Aspergillus spp., Penicillium spp. and Neurospora crassa. The two phytopathogenic fungi (Phaeosphaeria nodorum and Pyrenophora tritici-repentis) from Dothideomycetes contained 5 proteinase K-like genes each, whereas the other two phytopathogenic fungi (Botryotinia fuckeliana and Sclerotinia sclerotiorum) from Leotiomycetes contained 1 and 2 genes, respectively. Intriguingly, the only Orbiliomycetes fungi among the 83 species, Arthrobotrys oligospora, contained 19 duplicated proteinase K-like subtilases. All the yeast-like fungi shared a highly similar number of proteinase K-like subtilases except Yarrowia lipolytica, which contained 16 duplicate proteins. Similarily, all the Basidiomycota fungi contained 1 to 3 proteinase K-like subtilases except Coprinus cinereus which contained 6. Moreover, ten proteinase K-like subtilases were identified from the two Mucormycota fungi Phycomyces blakesleeanus and Rhizopus oryzae, while 1 gene was identified from the Chytridiomycota fungus Batrachochytrium dendrobatidis. For the three Microsporidia fungi, only 1 proteinase K-like subtilase was identified from Encephalitozoon cuniculi while no proteinase was found from the other two.
For pyrolisin, a large variation in its numbers was found in the Sordariomycetes fungi, ranging from 1 to 16. The pyrolisin genes in Verticillium spp., Metarhizium spp., and Magnaporthe spp. have significantly expanded and the rice blast fungus Magnaporthe grisea possessed 16 pyrolisin subfamily genes, to date the highest number of this gene subfamily from a single species. However, we did not find pyrolisin subfamily genes in most Eurotiomycetes fungi except for Aspergillus terreus and there was a complete absence of the pyrolisin genes in all yeast-like fungi (Saccharomycotina and Taphrinomycotina). Interestingly, this subfamily of genes also showed a large variation in Basidiomycota fungi: the phytopathogenic fungus Puccinia graminis contained 8 pyrolisins while Postia placenta, Sporobolomyces roseus, and Malassezia globosa contained none. In comparison, the ancestral Mucorales fungi R. oryzae and P. blakesleeanus contained 8 and 9 pyrolisin genes, respectively. Interestingly, regardless of taxonomic affiliation, none of the human/animal pathogenic fungi possessed pyrolisin genes.
Most fungi investigated in this study contained 1 to 3 kexin protease genes, with the exception of the Basidiomycota fungus Phanerochaete chrysosporium and the Saccharomycotina fungi S. kluyveri, Saccharomyces kudriavzevii and Saccharomyces mikatae, none of which contained any kexin genes. For the three fungi from Microsporidia, although 1 kexin-like subtilase was identified in Enterocytozoon bieneusi and Encephalitozoon intestinalis each, these two sequences showed ambiguous alignment aroud the three active catalytic residues (Asp-His-Ser). Moreover, no kexin-like sequence was identified in E. cuniculi.
In our results, genes from OSP, new 1, new 3 and new 4 groups were present in a few species of Pezizomycotina fungi. However, genes from new 2 were mainly found in Basidiomycota and Mucormycota fungi.The four Taphrinomycotina fungi contained one new 2 group gene each and the Chytridiomycota fungus B. dendrobatidis also contained one.
S53 family genes were commonly present in Pezizomycotina fungi with the exceptions of Verticillium spp. and M. poae. Like Pyrolisin genes, the S53 family genes was completely absent from all yeast-like fungi (Saccharomycotina and Taphrinomycotina). Some Basidiomycota fungi, such as Laccaria bicolor, P. chrysosporium, P. placenta and P. graminis, also contained S3 family genes and P. placenta contained the largest number (23) of S53 family genes.
Phylogenetic analysis
In our results, genes from the proteinase K, pyrolisin, kexin and S53 families were much more commonly identified in fungal genomes than other subtilase clusters (Fig. 1 and Table 1). Consequently, subsequent phylogenetic analyses were performed based on those four subfamilies. Moreover, the proteinase K subfamily genes from Sordariomycetes fungi showed significant variation compared with those from other species, suggesting that the proteinase K genes may have played important roles during the evolution of Sordariomycetes fungi. Thus, the proteinase K genes from Sordariomycetes fungi were used to construct phylogenetic trees and those genes identified from Saccharomycotina were used as outgroups. The best-fit models for each family, chosen by the program ProtTest, were provided in Supplementary Table S4.
Proteinase K-like subfamily
Phylogenetic relationships among the proteinase K-like subfamily genes were analyzed based on an alignment consisting of 184 amino acids from 138 genes (Supplementary data S5) from Sordariomycetes fungi and 8 outgroup genes from the four Taphrinomycotina fungi. Phylogenetic analyses grouped these genes into five distinct clades, which are abbreviated as SF1 to SF5 (Fig. 2 and Supplementary Fig. S6). Among these clades, SF1 (BS = 100% in both NJ and ML) consisted of the specialized vacuolar proteases from the Sordariomycetes fungi except for N. crassa and showed close relationships with the outgroup genes from Taphrinomycotina fungi. Clade SF2 (BS = 96% in NJ and BS = 92% in ML) to SF5(BS = 59% in NJ and BS = 52% in ML) were composed of the duplicated proteinase K-like subfamily genes in Sordariomycetes fungi, and inconsistent relationships among the four subfamilies were observed (Fig. 2 and Supplementary Fig. S6). However, given that duplicate genes were commonly represented in the four clades, it was assumed that four duplication events might have occurred at the beginning of the Sordariomycetes lineage. Moreover, within the SF3 and SF5 clades, there were several genus-specific subclades which were composed of the duplicate genes identical to those found inpathogenic fungi such as Fusarium spp., Metarhizium spp., Verticillium spp., and E. festucae, implying that the genes were duplicated before the divergence of the species within same genus.

