
Abstract
The Old World bollworm Helicoverpa armigera is now established in Brazil but efforts to identify incursion origin(s) and pathway(s) have met with limited success due to the patchiness of available data. Using international agricultural/horticultural commodity trade data and mitochondrial DNA (mtDNA) cytochrome oxidase I (COI) and cytochrome b (Cyt b) gene markers, we inferred the origins and incursion pathways into Brazil. We detected 20 mtDNA haplotypes from six Brazilian states, eight of which were new to our 97 global COI-Cyt b haplotype database. Direct sequence matches indicated five Brazilian haplotypes had Asian, African, and European origins. We identified 45 parsimoniously informative sites and multiple substitutions per site within the concatenated (945 bp) nucleotide dataset, implying that probabilistic phylogenetic analysis methods are needed. High diversity and signatures of uniquely shared haplotypes with diverse localities combined with the trade data suggested multiple incursions and introduction origins in Brazil. Increasing agricultural/horticultural trade activities between the Old and New Worlds represents a significant biosecurity risk factor. Identifying pest origins will enable resistance profiling that reflects countries of origin to be included when developing a resistance management strategy, while identifying incursion pathways will improve biosecurity protocols and risk analysis at biosecurity hotspots including national ports.
Similar content being viewed by others
Introduction
Global growth, food security and prosperity of agricultural communities are closely connected to international trade in agricultural and horticultural commodities. The volume and value of this trade is influenced by both climatic and pest/pathogen factors and there is interest in increasing the diversity and volume of trade whilst minimizing the impact of pests and pathogens. This conflict of interests poses a number of challenges, both before and after the incursion of a pest or pathogen into a new location.
When ascertaining the order of events that led to the incursion of a pest or pathogen, understanding the roles of propagule pressure and geographical origins is very important, especially in revealing likely incursion pathways. This, in turn, enables a more detailed assessment of factors that might have contributed to breaches in biosecurity protocols, and may assist the development of appropriate strategies for managing and reducing future incursions. Incursions of insect pests associated with international trade in agricultural and horticultural commodities are likely to increase in frequency due to globalisation and increased transport networks1. This highlights the importance of differentiating trade-related ‘unintended’ incursions from malicious introductions of pest species (sometimes described as agricultural bioterrorism).
Agricultural bioterrorism is defined as deliberate damage to plant crops and livestock or intentional introduction of pests or pathogens, with an aim to cause fear and negatively impact food security, human, animal and plant health, and the economy of the targeted country2,3. Agricultural bioterrorism could also involve the introduction of organisms with genes of biosecurity importance (i.e., those associated with insecticide/herbicide resistance). Developing guidelines and policies on agricultural bioterrorism is an area rapidly gaining attention due to its potential impact to global food security and socio-economic stability4. Accurate assessments that differentiate between natural and/or accidental introduction of pests and pathogens from that of malicious introductions will require analyses of international trade routes and activities, and a sound scientific knowledge of pests, including their invasive biology and evolutionary genetics.
The genetic diversity of a recently invaded pest species offers clues to the number of founder lineages (e.g.5,6,7), and incorporating information from other parts of its geographical range will allow more accurate inference of origins and incursion pathways (e.g.6,8,9,10). When a high number of individuals are introduced into a region (consecutively or concurrently; and/or over multiple occasions), the species is more likely to establish (i.e., high propagule pressure) and become invasive, for both demographic and genetic reasons11,12,13,14. The number of incursions preceding an invasion can be inferred using mitochondrial DNA (mtDNA), which allows us to estimate the number of genetically unique female founders (e.g.5,6,8,15). Usually, it is assumed that each unique mtDNA haplotype at an affected location represents a separate and independent incursion event (e.g.16,17); and assumption that seems sensible especially when the introduced populations are found in distant locations and each population has a unique mtDNA haplotype signature (e.g.17,18).
Helicoverpa armigera is a major polyphagous agricultural pest, with a propensity to develop insecticide resistance (e.g.19,20; see also21,22), and the ability to disperse over great distances under favourable conditions (e.g.23). Previously endemic to the Old World (Asia, Africa, Europe) and Australasia, it has now been reported in Argentina24 and Paraguay and Uruguay25, since its initial detection in Brazil26 with a likely arrival date of between 200627 and 200828. Long-distance dispersal may explain how ancestors of H. zea arrived in the New World29, and gene flow analyses of H. armigera (e.g., Israel and Turkey30; Europe (e.g., France, Portugal) and Africa (e.g., Tunisia, Morocco, Burkina Faso, Ivory Coast)31 suggest occurrence of long-distance migration between Africa, Europe and western Asia. Movements from Africa to Ascension Island (~1,600 km), and between Australia and New Zealand (∼2,200 km) are also known32,33. Genetic analysis of Brazilian H. armigera27 identified multiple female founders and indicated higher than expected genetic diversity among introduced populations, subsequently confirmed by other studies34,35. Economic losses in Brazil from this pest incursion have been estimated at US dollars (USD$) 2 billion for 2012 to 201436.
Pathways of incursion by H. armigera into the New World are hypothesised to be associated with international agricultural trade routes27, however this hypothesis remained untested. In this paper, we provide a detailed analysis of mtDNA gene diversity in Brazilian H. armigera populations sampled during the 2012/13 early incursion period. We examined the concatenated partial mtDNA COI-Cyt b genes in previously studied samples from Australia, China, India, Pakistan, Uganda, Burkina Faso29,37, and include also new material from New Zealand, France, Spain, Madagascar, Ghana, Cameroon and Senegal, with the aim of consolidating relevant mtDNA data at the individual and population levels, for the purpose of identifying potential geographical origin(s) of the Brazilian incursion(s). We then consider international agricultural and horticultural trade data into Brazil to identify patterns that might enable testing of the ‘incursion pathways via international trade routes hypothesis’ and to corroborate inferred incursion pathways. These data are discussed in terms of the biological invasion processes and the potential of elucidating appropriate insecticide and Bt resistance management practices for H. armigera in Brazil.
Material and Methods
Sample collection and DNA extraction
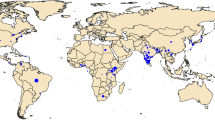
Helicoverpa armigera samples were collected either as larvae (from host plants) or adults (by light/pheromone traps) from Asia, Australasia, Africa, and Europe (n = 329; collected between 2001–2014), and from Brazil (n = 114; collected between March and August, 2013). Samples were preserved in ethanol (>95%) prior to DNA extraction using the Qiagen Blood and Tissue Kit. Taxonomic identity of each specimen was confirmed using molecular DNA markers i.e., using the mtDNA COI and Cyt b genes, as per37 (Fig. 1; Suppl. Table 1).

Map created using Mapchart https://mapchart.net. See Supplementary Table 1 for sampling details].
Molecular characterisation of the samples
PCR amplification of partial mtDNA COI and Cyt b genes was done as reported27,37. Amplicons were confirmed in 1.5% agarose gels containing 1% w/v GelRed prior to Sanger sequencing (Macrogen Inc., Seoul, Rep. of Korea; Biological Resources Facility, Australian National University, Canberra, Australia). DNA trace sequences were assembled using Staden Pregap 4 and Gap 438. Species identity was confirmed through BLASTN searches against the non-redundant DNA database in GenBank39. Estimates of haplotype (h) and nucleotide (π) diversity for Brazil, Old World, and at the global level were inferred using DnaSP40. A haplotype network for the 97 unique mtDNA haplotypes identified from this study was inferred using TCS41 within PopART42.
Molecular data survey and phylogenetic analysis
Partial COI and Cyt b gene sequences were aligned prior to concatenation to form a master alignment (443 sequences; 945 base pairs (bp)), and surveyed for evidence of multiple substitutions at the same sites to minimise biases to phylogenetic estimates. Evidence of multiple substitutions was analysed using Reticulate43 to yield a compatibility plot (Suppl. Fig. 3), and the program dnapars44 was used to infer the most parsimonious number of substitutions that might have occurred at the parsimony-informative sites (Suppl. Fig. 4). Analysis was repeated 10,000 times to increase the chance of finding the most parsimonious estimate.
Phylogenetic analysis based on 134 aligned sequences (after removal of surplus identical sequences from the master alignment) involved a probabilistic maximum-likelihood approach to enable handling of detected multiple substitutions. ModelFinder45 and the AICc optimality criterion were used to find the optimal sequence evolution model. IQ-TREE46 was used to infer the most likely phylogeny with 10,000 replications, and the consistency of the phylogeny estimated using ultrafast bootstrap47 with default settings. This enabled assessment of whether different sets of sites, drawn at random with replacement from the sub-alignment, generated a consistent phylogenetic result.
As geographical locations were unevenly sampled (i.e., ranging from 3 to 58 sequences/location), the potential impact of this uneven sampling effort on the phylogeny was evaluated using jackknifing. First, 200,000 sub-alignments that consisted of three sequences from each location were generated randomly from the master alignment using AliJack http://github.com/thomaskf/AliJack to produce sub-alignments with 104 sequences. Next, the most likely trees from these sub-alignments were inferred using IQ-TREE and the HKY + R2 model. Finally, from the inferred trees we computed the ratio, J, between (i) the number of trees with selected sequences forming a clade, and (ii) the number of trees with the selected sequences included. The jackknife score (i.e., product 100 × J) was obtained using CheckJack http://github.com/thomaskf/CheckJack.
Trade data
Trade data (value in USD$), were obtained from the Observatory of Economic Complexity (OEC) website48. The OEC compiled its source data from the United Nations Statistical Division (COMTRADE) for the period 2001–2013 using repository of official trade statistics, and as compiled by the International Merchandise Trade Statistics Section of COMTRADE. 2002–2013 trade data were extracted for live horticultural and agricultural commodities imported from the Old World to Brazil, to quantify the movement of potentially contaminated plant material and agricultural products. Trade data classified as ‘Live Trees and Other Plants’ (Harmonized System [HS] code 06; Suppl. Table 3), ‘Edible Vegetables and certain roots and tubers’ (HS code 07; Suppl. Table 4), and ‘Edible Fruits and Nuts, Peel of Citrus/Melons’ (HS code 08; Suppl. Table 5) were used. Countries were grouped into respective continents (Africa, Asia, Australasia, Europe, North America, South America) as per the OEC website (see Suppl. Tables 3, 4 and 5). Data excluded import activities from North and South American countries into Brazil (see Suppl. Tables 3, 4 and 5) due to the presumed pre-2006 absence of H. armigera in these regions.
Import risk factor analysis
We assessed the biosecurity import risks of H. armigera with respect to trade data, the import risk/likelihood of entry, population establishment and its subsequent spread against suitable commodities (i.e., HS codes 06, 07, 08) using Biosecurity Australia’s import risk analyses method49. This method considered trade volume (as measured by value) in a year to estimate pest entry likelihood49. Based on eight specific criteria (i.e., (i) ecological specificity, (ii) plant host availability and suitability, (iii) survey methodology, (iv) taxonomic recognition, (v) entry potential, (vi) destination of infested material, (vii) potential economic impact, and (viii) establishment potential), H. armigera has been rated as a ‘High’ risk pest to the New World50. We assessed H. armigera’s import risk on a per-year basis for the period 2002–2013. As the overall trade volumes are expected to grow over time, this in turn would lead to the increase in the likelihood for both pest entry and population establishment49. Equation (1) (see from49) was applied with time (t) replaced by volume (ϑ), to estimate the likelihood of incursion (‘incursion’ as per ‘incursion’ as per49) and establishment by H. armigera:

where L is the likelihood of incursion, p is the probability of incursion when importing one unit of volume of the product51, and ϑ is product units (note that 1−p represents the probability that H. armigera will not invade a location in a particular year of trade).