Phylogenetic analyses were performed using maximum likelihood (ML) methods as implemented in PHYML 3.0 with GTR+I+G model based on an alignment of 184 amino acids from 138 proteinase K-like genes from Sordariomycetes fungi and 8 outgroup genes from the four Taphrinomycotina fungi. Five clades were designated (SF1-SF5). The bootstrap support value for each clade is shown. The 10 proteinase K-like subfamily genes Pr1A, Pr1B, Pr1D to Pr1K and the duplicated genes newly identified in Metarhizium spp. are highlighted.
Also, several gene loss events were identified in some species. For example, the duplicate genes formed into SF1 (BS = 100% in both NJ and ML) all came from the Sordariomycetes fungi considered in this study except Fusarium solani and Verticillium albo-atrum. In contrast, the genes from N. crassa, E. festucae, M. grisea, M. poae, and Trichoderma reesei were not included in SF4. These results suggest a subsequent loss of proteinase K-like genes in these fungi. The duplicate genes from the endophytic fungus E. festucae always showed close relationships with the duplicated genes from Metarhizium spp., suggesting the genes duplicated before the E. festucae and Metarhizium spp. split. However, in some subclades, no E. festucae genes were found to cluster with the duplicated genes of Metarhizium spp., suggesting subsequent gene losses in E. festucae. In the clade SF3 (BS = 78% in NJ and BS = 65% in ML) and SF5 (BS = 59% in NJ and BS = 52% in ML), the genes belonging to genus Metarhizium have largely expanded, and some genes have also been lost in Metarhizium acridum. Moreover, in clade SF5, at least five duplications occurred before the divergence of Fusarium species but F. solani retains only two of the duplications.
Pyrolisin subfamily
Our phylogenetic analysis based on an alignment of 282 amino acids from 136 Pyrolisin sequences (Supplementary data S7) suggested that the pyrolisin genes are mainly grouped into seven clusters which were designated sf1 to sf7 though inconsistent relationships among the six clades were also observed (Fig. 3 and Supplementary Fig. S8). Among these seven clades, sf1 (BS = 88% in NJ and BS = 90% in ML) is the Mucorales-specific lineage which contains the duplicated genes from R. oryzae and P. blakesleeanus. The pyrolisin genes from Basidiomycota fungi fall into the sf2 clade (BS = 94% in NJ and BS = 97% in ML). Within this clade, the 8 duplicated genes of P. graminis formed one species-specific subcluster, suggesting the duplication occurred after the speciation of P. graminis. The gene identified from Pichia pastoris (CAY68779) and Cryptococcus neoformans (CNAG_00150T0) is clustered into sf3 (BS = 95% in NJ and BS = 94% in ML) with the genes from Verticillium spp. (VDAG_09626T0 in Verticillium dahliae and VDBG_07757T0 in V. albo-atrum) and F. solani (fsol_84201). Clade sf4 (BS = 63% in NJ and BS = 55% in ML) consisted of the six genes from B. fuckeliana (BCIG_12343.t1), S. sclerotiorum (SSIG_09060), T. reesei (tree_109276), M. grisea (MGG_07358.t1 and MGG_00282.t1), and M. poae (MAPG_03852T0). Clade sf5 (BS = 100% in both NJ and ML) was composed of the two duplicated genes from the nematophagous fungus Arthobotrys oligospora (AOL_s0054g992 and AOL_ s00215g551). In NJ analyses, the gene from P. tritici-repentis (Ptri_Pt-1C-BFP:PTRG_07828.t1) clustered together with the genes in clade sf5, while it showed close relationships with the genes in clade sf7 in ML analyses. Clade sf6 (BS = 55% in NJ and BS = 73% in ML) was a lineage-specific clade which consisted of the genes identified from Sordariomycetes fungi. The duplicated genes within this clade are almost all from phytopathogenic fungi except one gene from the saprophytic fungus T. reesei (tree_60791) and one gene from E. festucae(ACN30268). Clade sf7 (BS = 52% in NJ and BS = 56% in ML) is composed of the genes from Pezizomycotina fungi. The three duplicated genes from the Dothideomycetes fungus P. tritici-repentis within this clade did not clustered together and the two duplicated genes from the nematophagous fungi Arthrobotrys oligospora are clustered at the base of this clade.

Phylogenetic analyses were performed using maximum likelihood (ML) methods as implemented in PHYML 3.0 with GTR+I+G model based on an alignment of 282 amino acids from 136 Pyrolisin sequences. Seven clades were designated (sf1-sf7) and the bootstrap support value for each clade is shown. The Pyrolisin subfamily genes Pr1C and the duplicated genes newly identified in Metarhizium spp. are highlighted.
Kexin subfamily
Based on an alignment of 465 amino acids from 84 kexin sequences (Supplementary data S9), the observed tree topology of kexin subfamily subtilase genes is largely consistent with the taxonomical relationships of Mucormycota, Basidiomycota and Ascomycota at the main nodes (Fig. 4 and Supplementary Fig. S10). However, several exceptions were noted. In the first, the kexin genes from Taphrinomycotina fungi showed a close relationship with those from Basidiomycota fungi but not with Saccharomycotina fungi. In the second, the genes from the two Leotiomycetes fungi were clustered together with those of Sordariomycetes fungi. Finally, one kexin gene of the Sordariomycetes fungus E. festucae was clustered into the clade of Eurotiomycetes fungi.