We inferred the volume units by calculating the ratio per commodity (i.e., HS codes 06, 07, 08) and for overall trade volume (HS codes 06 + 07 + 08), by dividing individual trade volumes per region (i.e., Asia, Africa, Europe, Australasia) per year by the highest trade volume for the HS code commodity in question (i.e., such that the highest volume for the particular HS code is equal to 1). This assessment of likelihood based on a semi-quantitative method relies on language-based likelihoods by intervals of probability (i.e., ‘high’ = 0.7–1.0; ‘Moderate’ = 0.3–0.7; ‘Low’ = 0.05–0.3)49,51. For H. armigera, we used the probability threshold of 0.7 to conservative represent the ‘lower end’ of high-risk likelihood of an incursion.
Results
MtDNA diversity
We analysed 945 bp (i.e., 511 bp COI; 434 bp Cyt b) from 443H. armigera individuals (Brazil: N = 114; Old world/Australasia: N = 329) and identified 97 haplotypes, including 20 from Brazil (Suppl. Table 1). These samples were compared to those reported previously27,29, with additional samples from Chad (N = 6), Cameroon (N = 13), Senegal (N = 11), Ghana (N = 4), Madagascar (N = 14), Spain (N = 6), France/French Corsica (N = 17), and New Zealand (N = 9) (Suppl. Table 1) also included in our analyses. 21 new mtDNA COI (GenBank: KX494879-KX494899) and 10 new Cyt b haplotypes (GenBank: KX494900-KX494909) were identified in Senegal, Madagascar, Chad, Cameroon, Spain, French Corsica, New Zealand, and Brazil (Suppl. Table 2). Concatenating the partial gene haplotypes generated the 97H. armigera haplotypes mentioned above, and eight of the 20 Brazilian haplotypes have never been reported (Suppl. Table 2).
Mitochondrial DNA haplotype diversity of H. armigera
Estimates of haplotype and nucleotide diversity among samples of H. armigera from Brazil, the Old World, and the globe are provided in Table 1. The haplotype diversity estimate for Brazil was 0.753, while that for the Old World was 0.820. However, these values overlap when their standard deviations are taken into account. The haplotype network showed a high level of complexity, but lacked obvious population/haplotype cluster patterns (Suppl. Fig. 1). Five haplotypes were found in Brazil and one other country (i.e., Madagascar, Chad, India, China, France), implying five possible sources for incursions into Brazil. The lack of distinct clusters within Brazil is consistent with earlier studies (e.g.27,29,35,36) involving the new Brazilian populations and indicating high mobility in this species.
Trade data on horticultural and agricultural commodities into Brazil showed that we lacked samples from countries with significant export or re-export activities to Brazil (e.g., Israel, Tunisia, Turkey, Thailand, Egypt, South Africa, Nigeria, Italy, The Netherlands, Greece), with these countries being possible origins of novel and unique Brazilian haplotypes. Our mtDNA analysis also identified globally distributed haplotypes (e.g., Hap0101, Hap0108, Hap0111, Hap0201; Suppl. Tables 1 and 2; Suppl. Figs 1 and 2), suggesting that while some haplotypes might allow us to identify origins of introduction, others are too widespread to offer an insight into incursion sources.
Phylogenetic analysis of the molecular data
The inferred phylogeny (Fig. 2; HKY + R2 optimal sequence evolution model) is characterized by an abundance of polytomies, likely due to the low number of parsimony-informative sites. In some cases, the bootstrap scores (indicated for selected edges) are high (e.g., 321_Hap5131; 314_Hap5131), implying that the data are phylogenetically consistent for these sequences, but in the majority of cases are far below 100% due to phylogenetically inconsistent sequence data. The jackknife scores (i.e., for assessing the impact of incomplete taxon sampling) were generally high (Fig. 2), suggesting that our taxon sampling for this study is unlikely to be an issue.

Names of sequences are represented by individual ID (see Suppl. Table 1) followed by haplotype ID (see Suppl. Table 2). Sequence discussed in the main text are in bold. Blue edges indicate Brazilian unique haplotypes; red edges indicate haplotypes found in Brazil and one other country; grey edges indicate haplotypes found in Brazil and more than one other country. Bootstrap scores (from 10,000 replicates) for selected edges are shown in a light font; jackknife scores (from 200,000 replicates) for selected edges are shown in a bold font.
The phylogeny reveals that the eight unique Brazilian haplotypes are often not as closely related to one another as they are to a haplotype found in Australia (Hap1001, Hap3001, Hap0404 or Hap0105), New Zealand (Hap4701), China (Hap1001), India (Hap1001 or Hap1009), Burkina Faso (Hap1001), Uganda (Hap0404) or Senegal (Hap3401). Of these seven non-Brazilian haplotypes (i.e., Hap1001, Hap3001, Hap0404, Hap0105, Hap4701, Hap1009, Hap3401) found in distant locations, five are reported from only one country (i.e., Australia, New Zealand, India, Senegal). Therefore, it is possible that these eight uniquely Brazilian haplotypes could have originated from these populations, and perhaps also populations in Uganda, Burkina Faso, and China (Suppl. Table 1), as supported also by genome-wide SNP data and pyrethroid resistance gene analysis52.