Phylogenetic analyses were performed using maximum likelihood (ML) methods as implemented in PHYML 3.0 with TrN+I+G model based on an alignment of 465-bp amino acids from 84 kexin sequences.
S53 family
Phylogenetic analyses based on an alignment of 363 amino acids from 126 S53 family sequences (Supplementary data S11) revealed that the S53 family genes in fungi seemed to have evolved by a series of duplication events after the split of the main lineages. These sequences were mainly grouped into five clusters designated S53-1 to S53-5 (Fig. 5 and Supplementary Fig. S12). As seen in Fig. 5, clade S53-1 (BS = 70% in NJ and BS = 58% in ML) was mainly composed of the genes from the Basidiomycota fungi and showed early divergence. However, it also included the duplicated genes from the three Onygenales fungi (Blastomyces dermatitidis, Histoplasma capsulatum, and Paracoccidioides brasiliensis). It seemed that the duplications occurred before the divergence of these three fungi. Moreover, duplications must have occurred in the Leotiomycetes lineage, except that B. fuckeliana retained only one of the duplications. Clade S53-2 (BS = 100% in both NJ and ML) was a lineage-specific clade which contained 22 duplicated genes from the Basidiomycota fungus P. blakesleeanu and 3 duplicated genes from the Basidiomycota fungus P. chrysosporium. Clade S53-3 (BS = 100% in NJ and BS = 84% in ML) was comprised of genes from several Ascomycota fungi and two Basidiomycota fungi. S53-4 (BS = 98% in NJ and BS = 88% in ML) was a lineage-specific clade which consisted of the duplicated genes from Sordariomycetes fungi. Moreover, the genes from Fusarium spp. and Metarhizium spp. were duplicated after the split of Fusarium and Metarhizium but before the speciation within each genus. S53-4 (BS = 64% in NJ and BS = 58% in ML) was a large clade which was composed of the duplicated genes from the main lineages of Pezizomycotina fungi. Within this clade, the genes from the two Dothideomycetes fungi duplicated at least three times and the genes from the Aspergillus spp., Trichophyton spp. and Onygenales fungi duplicated before the deep radiation of each genus/family.

Phylogenetic analyses were performed using maximum likelihood (ML) methods as implemented in PHYML 3.0 with TrN+I+G model based on an alignment of 363 amino acids from 126 S53 family sequences. Five clades were designated (S53-1 to S53-5) and the bootstrap support value for each clade is shown.
Discussions
Previously, several studies revealed substantial variations in the numbers of subtilase genes in different fungi1,3,4,15. In this study, we conducted a large-scale phylogenomic survey of subtilase protease genes from 83 fungal genomes in order to identify the evolutionary patterns and subsequent functional divergences of different subtilase subfamilies/families in the fungal kingdom. Besides the major clusterssuch as proteinase K-like and Pyrolisin subfamilies, and S53 family, Muszewska et al. recently found six new groups of subtilase genes (new 1 to new 6). However, only four new groups were identified in our study (new 1, new 3 and new 4 groups are the same as the classification of Muszewska et al., and new 2 group in our study corresponds to the new 6 group proposed by Muszewska et al.15) and the new 2 and new 5 groups proposed by Muszewska et al. were not identified in our study. We initial speculated that the inconsistency between our study and that of Muszewska et al. may be due to the deletion of the ambiguous alignment around the three active catalytic residues. We tried to find those missing groups from our initial alignment without any deletion, however, no new groups were discovered. Thus we speculated that the missing groups were likely artifacts in previous studies and were removed in our newer assemblies.
Firstly, during the evolution of the main fungal lineages, both gene duplication and loss events have occurred in subtilase genes, leading to the diversification of subtilase genes in different fungi. For example, 19, 19 and 15 duplicated proteinase K-like subfamily genes were characterized in M. anisopliae, M. robertsii, and M. acridum in our study, respectively, including the 10 subtilases which have been identified previously based on the expressed sequence tag (EST) analyses during growth on insect cuticle (Pr1A, Pr1B, Pr1D to Pr1K)37. The smaller numbers in M. acridum suggested that several proteinase K-like genes have been lost in this fungus. In the case of M. anisopliae, our phylogenetic analyses revealed that these proteinase K-like subfamily genes clustered into at least five clusters: the vacuolar proteases Pr1H clustered with other vacuolar proteases from Sordariomycetes fungi into SF1, while other duplicated genes clustered into SF2 and SF5, respectively. Considering that gene duplication and subsequent functional divergence is an important mechanism for generating the functional diversification necessary for adaptation, the presence of multiple subtilases in one pathogenic fungus may reflect their expanded ecological roles in pathogenesis. Specifically, the duplication and diversification events have likely contributed to increased adaptability, host range, and/or survived in various ecological habitats outside the hosts38,39. During such events, the duplicated genes within a species/genus that were clustered into one group might have similar functions, whereas novel functions might have formed among the duplicated genes which clustered into separate clades. Previous researches suggested that Pr1A was the key virulence factor involved in the degradation of insect cuticles, while other Pr1’s are minor components for cuticle degradation37. However, from our phylogenetic results, the functionally confirmed insect cuticle-degrading PR1A (accession GenBank number of KFG86683) was grouped into SF3 with the other 7 paralogous genes (Pr1B, Pr1G, Pr1I, Pr1K and other three genes with accession GenBank numbers: KFG79696, KFG79277, and KFG81372). It is likely that those subtilase proteases clustered within SF3 may serve a similar function during the degradation of insect cuticles. Moreover, SF5 included the genes Pr1D, Pr1E, Pr1F, Pr1J and other three genes with GenBank numbers of KFG79137, KFG81922, and KFG85392. Although not all the functions of the genes have been characterized, the duplicated subtilase genes in the entomopathogenic fungi Metarhizium spp. likely play different roles in pathogenesis such as increasing adaptability and host range, or have different functions in survival in various ecological habitats outside the host. Our analyses offer a promising basis for further understanding the gene duplication of subtilase genes in fungi. It should be noted that gene duplication and loss events of proteins are common in fungi. Many functional proteinases such as chitinases or Zincin metalloproteinases also experienced complicated gene duplications and losses during fungal evolution. For instance, Karlsson and Stenlid identified the glycoside hydrolase 18 (GH18) family of chitinases in fungi and found that many duplication and loss events have occurred for this group of genes in fungi40. Also, the M35 family (deuterolysin) and M36 family (fungalysin) genes of Zincin metalloproteinases showed large variations in gene numbers among Ascomycota fungi. Strikingly different gene duplication and loss events of metalloproteinases have been observed in Onygenales fungi and the duplicated genes have likely diverged functionally to play important roles during the evolution of pathogenicity of dermatophytic and Coccidioides fungi41. All these studies suggested that gene duplication and loss play an important role in the evolution of novel functions and for shaping an organism’s gene content.
Secondly, subtilase genes belonging to different families have distinct evolutionary histories, with the proteinase K-like subfamily displaying the most complicated evolutionary history while the kexin subfamily shows the most steady evolution. For proteinase K-like subfamily genes, the vacuolar proteases of Sordariomycetes fungi formed SF1 and showed close relationships with the outgroup genes (Fig. 2), suggesting the primitive character of the vacuolar genes in Sordariomycetes fungi. Vacuolar proteases play an essential role in the breakdown of autophagic bodies in the vacuole during autophagy, allowing recycling of macromolecules during nutrient starvation42,43. Thus, the genes within SF1 are likely the main protease enzymes involved in autophagy in fungi. This result is consistent with our previous studies which hypothesized that the proteinase K-like serine proteases of Pezizomycotina fungi most likely evolved from endocellular to extracellular proteases4. Subsequently, extensive duplications and functional divergences of proteinase K-like genes have occurred especially in Sordariomycetes pathogenic fungi (Table 1 and Fig. 2), which may have facilitated the adaptation of these fungi to a broad array of ecological niches.
The pyrolisin subfamily genes also showed variable numbers among the fungal species. In our analyses, 8 and 9 Pyrolisin genes were identified in the two Mucorales fungi R. oryzae and P. blakesleeanus, while only 1 to 2 Pyrolisins were observed in the Basidiomycota fungi, and no pyrolisin was detected in the Basidiomycota fungi M. globosa, Sporobolomyces roseus, and P. placenta, suggesting that Pyrolisins have contracted or lost in Basidiomycota fungi. However, the phytopathogenic Basidiomycota fungus P. graminis possessed 8 Pyrolisin genes, suggesting that gene duplication of Pyrolisin occurred in this fungus. Interestingly, no pyrolisin gene was identified in any yeast-like fungus (Saccharomycotina and Taphrinomycotina) but pyrolisin genes were extensively found in Basidiomycota fungi, suggesting that the gene loss may have taken place in the common ancestor of the Saccharomycotina and Taphrinomycotina lineages. All of the Eurotiomycetes fungi except for A. terreus did not possess pyrolisin genes, hence the pyrolisin gene may also have been lost at an early stage in the common ancestor of Eurotiomycetes fungi. Although gene loss events have frequently occurred in Basidiomycota, Saccharomycotina, Taphrinomycotina and Eurotiomycetes fungi, it seems that duplication of Pyrolisin genes has commonly occurred in Sordariomycetes fungi, especially in the phytopathogenic and entomopathogenic fungi, such as Magnaporthe spp. Metarhizium spp., and Verticillium spp. In Metarhizium spp., a Pyrolisin, Pr1C, was identified previously based on expressed sequence tag (EST) analyses during growth on insect cuticle37. In our study, 9, 11 and 7 duplicated Pyrolisin sequences were identified in M. anisopliae, M. robertsii, and M. acridum, respectively. Phylogenetic analyses revealed that these duplicated Pyrolisin genes clustered into two clades: one duplicated Pyrolisin gene is clustered into sf6 while Pr1C clustered into sf7 with seven other duplicated Pyrolisin genes (Fig. 3). Thus, although pyrolisin has not been extensively studied in fungi, it may play an important role in interactions with the environment, especially in pathogenic fungi. Previously, phylogenetic analysis based on a relatively small dataset of sequenced fungal genomes grouped the Pyrolisin subfamily genes into 3 distinct clusters in fungi: one was Basidiomycota and two were subfamilies of Ascomycota (abbreviated as sf1 and sf2)3. However, our phylogenetic analysis of the Pyrolisin subfamily identified three additional clades: the Mucorales fungi R. oryzae and P. blakesleeanus were clustered together and the Ascomycota fungi formed at least 4 clades.