The phylogeny also reveals five haplotypes found in Brazil and one other country (i.e., France, Senegal, Ghana, India, and China). This result again suggests separate incursion events in addition to those mentioned above. Similarly, seven haplotypes were found in Brazil and in more than one other country (Suppl. Table 2). The geographical distributions of these seven haplotypes also hint of multiple incursions (i.e., ≥three countries), however, the evolutionary relationship among these seven haplotypes is unclear (Fig. 2). Taken as a whole, mtDNA haplotype analyses support multiple and perhaps continuous incursions of H. armigera into Brazil. At this early incursion stage, there exist at least 20H. armigera maternal lineages in Brazil. Of these, eight are unique to Brazil and their close evolutionary relationship with haplotypes found in other countries suggests that they are unlikely to be the result of recent divergence (c.f.35).
Trade data
Trade data from 2002–2013 indicate an approximately fourfold increase in activity from Asia into Brazil with limited imports originating from Australasia (Australia, New Zealand). Within Asia; India, Thailand, Israel, Turkey and China were the main trading partners although others (e.g., Japan, Malaysia, South Korea, Sri Lanka, Lebanon, Saudi Arabia, United Arab Emirates, Syria) also had significant trade activity with Brazil over the surveyed period.
Significant exports to Brazil of HS code 06 commodities (Living Trees and Other Plants; Suppl. Table 3) were identified from European countries (Suppl. Fig. 5), including The Netherlands, France, Italy, Spain, Germany, Belgium, Luxemburg, Denmark, Greece, Portugal, Great Britain, and Ireland. During the 12-year period surveyed, the African continent exported high volume of HS code 07 commodities (Edible Vegetables, including Certain Roots and Tubers; Suppl. Table 4). Over the period 2007–2012, there were significant imports of fresh or chilled vegetables from Egypt (Suppl. Fig. 6), with a drop in (or absence of) this trade from Europe and Asia. European countries exported high volumes of HS code 08 commodities (Edible Fruits and Nuts, Peel of Citrus/Melons; Suppl. Table 5) to Brazil, with a fourfold increase in trade since 2002 from Asia and a sevenfold increase in trade volume from New Zealand (Suppl. Fig. 7).
Trade volume and associated biosecurity import risk analysis
The biosecurity import risks of H. armigera increased expectedly with increasing trade volume (Fig. 3; Suppl. Figs 5, 6 and 7). For HS code 6 commodities, there was a moderate-to-high import risk (i.e., likelihood from 0.42 to 0.70) between 2008 and 2012 for products from Europe, followed by products from Asia with a likelihood of import risk that rapidly increased from ‘low’ (i.e., 0.07) in 2008 to moderate (i.e., 0.37) in 2012. For HS code 07 commodities, the likelihood of the H. armigera import risk from African products spiked at 0.7 (i.e., high) in 2010, and at ‘moderate risk levels’ (i.e., 0.3, 0.4 and 0.5) in 2002, 2007, and 2012, respectively, from Asian products. Import risks from H. armigera for HS code 08 commodities was the greatest from Europe, rapidly increasing from 0.15 (i.e., ‘low’) to 0.7 (i.e., ‘high’) over the 12 years surveyed, while those for the other three continents also grew from ≤0.01 (i.e., negligible/very low) to approximately 0.16 (i.e., low). When combining all three HS codes of commodities (Fig. 3), the biosecurity import risk likelihood was similar to that for HS code 08, although the likelihood was slightly higher (but still ‘low’) for Asia (0.03 in 2002, to 0.23 in 2012), Australasia (0.0003 in 2002, to 0.09 in 2012) and Africa (0.008 in 2002, to 0.05 in 2012).

Data are for 12-years period (2002–2013; X-axis) for Harmonized commodity description and coding system (HS) codes 06, 07 and 08 (Suppl. Tables 3, 4 and 5) in USD$ (left Y-axis). Biosecurity entry risk factor (orange doted lines, right Y-axis) for H. armigera that considered introduction, establishment and population spread as calculated for trade volume per year by continental regions are also shown.
Discussion
By combining multiple mtDNA markers and increasing global sampling sites, we identified Asia (i.e., China, India), Africa (i.e., Madagascar, Senegal) and Europe (i.e., France) as potential origins of Brazilian H. armigera, and inferred incursion pathways by combining genetic and trade data. This approach may be relevant to other invasive organisms with high effective population sizes, such as the soybean stem fly (SSF) Melanagromyza sojae53, the whitefly Bemisia tabaci cryptic species complex (e.g.54), the spotted-wing vinegar fruit fly Drosophila suzukii55, the European Grapevine moth Lobesia botrana56,57, and the Russian Wheat Aphid Diuraphis noxia and incorporating genetic signatures of endosymbiont bacteria (e.g.58), amongst other.
Leite et al.35 compared the standard barcode mtDNA COI region of Brazilian H. armigera to publicly available mtDNA COI haplotypes from the Old World, and identified Asian (i.e., China) and European maternal lineages in Brazil, based on shared haplotypes. Africa, Australia and Pakistan were not identified as potential origins due to poor availability of sequence data from these countries. Furthermore, common global haplotypes shared widely between Brazil and Old World locations also reduced the power of detecting potential origins. We showed that these regions, especially China, India, and France, were significant export countries of live host material capable of supporting H. armigera into Brazil, thereby supporting the notion of increased international trade activities as risks factors1,59.
Rumours of ‘agricultural bioterrorism’ have surfaced periodically in Brazil (e.g., Jornal diz que ocorrência de lagarta pode ser bioterrorismo; New threat to Brazil’s breadbasket: a pesky caterpillar; The Helicoverpa armigera). The diverse origins of H. armigera detected in Brazil would mean it is highly unlikely there can have been a deliberate release of this pest. Previously, Spanish plant products were identified as the commodity category where most H. armigera interceptions occurred in the EU60. This is in general agreement with our trade data patterns, although we also identified France, Italy and The Netherlands as countries with significant export activities to Brazil. Since 2008H. armigera has been ‘deregulated’ in Europe for trade involving cut flowers from Africa61. Much of these horticultural/agricultural goods are exported to third countries including Brazil. This suggests that Europe might also be the route of introduction, and highlights the potential global impact of regional phytosanitary policies. The general lack of agricultural trade with Australia could explain the absence of unique Australian haplotypes in our Brazilian data. Interestingly, H. armigera was detected in 2014 in Capsicum sp. from the Dominican Republic into the EU62, a first reported incidence of a New World to Old World reintroduction.