There is little evidence for gene duplication and loss in the kexin subfamily, which may be related to the functional conservation of kexins in fungi (Fig. 4). The kexins displayed the lowest variations in gene numbers among the analyzed fungal taxa and most fungi contained at least one kexin except for four fungi: P. chrysosporium, S. kluyveri, S. kudriavzevii and S. mikatae. However, considering that kexins play essential roles in fungi, the absence of kexin proteins in these four genomes may be related to the low quality of their genome sequences. However, we still can not exclude the possibility that these fungi may have lost their kexin during evolution. The identification of kexin-like proteins in Microsporidia E. bieneusi and E. intestinalis suggested that kexin proteins might have originated from a common eukaryotic ancestor, but was discarded by Microsporidia during evolution. Moreover, the evolutionary history of kexin is similar to the phylogeny of fungi because the observed tree topology of the kexin subfamily shows a general agreement with taxonomic relationships at the main nodes as revealed by other molecular markers. In general, kexins are thought to play an important role in post-translational modification in eukaryotes14,44. Secreted proteins in eukaryotes are often synthesized as preproproteins, which undergo two proteolytic processing events to become mature proteins. The prepeptide is normally removed by a signal peptidase in the endoplasmic reticulum. The resulting pro-protein is then transferred to the Golgi, where kexin cleaves the propeptide to produce the mature protein45. Therefore, the functional conservation of kexins is consistent with the low frequency of duplication and loss of kexin in fungi.
S53 family genes were retained by only a few Basidiomycota fungi. However, the fungus P. placenta contained the largest number (23) of S53 family genes, 22 of which formed a lineage-specific clade with the three duplicated genes of its relative P. chrysosporium. The unusual number of S53 genes in P. placenta implied some particular adaptive significance for this fungus. Intriguingly, S53 genes have been totally discarded in yeast-like fungi but commonly retained in Pezizomycotina fungi. Phylogenetic analyses of S53 family genes revealed that clade S53-2 contained the orthologous S53 genes from most fungal species and showed close relationships to the genes from P. blakesleeanus, suggesting that the genes within this clade may be the ancestral orthologous genes of this family (Fig. 5). Subsequently, duplicated S53 family genes formed other clades. The duplicated genes from Eurotiomycetes fungi mainly clustered in S53-8, while the genes from Sordariomycetes fungi clustered as clade S53-4 (Fig. 5). The existence of lineage-specific clades suggested that duplications occurred before the radiation of the fungi within the same class. Although the functions of S53 family genes are still unknown, their common presence and frequent duplications in fungi, especially in filamentous fungi, imply their functional importance in fungi.
Thirdly, although subtilase genes have been considered as key virulence factors for some pathogens to infect hosts, our study revealed no association between gene expansions and pathogenicity in some fungi. For example, several opportunistic fungal pathogens such as A. fumigatus and A. clavatus share similar and lower subtilase gene copy numbers than those of other saprophytic Aspergillus fungi. The yeast-like human/animal pathogenic fungi Candida spp. also share similar subtilase gene numbers with other saprophytic Saccharomycotina fungi. The two phytopathogenic fungi (S. sclerotiorum and B. fuckeliana) belonging to Leotiomycetes also contain low copy numbers of subtilase genes. In comparison, several saprophytic fungi such as Uncinocarpus reesei, T. reesei and Podospora anserina showed extreme subtilase gene family expansions, suggesting that gene expansion is not associated with pathogenicity in some filamentous fungi. However, it may be difficult to define the exact functions of the expanded subtilase genes in a species because the deletion of one pathogenic factor may not significantly weaken the pathogenicity of the pathogen. Moreover, for pathogenic fungi to infect hosts, the collaboration of several pathogenicity factors are often needed which may also include multiple subtilase genes.
Conclusions
The present study provides new insights into the evolution of the subtilase superfamily genes in fungi. Our comparative genomic analysis indicated that considerable duplication, loss and functional diversification of these genes have occurred in fungi. The four families of subtilase in fungi have different evolutionary patterns. These evolutionary dynamics may be related to the functional diversity of these subtilases. Our analysis provides useful information to understand the evolution and functional importance of the important subtilase superfamily in fungi.
Additional Information
How to cite this article: Li, J. et al. Phylogenomic evolutionary surveys of subtilase superfamily genes in fungi. Sci. Rep. 7, 45456; doi: 10.1038/srep45456 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Bryant, M. K., Schardl, C. L., Hesse, U. & Scott, B. Evolution of a subtilisin-like protease gene family in the grass endophytic fungus Epichloe festucae . BMC Evol Biol 9, 168 (2009).
Gunkel, F. A. & Gassen, H. G. Proteinase K from Tritirachium album Limber. Characterization of the chromosomal gene and expression of the cDNA in Escherichia coli . Euro J Biochem 179, 185–194 (1989).
Hu, G. & St Leger, R. A phylogenomic approach to reconstructing the diversification of serine proteases in fungi. J Evol Biol 17, 1204–1214 (2004).
Li, J. et al. New insights into the evolution of subtilisin-like serine protease genes in Pezizomycotina. BMC Evol Biol 9, 68 (2010).
Huang, X., Zhao, N. & Zhang, K. Extracellular enzymes serving as virulence factors in nematophagous fungi involved in infection of the host. Res Microbiol 155, 811–816 (2004).
St Leger, R. The role of cuticle-degrading proteases in fungal pathogenesis of insects. Can J Bot 73, 1119–1125 (1995).
Wang, B., Liu, X., Wu, W., Liu, X. & Li, S. Purification, characterization, and gene cloning of an alkaline serine protease from a highly virulent strain of the nematode-endoparasitic fungus Hirsutella rhossiliensis . Microbiol Res 164, 665–673 (2009).