A common finding between previous (e.g.27,34,35) and this study is the high genetic mtDNA haplotype diversity in the Brazilian populations, which is unusual for a recently introduced pest. Eight haplotypes (Suppl. Tables 1 and 2) represented unique maternal lineages not currently known from Old World populations. Matching of these to better identify additional potential origins of H. armigera in Brazil will require sampling countries with substantial agricultural commodity exports to Brazil. Despite the lack of substantial trade in agricultural and horticultural commodities with Brazil, a Malagasy haplotype (Hap0208) was identified. Population movement of H. armigera between Madagascar and neighbouring African nations is currently unknown but significant trade does occur between South Africa and Brazil. It may be that this haplotype represents a general ‘southern African’ haplotype and is present in both South Africa and Madagascar.
The discovery of Asian and African/European haplotypes offers unique insights into two potential introduction pathways in Brazil: natural (chance) dispersals, and human-mediated dispersal. Our finding that Senegal, Madagascar, French Corsica and Brazilian populations share the same unique haplotypes could indicate chance dispersal from the African continent, as well as human mediated introductions from Asia and Europe. Insect dispersals from Africa to the New World are hypothesised to associate with easterly winds (e.g.63). Knowing the potential origins of H. armigera in Brazil will provide valuable insights for its managing in the New World. In Cameroon, China and India, H. armigera carries resistance to synthetic pyrethroids64,65,66, organophosphates67,68 and carbamates69,70. Resistance to diverse classes of pyrethroids has also been reported in populations from Benin (west Africa)71. In southern France, pyrethroid-resistance H. armigera was also reported72,73, while populations resistant to pyrethroids, organophosphates and carbamates were identified in Spain74,75. Significant reduced susceptibility to Cry1Ac has also been detected in some Chinese H. armigera populations76, and H. armigera larvae on Bt cotton (BG-II) which has cry1Ac+ cry2Ab genes exists in India77.
In Brazil, farmers reported difficulties managing H. armigera and overused insecticides to restrict its outbreak78, potentially reflecting insecticide resistances in the introduced populations. This is supported by Anderson et al.52 where Brazilian H. armigera grouped with Asian individuals for an allele for pyrethroid resistance in H. armigera sampled during this early stage of incursion. Population-wide studies (e.g.52) will therefore provide a starting point for prioritising screens for resistance in chemistries (organophosphates, carbamates, Cry1Ac, Cry2Ab) that are likely to be present in source populations and which have high use in the New World. This information will be critical for developing strategies to manage the further evolution of insecticide resistance and for the management of H. armigera elsewhere, including the re-introduction of H. armigera back into the Old World with heightened resistance acquired in the New World, and the potential spread into the North American final frontier.
While it may have been possible to eliminate the initial H. armigera population(s) in Brazil (assuming single incursion event), this is now unlikely given shared geographical boundaries and similar economic activities. H. armigera’s presence in the New World may also have been as early as the mid-2000’s, with individuals intercepted in 2006 in a Peruvian “mange-tout” bean consignment found to possess COI haplotypes (630 bp) that matched those from China, Spain, Uganda and Egypt79. This suggests that incursions into Brazil may well have involved neighbouring countries, and reinforcing the need to coordinate and co-operate in the area of biosecurity. Indeed, the study of Lopes-da-Silva et al.36 has shown that at least in Brazil, there has been an increase in frequencies of incursions by exotic agricultural insect pests since the early 1900’s, with at least two more exotic insect pest incursions being reported in recent times (e.g.80,81). On-going monitoring of New World H. armigera populations though genetic analysis of quarantine-intercepted material will contribute to mapping its New World spread, thereby enabling early intervention via regional-specific pest management strategies to achieve their intended efficacies.
Additional Information
How to cite this article: Tay, W. T. et al. Mitochondrial DNA and trade data support multiple origins of Helicoverpa armigera (Lepidoptera, Noctuidae) in Brazil. Sci. Rep. 7, 45302; doi: 10.1038/srep45302 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Hulme, P. E. Trade, transport and trouble: managing invasive species pathways in an era of globalization. J Appl Ecol 46, 10–18, doi: 10.1111/j.1365-2664.2008.01600.x (2009).
Monke, J. Agroterrorism: Threats and Preparedness. (ed. Science Resources, and Industry Division) 63 (Congressional Reserach Service, 2007).
Monthei, D., Mueller, S., Lockwood, J. & Debboun, M. Entomological terrorism: A tactic in asymmetrical warfare. The United States Army Medical Department Journal April - June 2010, 11–20 (2010).
Callaway, E. Bioterror: The green menace. Nature 452, 148–150, doi: 10.1038/452148a (2008).
Vincek, V. et al. How large was the founding population of Darwin’s finches? P Roy Soc B-Biol Sci 264, 111–118 (1997).
Ficetola, G. F., Bonin, A. & Miaud, C. Population genetics reveals origin and number of founders in a biological invasion. Mol Ecol 17, 773–782 (2008).
Frantz, A. C. et al. Limited mitochondrial DNA diversity is indicative of a small number of founders of the German raccoon (Procyon lotor) population. Eur J Wildlife Res 59, 665–674, doi: 10.1007/s10344-013-0719-6 (2013).
Dickey, A. M., Osborne, L. S., Shatters, R. G., Hall, P. M. & Mckenzie, C. L. Population Genetics of Invasive Bemisia tabaci (Hemiptera: Aleyrodidae) Cryptic Species in the United States Based on Microsatellite Markers. J Econ Entomol 106, 1355–1364, doi: 10.1603/Ec12512 (2013).