Wang, R. B., Yang, J. K., Lin, C., Zhang, Y. & Zhang, K. Q. Purification and characterization of an extracellular serine protease from the nematode-trapping fungus Dactylella shizishanna . Lett Appl Microbiol 42, 589–94 (2006).
Yang, J., Liang, L., Li, J. & Zhang, K.-Q. Nematicidal enzymes from microorganisms and their applications. Appl Microbiol Biotechnol 97, 7081–7095 (2013).
Rawlings, N. D., Barrett, A. J. & Finn, R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucl Acids Res 44, D343–50 (2016).
Schultz, R. & Liebman, M. Structure-function relationship in protein families. In Textbook of Biochemistry with Clinical Correlations(ed. Devlin, T. ) 1–2 (New York, USA: Wiley-Liss, Inc., 1997).
Siezen, R. & Leunissen, J. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Science 6, 501–523 (1997).
de Vos, W. M. et al. Purification, characterization, and molecular modeling of pyrolysin and other extracellular thermostable serine proteases from hyperthermophilic microorganisms. Methods Enzymol 330, 383–93 (2001).
Rogers, D., Saville, D. & Bussey, H. Saccharomyces cerevisiae killer expression mutant kex2 has altered secretory proteins and glycoproteins. Biochem Bioph Res Co 90, 187–193 (1979).
Muszewska, A., Taylor, J. W., Szczesny, P. & Grynberg, M. Independent subtilases expansions in fungi associated with animals. Mol Biol Evol 28, 3395–3404 (2011).
St Leger, R. J., Joshi, L. & Roberts, D. W. Adaptation of proteases and carbohydrates of saprophytic, phytopathogenic and entomopathogenic fungi to the requirements of their ecological niches. Microbiology 143 (Pt 6), 1983–92 (1997).
Pinan-Lucarre, B., Paoletti, M., Dementhon, K., Coulary-Salin, B. & Clave, C. Autophagy is induced during cell death by incompatibility and is essential for differentiation in the filamentous fungus Podospora anserina . Mol Microbiol 47, 321–33 (2003).
Fuller, R. S. & Kexin, H. In Handbook of Proteolytic Enzymes(eds Barrett, A. J., Rawlings, N. D. & Woessner, J. F. ) 1849–1854 (Elsevier, London 2004).
Freeman, J. et al. Copy number variation: new insights in genome diversity. Genome Res 16, 949–961 (2006).
Lynch, M. & Conery, J. The origins of genome complexity. Science 302, 1401–1404 (2003).
Wapinski, I., Pfeffer, A., Friedman, N. & Regev, A. Natural history and evolutionary principles of gene duplication in fungi. Nature 449, 54–61 (2007).
Graur, D. & Li, W. Fundamentals of molecular evolution(Sinauer, Sunderland, Massachusetts, 2000).
James, T. Y. et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443, 818–822 (2006).
Spatafora, J. W. et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108, 1028–1046 (2016).
Frickey, T. & Lupas, A. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics 20, 3702–4 (2004).
Edgar, R. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32, 1792–1797 (2004).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2, 28–36 (1994).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–40 (2014).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17, 540–552 (2000).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56, 564–577 (2007).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30, 2725–2729 (2013).
Galgiani, J. N. Coccidioidomycosis: a regional disease of national importance: rethinking approaches for control. Ann Intern Med 130, 293–300 (1999).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52, 696–704 (2003).
Felsenstein, J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 (1985).
Takagi, H. et al. Functional analysis of the propeptides of subtilisin E and aqualysin I as intramolecular chaperones. FEBS Lett 508, 210–4 (2001).
Muhle-Goll, C. et al. Structural and functional studies of titin’s fn3 modules reveal conserved surface patterns and binding to myosin S1–a possible role in the Frank-Starling mechanism of the heart. J Mol Biol 313, 431–47 (2001).
Bagga, S., Hu, G., Screen, S. & St Leger, R. Reconstructing the diversification of subtilisins in the pathogenic fungus Metarhizium anisopliae . Gene 324, 159–169 (2004).
Raes, J. & Van de Peer, Y. Gene duplication, the evolution of novel gene functions, and detecting functional divergence of duplicates in silico. Appl Bioinformatics 2, 91–102 (2003).
Zhang, J. Evolution by gene duplication: an update. Trends Ecol Evol 18, 292–298 (2003).
Karlsson, M. & Stenlid, J. Comparative evolutionary histories of the fungal chitinase gene family reveal non-random size expansions and contractions due to adaptive natural selection. Evol Bioinform Online 4, 47–60 (2008).
Li, J. & Zhang, K. Q. Independent expansion of zincin metalloproteinases in Onygenales fungi may be associated with their pathogenicity. PLoS One 9, e90225 (2014).
Takeshige, K., Baba, M., Tsuboi, S., Noda, T. & Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 119, 301–311 (1992).
Pinan-Lucarré, B., Paoletti, M., Dementhon, K., Coulary-Salin, B. & Clave, C. Autophagy is induced during cell death by incompatibility and is essential for differentiation in the filamentous fungus Podospora anserina . Mol Microbiol 47, 321–333 (2003).
Wilcox, C. A. & Fuller, R. S. Posttranslational processing of the prohormone-cleaving Kex2 protease in the Saccharomyces cerevisiae secretory pathway. J Cell Biol 115, 297–307 (1991).
Conesa, A., Punt, P. J., van Luijk, N. & van den Hondel, C. A. The secretion pathway in filamentous fungi: a biotechnological view. Fungal Genet Biol 33, 155–171 (2001).
Hane, J. K. et al. Dothideomycete plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum . Plant Cell 19, 3347–68 (2007).
Aboukhaddour, R., Cloutier, S., Ballance, G. M. & Lamari, L. Genome characterization of Pyrenophora tritici-repentis isolates reveals high plasticity and independent chromosomal location of ToxA and ToxB. Mol Plant Pathol 10, 201–12 (2009).
Qiao, K., Chooi, Y. H. & Tang, Y. Identification and engineering of the cytochalasin gene cluster from Aspergillus clavatus NRRL 1. Metab Eng 13, 723–32 (2011).
Nierman, W. C. et al. Genome sequence of Aspergillus flavus NRRL 3357, a strain that causes aflatoxin contamination of food and feed. Genome Announc 3 (2015).
Fedorova, N. D. et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus . PLoS Genet 4, e1000046 (2008).
Galagan, J. E. et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae . Nature 438, 1105–15 (2005).
Andersen, M. R. et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21, 885–97 (2011).
Umemura, M. et al. Comparative genome analysis between Aspergillus oryzae strains reveals close relationship between sites of mutation localization and regions of highly divergent genes among Aspergillus species. DNA Res 19, 375–82 (2012).
Askenazi, M. et al. Integrating transcriptional and metabolite profiles to direct the engineering of lovastatin-producing fungal strains. Nat Biotechnol 21, 150–6 (2003).
Wang, F. Q. et al. Genome sequencing of high-penicillin producing industrial strain of Penicillium chrysogenum . BMC Genomics 15 Suppl 1, S11 (2014).
Woo, P. C. et al. Draft genome sequence of Penicillium marneffei strain PM1. Eukaryot Cell 10, 1740–1 (2011).
Sharpton, T. et al. Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res 19, 1722–1731 (2009).
Coelho, L. M., Cursino-Santos, J. R., Persinoti, G. F., Rossi, A. & Martinez-Rossi, N. M. The Microsporum canis genome is organized into five chromosomes based on evidence from electrophoretic karyotyping and chromosome end mapping. Med Mycol 51, 208–13 (2013).
Desjardins, C. A. et al. Comparative genomic analysis of human fungal pathogens causing paracoccidioidomycosis. PLoS Genet 7, e1002345 (2011).
Munoz, J. F. et al. The dynamic genome and transcriptome of the human fungal pathogen Blastomyces and close relative emmonsia. PLoS Genet 11, e1005493 (2015).
Magrini, V. et al. Fosmid-based physical mapping of the Histoplasma capsulatum genome. Genome Res 14, 1603–9 (2004).
Martinez, D. A. et al. Comparative genome analysis of Trichophyton rubrum and related dermatophytes reveals candidate genes involved in infection. MBio 3, e00259–12 (2012).
Cuomo, C. A., Untereiner, W. A., Ma, L. J., Grabherr, M. & Birren, B. W. Draft Genome sequence of the cellulolytic fungus Chaetomium globosum . Genome Announc 3 (2015).
Li, X. et al. Genome-wide analysis of codon usage bias in Epichloe festucae . Int J Mol Sci 17 (2016).
Ma, L. J. et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium . Nature 464, 367–73 (2010).
Cuomo, C. A. et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317, 1400–2 (2007).
Williams, A. H. et al. Comparative genomics and prediction of conditionally dispensable sequences in legume-infecting Fusarium oxysporum formae speciales facilitates identification of candidate effectors. BMC Genomics 17, 191 (2016).
Shirke, M. D., Mahesh, H. B. & Gowda, M. Genome-Wide Comparison of Magnaporthe Species Reveals a Host-Specific Pattern of Secretory Proteins and Transposable Elements. PLoS One 11, e0162458 (2016).
Gao, Q. et al. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum . PLoS Genet 7, e1001264 (2011).
Klosterman, S. J. et al. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog 7, e1002137 (2011).
Galagan, J. E. et al. The genome sequence of the filamentous fungus Neurospora crassa . Nature 422, 859–68 (2003).
Espagne, E. et al. The genome sequence of the model ascomycete fungus Podospora anserina . Genome Biol 9, R77 (2008).
Martinez, D. et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26, 553–60 (2008).
Amselem, J. et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea . PLoS Genet 7, e1002230 (2011).
Yang, J. et al. Genomic and proteomic analyses of the fungus Arthrobotrys oligospora provide insights into nematode-trap formation. PLoS Pathog 7, e1002179 (2011).
Mattanovich, D. et al. Genome, secretome and glucose transport highlight unique features of the protein production host Pichia pastoris. Microb Cell Fact 8, 29 (2009).
Love, K. R. et al. Comparative genomics and transcriptomics of Pichia pastoris . BMC Genomics 17, 550 (2016).
de Montigny, J. et al. Genomic exploration of the hemiascomycetous yeasts: 8. Zygosaccharomyces rouxii. FEBS Lett 487, 52–5 (2000).
Souciet, J. L. et al. Comparative genomics of protoploid Saccharomycetaceae. Genome Res 19, 1696–709 (2009).
Souciet, J. et al. Genomic exploration of the hemiascomycetous yeasts: 1. A set of yeast species for molecular evolution studies. FEBS Lett 487, 3–12 (2000).
Dietrich, F. S. et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304, 304–7 (2004).
Butler, G. et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–62 (2009).