Pisanu, B., Obolenskaya, E. V., Baudry, E., Lissovsky, A. A. & Chapuis, J. L. Narrow phylogeographic origin of five introduced populations of the Siberian chipmunk Tamias (Eutamias) sibiricus (Laxmann, 1769) (Rodentia: Sciuridae) established in France. Biol Invasions 15, 1201–1207, doi: 10.1007/s10530-012-0375-x (2013).
Praebel, K., Gjelland, K. O., Salonen, E. & Amundsen, P. A. Invasion genetics of vendace (Coregonus albula (L.)) in the Inari-Pasvik watercourse: revealing the origin and expansion pattern of a rapid colonization event. Ecol Evol 3, 1400–1412, doi: 10.1002/ece3.552 (2013).
Kolar, C. S. & Lodge, D. M. Progress in invasion biology: predicting invaders. Trends Ecol Evol 16, 199–204, doi: 10.1016/S0169-5347(01)02101-2 (2001).
Lockwood, J. L., Cassey, P. & Blackburn, T. The role of propagule pressure in explaining species invasions. Trends Ecol Evol 20, 223–228, doi: 10.1016/j.tree.2005.02.004 (2005).
Von Holle, B. & Simberloff, D. Ecological resistance to biological invasion overwhelmed by propagule pressure. Ecology 86, 3212–3218 (2005).
Colautti, R. I., Grigorovich, I. A. & MacIsaac, H. J. Propagule pressure: A null model for biological invasions. Biol Invasions 8, 1023–1037, doi: 10.1007/s10530-005-3735-y (2006).
Tay, W. T., Kerr, P. J. & Jermiin, L. S. Population genetic structure and potential incursion pathways of the bluetongue virus vector Culicoides brevitarsis (Diptera: Ceratopogonidae) in Australia. PLoS One 11 (1), e0146699, doi: 10.1371/journal.pone.0146699 (2016).
Ascunce, M. S. et al. Global invasion history of the fire ant Solenopsis invicta . Science 331, 1066–1068, doi: 10.1126/science.1198734 (2011).
Goldstien, S. J. et al. Global Phylogeography of the Widely Introduced North West Pacific Ascidian Styela clava . PLoS One 6, doi: 10.1371/journal.pone.0016755 (2011).
Castalanelli, M. A. et al. Multiple incursions and putative species revealed using a mitochondrial and nuclear phylogenetic approach to the Trogoderma variabile (Coleoptera: Dermestidae) trapping program in Australia. B Entomol Res 101, 333–343, doi: 10.1017/S0007485310000544 (2011).
Downes, S. & Mahon, R. Evolution, ecology and management of resistance in Helicoverpa spp. to Bt cotton in Australia. J Invertebr Pathol 110, 281–286, doi: 10.1016/j.jip.2012.04.005 (2012).
Downes, S., Walsh, T. & Tay, W. T. Bt resistance in Australian insect pest species. Current Opinion in Insect Science 15, 78–83 (2016).
Tabashnik, B. E. ABCs of insect resistance to Bt. PLoS Genet 11, e1005646, doi: 10.1371/journal.pgen.1005646 (2015).
Tay, W. T. et al. Insect Resistance to Bacillus thuringiensis Toxin Cry2Ab Is Conferred by Mutations in an ABC Transporter Subfamily A Protein. PLoS Genet 11, e1005534, doi: 10.1371/journal.pgen.1005534 (2015).
Fitt, G. P. The ecology of Heliothis species in relation to agroecosystems. Annu Rev Entomol 34, 17–52 (1989).
Murúa, M. G. et al. First Record of Helicoverpa armigera (Lepidoptera: Noctuidae) in Argentina. Florida Entomologist 97, 854–856 (2014).
Arnemann, J. A. et al. Mitochondrial DNA COI characterization of Helicoverpa armigera (Lepidoptera: Noctuidae) from Paraguay and Uruguay. Genet Mol Res 15 (2), gmr.15028292, doi: 10.4238/gmr.15028292 (2016).
Czepak, C., Albernaz, C., Vivan, L. M., Guimarães, H. O. & Carvalhais, T. First reported occurrence of Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae) in Brazil. Pesq. Agropec. Trop., Goiânia 43, 110–113 (2013).
Tay, W. T. et al. A Brave New World for an Old World Pest: Helicoverpa armigera (Lepidoptera: Noctuidae) in Brazil. PLoS One 8(11), e80134, doi: 10.1371/journal.pone.0080134 (2013).
Sosa-Gómez et al. Timeline and geographical distribution of Helicoverpa armigera (Hübner) (Lepidoptera, Noctuidae: Heliothinae) in Brazil. Revista Brasileira de Entomologia 60, 101–104 (2015).
Behere, G. T. et al. Mitochondrial DNA analysis of field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) and of its relationship to H-zea. Bmc Evol Biol 7, 117, doi: 10.1186/1471-2148-7-117 (2007).
Zhou, X. F., Faktor, O., Applebaum, S. W. & Coll, M. Population structure of the pestiferous moth Helicoverpa armigera in the Eastern Mediterranean using RAPD analysis. Heredity 85, 251–256, doi: 10.1046/j.1365-2540.2000.00738.x (2000).
Nibouche, S., Bues, R., Toubon, J. F. & Poitout, S. Allozyme polymorphism in the cotton bollworm Helicoverpa armigera (Lepidoptera: Noctuidae): comparison of African and European populations. Heredity 80, 438–445, doi: 10.1046/j.1365-2540.1998.00273.x (1998).
Widmer, M. W. & Schofield, P. Heliothis dispersal and migration. In TDRI (College House) Information Service Annotated Bibliographies Series No. 2 i + 41pp (Tropical Development and Research Institute, London, U.K., 1983).
Hartnett, D. E. et al. In Surveillance for Biosecurity: Pre-boarder to pest management (eds Froud, K. J., Popay, A. I. & Zydenbos, S. M. ) 111–119 (The New Zealand Plant Protection Society (Incorporated), Paihia, New Zealand, 2008).
Mastrangelo, T. et al. Detection and genetic diversity of a heliothine invader (Lepidoptera: Noctuidae) from north and northeast of Brazil. J Econ Entomol 107, 970–980 (2014).
Leite, N. A., Alves-Pereira, A., Correa, A. S., Zucchi, M. I. & Omoto, C. Demographics and Genetic Variability of the New World Bollworm (Helicoverpa zea) and the Old World Bollworm (Helicoverpa armigera) in Brazil. PLoS One 9, e113286, doi: 10.1371/journal.pone.0113286 (2014).
Lopes-da-Silva, M., Sanches, M., Stancioli, A., Alves, G. & Sugayama, R. The Role of Natural and Human-Mediated Pathways for Invasive Agricultural Pests: A Historical Analysis of Cases from Brazil. Agricultural Sciences 5, 634–646, doi: 10.4236/as.2014.57067 (2014).
Behere, G. T., Tay, W. T., Russell, D. A. & Batterham, P. Molecular markers to discriminate among four pest species of Helicoverpa (Lepidoptera: Noctuidae). Bull Entomol Res 98, 599–603, doi: 10.1017/S0007485308005956 (2008).
Staden, R., Beal, K. F. & Bonfield, J. K. The Staden package, 1998. Methods Mol Biol 132, 115–130 (2000).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Rozas, J. & Rozas, R. DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15, 174–175 (1999).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol Ecol 9, 1657–1659 (2000).
French, N. et al. In Campylobacter Ecology and Evolution in New Zealand. In Campylobacter Ecology and Evolution (eds Sheppard, S. K. & Méric, G. ) pp. 221–240 (Caister Academic Press, 2014).
Jakobsen, I. B. & Easteal, S. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. CABIOS 12, 291–295 (1996).
PHYLIP (Phylogeny Inference Package) version 3.7a (2009).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. F. K., von Haeseler, A. & Jermiin, L. S. ModelFinder: A Model-selection method that greatly improves the accuracy of phylogenetic estimates. Nature Methods (In press). (2017).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Minh, B. Q., Nguyen, M. A. T. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013).
Simoes, A. J. G. & Hidalgo, C. A. In Workshops at the Twenty-Fifth AAAI Conference on Artificial Intelligence.
McCarthy, M., Burgman, M. & Gordon, I. ACERA project 0702. Review of the use of period of trade and trade volume in import risk analysis. 34 (The University of Melbourne, Melbourne, 2007).
Venette, R. C., Davis, E. E., Zaspel, Z., Heisler, H. & Larson, M. Mini risk assessment, Old World bollworm, Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae). (US Department of Agriculture, Animal and Plant Health Inspection Service, 2003).
Commonwealth of Australia. Guidelines for Import Risk Analysis. (ed. Agriculture, Fisheries and Forestry) (Canberra, Australia, 2001).
Anderson, C. J., Tay, W. T., McGaughran, A., Gordon, K. & Walsh, T. K. Population structure and gene flow in the global pest, Helicoverpa armigera . Mol Ecol 25, 5296–5311, doi: 10.1111/mec.13841 (2016).
Arnemann, J. A. et al. Complete mitochondrial genome of the soybean stem fly Melanagromyza sojae (Diptera: Agromyzidae). Mitochondrial DNA 1, 2, doi: 10.3109/19401736.2015.1101550 (2015).
Barbosa, L. D. et al. First report of Bemisia tabaci Mediterranean (Q biotype) species in Brazil. Pest Manag Sci 71, 501–504, doi: 10.1002/ps.3909 (2015).
Cini, A., Ioriatti, C. & Anfora, G. A review of the invasion of Drosophila suzukii in Europe and a draft research agenda for integrated pest management. B Insectol 65, 149–160 (2012).
Gilligan, T. M. et al. Discovery of Lobesia Botrana ([Denis & Schiffermuller]) in California: An Invasive Species New to North America (Lepidoptera: Tortricidae). P Entomol Soc Wash 113, 14–30, doi: 10.4289/0013-8797.113.1.14 (2011).
Tay, W. T. Rapid molecular DNA identification method for the European invasive grapevine moth Lobesia botrana. Report No. ISBN: 978-1-4863-0729-6, 42 (CSIRO, Australia, 2016).
Zhang, B., Edwards, O. R., Kang, L. & Fuller, S. J. Russian wheat aphids (Diuraphis noxia) in China: native range expansion or recent introduction? Mol Ecol 21, 2130–2144, doi: 10.1111/j.1365-294X.2012.05517.x (2012).
Hulme, P. E. Beyond control: wider implications for the management of biological invasions. J Appl Ecol 43, 835–847, doi: 10.1111/j.1365-2664.2006.01227.x (2006).
Lammers, W. & MacLeod, A. M. Report of a Pest Risk Analysis, Helicoverpa armigera (Hübner, 1808). 18 (Plant Protection Service, the Netherlands, and Central Science Laboratory, United Kingdom, 2007).
European Union. EUROPHYT European Union Notification System for Plant Health Interceptions Annual Report 2014. 70 pp. (Food and Veterinary Office, Luxembourg, 2015).
ILEI Wageningen U. R. & Rikken M. 2015 CBI Trade Statistics: Cut Flowers and Foliage. (p. 9. The Netherlands, CBI Ministry of Foreign Affairs.
Rosenberg, J. & Burt, P. J. A. Windborne displacements of Desert Locusts from Africa to the Caribbean and South America. Aerobiologia 15, 167–175 (1999).