Jackson, A. P. et al. Comparative genomics of the fungal pathogens Candida dubliniensis and Candida albicans . Genome Res 19, 2231–44 (2009).
Dujon, B. et al. Genome evolution in yeasts. Nature 430, 35–44 (2004).
Logue, M. E., Wong, S., Wolfe, K. H. & Butler, G. A genome sequence survey shows that the pathogenic yeast Candida parapsilosis has a defective MTLa1 allele at its mating type locus. Eukaryot Cell 4, 1009–17 (2005).
Jeffries, T. W. et al. Genome sequence of the lignocellulose-bioconverting and xylose-fermenting yeast Pichia stipitis. Nat Biotechnol 25, 319–26 (2007).
Wei, W. et al. Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci USA 104, 12825–30 (2007).
Cliften, P. et al. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301, 71–6 (2003).
Scannell, D. R. et al. Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proc Natl Acad Sci USA 104, 8397–402 (2007).
Pomraning, K. R. & Baker, S. E. Draft genome sequence of the dimorphic yeast Yarrowia lipolytica Strain W29. Genome Announc 3 (2015).
Magnan, C. et al. Sequence assembly of Yarrowia lipolytica strain W29/CLIB89 shows transposable element diversity. PLoS One 11, e0162363 (2016).
Rhind, N. et al. Comparative functional genomics of the fission yeasts. Science 332, 930–6 (2011).
Wood, V. et al. The genome sequence of Schizosaccharomyces pombe. Nature 415, 871–80 (2002).
Loftus, B. J. et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans . Science 307, 1321–4 (2005).
Martin, F. et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 452, 88–92 (2008).
Martinez, D. et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol 22, 695–700 (2004).
Martinez, D. et al. Genome, transcriptome, and secretome analysis of wood decay fungus Postia placenta supports unique mechanisms of lignocellulose conversion. Proc Natl Acad Sci USA 106, 1954–9 (2009).
Duplessis, S. et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci USA 108, 9166–71 (2011).
Xu, J. et al. Dandruff-associated Malassezia genomes reveal convergent and divergent virulence traits shared with plant and human fungal pathogens. Proc Natl Acad Sci USA 104, 18730–5 (2007).
Kamper, J. et al. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature 444, 97–101 (2006).
Stajich, J. E. et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc Natl Acad Sci USA 107, 11889–94 (2010).
Ma, L. J. et al. Genomic analysis of the basal lineage fungus Rhizopus oryzae reveals a whole-genome duplication. PLoS Genet 5, e1000549 (2009).
Corrochano, L. M. et al. Expansion of signal transduction pathways in fungi by extensive genome duplication. Curr Biol 26, 1577–84 (2016).
Rosenblum, E. B. et al. Complex history of the amphibian-killing chytrid fungus revealed with genome resequencing data. Proc Natl Acad Sci USA 110, 9385–90 (2013).
Pombert, J. F. et al. Complete genome sequences from three genetically distinct strains reveal high intraspecies genetic diversity in the microsporidian Encephalitozoon cuniculi. Eukaryot Cell 12, 503–11 (2013).
Akiyoshi, D. E. et al. Genomic survey of the non-cultivatable opportunistic human pathogen, Enterocytozoon bieneusi . PLoS Pathog 5, e1000261 (2009).
Corradi, N. et al. Patterns of genome evolution among the microsporidian parasites Encephalitozoon cuniculi, Antonospora locustae and Enterocytozoon bieneusi . PLoS One 2, e1277 (2007).
Galvan, A. et al. Variability in minimal genomes: analysis of tandem repeats in the microsporidia Encephalitozoon intestinalis . Infect Genet Evol 20, 26–33 (2013).
Acknowledgements
We are grateful to Prof. Jianping Xu (McMaster University, Canada) for his valuable comments and critical discussions. We also gratefully acknowledge the genome sequencing platform provided by the Broad Institute of MIT and Harvard (http://www.broad.mit.edu/annotation/fungi/fgi/index.html). This work is jointly funded by National Basic Research Program of China (approved no. 2013CB127503), the National Natural Science Foundation of China (approved nos 31560025 and 31100894), the NSFC-Yunnan Joint Fund (U1402265), General Program of the Applied Basic Research Programs of Yunnan Province (approval no. 2016FB044), and the Program for Excellent Young Talents of Yunnan University (to Jinkui Yang).
Author information
Authors and Affiliations
Contributions
J.L., J.Y. and K.Z. conceived this study. J.L. conducted all experimental work. J.L. collected data, carried out analyses and wrote the draft manuscript. F.G. and R.W. contributed to data analyses. All other authors helped in interpretation of data and discussion of results. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, J., Gu, F., Wu, R. et al. Phylogenomic evolutionary surveys of subtilase superfamily genes in fungi. Sci Rep 7, 45456 (2017). https://doi.org/10.1038/srep45456
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep45456
This article is cited by
-
Insights into the Role of Biochar as Potential Agent in the Management of Disease Caused by Phytopathogens: a Review
Journal of Soil Science and Plant Nutrition (2023)
-
New perception about the use of nanofungicides in sustainable agriculture practices
Archives of Microbiology (2023)
-
Identification of Extracellular Proteases Induced by Nitrogen-Limited Conditions in the Thraustochytrids Schizochytrium aggregatum ATCC 28209
Marine Biotechnology (2022)
-
Co-option of an extracellular protease for transcriptional control of nutrient degradation in the fungus Aspergillus nidulans
Communications Biology (2021)
-
Evolution and comparative genomics of the most common Trichoderma species
BMC Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