Achaleke, J., Martin, T., Ghogomu, R. T., Vaissayre, M. & Brevault, T. Esterase-mediated resistance to pyrethroids in field populations of Helicoverpa armigera (Lepidoptera: Noctuidae) from Central Africa. Pest Manag Sci 65, 1147–1154, doi: 10.1002/ps.1807 (2009).
Wu, Y., Shen, J., Tan, F. & You, Z. Resistance monitoring of Helicoverpa armigera in Yanggu county of Shandong province of China. Journal Nanjing Agriculture University 18, 48–53 (1995).
Kranthi, K. R. et al. Insecticide resistance in five major insects pests of cotton in India. Crop Protection 21, 449–460 (2002).
Kranthi, K. R., Jadhav, D. R., Wanjari, R. R., Ali, S. S. & Russell, D. Carbamate and organophosphate resistance in cotton pests in India, 1995 to 1999. B Entomol Res 91, 37–46 (2001).
Wu, Y., Shen, J., Chen, J., Lin, X. & A., L. Evaluation of two resistance monitorings methods in Helicoverpa armigera: topical application method and leaf dipping method. Plant Protection 22, 3–6 (1996).
Armes, N. J., Jadhav, D. R. & DeSouza, K. R. A survey of insecticide resistance in Helicoverpa armigera in the Indian subcontinent. B Entomol Res 86, 499–514 (1996).
Cheng, G. & Liu, Y. Cotton bollworm resistance and its development in northern cotton region of China 1984–1985. Resistance Pest Management 8, 32–33 (1996).
Djihinto, A. C., Katary, A., Prudent, P., Vassal, J. M. & Vaissayre, M. Variation in resistance to pyrethroids in Helicoverpa armigera from Benin Republic, West Africa. J Econ Entomol 102, 1928–1934 (2009).
Buès, R. & Boudinhon, L. Résistance aux insecticides de Helicoverpa armigera (Lépidoptère: Noctuidae) dans le sud de la France. Cah. Agric. 12, 1–7 (2003).
Buès, R., Bouvier, J. C. & Boudinhon, L. Insecticide resistance and mechanisms of resistance to selected strains of Helicoverpa armigera (Lepidoptera: Noctuidae) in the south of France. Crop Protection 24, 814–820, doi: 10.1016/j.cropro.2005.01.006 (2005).
Torres-Vila, L. M., Rodriguez-Molina, M. C., Lacasa-Plasencia, A., Bielza-Lino, P. & Rodriguez-del-Rincon, A. Pyrethroid resistance of Helicoverpa armigera in Spain: current status and agroecological perspective. Agr Ecosyst Environ 93, 55–66, doi: Pii S0167-8809(02)00003-8 10.1016/S0167-8809(02)00003-8 (2002).
Torres-Vila, L. M., Rodriguez-Molina, M. C., Lacasa-Plasencia, A. & Bielza-Lino, P. Insecticide resistance of Helicoverpa armigera to endosulfan, carbamates and organophosphates: the Spanish case. Crop Protection 21, 1003–1013, doi: Pii S0261-2194(02)00081-9 10.1016/S0261-2194(02)00081-9 (2002).
Zhang, H. N. et al. Diverse genetic basis of field-evolved resistance to Bt cotton in cotton bollworm from China. P Natl Acad Sci USA 109, 10275–10280, doi: 10.1073/pnas.1200156109 (2012).
Kranthi, K. R. Technologies are breaking down - What next? 12 (Cotton Association of India, 2015).
Pomari-Fernandes, A., de Freitas Bueno, A. & Sosa-Gómez, D. R. Helicoverpa armigera: current status and future perspectives in Brazil. Current Agricultural Science and Technology 21, 1–7 (2015).
Collins, D. et al. Defra SID5 Research Project Final Report PH0311. Molecular and morphological identification of the eggs and caterpillars of quarantine listed Noctuidae 21 (Department for Environment, Food and Rural Affairs, 2007).
Arnemann, J. A. et al. Soybean Stem Fly, Melanagromyza sojae (Diptera: Agromyzidae), in the New World: detection of high genetic diversity from soybean fields in Brazil. Genet Mol Res 15, doi: 10.4238/gmr.15028610 (2016).
Costa-Lima, T. C., Moreira, G. R., Goncalves, G. L. & Specht, A. Lasiothyris luminosa (Razowski & Becker) (Lepidoptera: Tortricidae): a New Grapevine Pest in Northeastern Brazil. Neotrop Entomol 45, 336–339, doi: 10.1007/s13744-016-0379-9 (2016).
Acknowledgements
ESC and IBM were supported by CSIRO Health and Biosecurity, the Brazilian Government’s, Science without Boarders (Ciência sem Fronteiras) summer internship program (242166/2012–1 (ESC); 209297/2013-1 (IBM)), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) scholarships. CA was supported by a CSIRO OCE Post Doctoral Fellowship (R-03255-01). WTT and TKW acknowledged funing support from CSIRO Health & Biosecurity ‘Genes of Biosecurity Significance’ (R-8681-1). CC acknowledges funding support from FAPEG (Fundação de amparo a pesquisa do estado de Goiás) (Grant number: Helicoverpa/2013102670001419). Ghana samples were provided by Ibrahim Atokple. William James (CSIRO) assisted in the laboratory. Dean Paini, John Oakeshott (CSIRO) and Jonas Arnemann (UFSM, Brazil) provided helpful discussion.
Author information
Authors and Affiliations
Contributions
Project design: W.T.T., T.K.W., S.D., C.C., K.H.J.G. Laboratory work: W.T.T., T.K.W., E.S.C., I.B.M., C.A., M.C.P., G.T.B. Field data collection: M.F.S., M.F., C.C. Genetic data analysis: W.T.T., T.W., E.S.C., I.B.M., L.S.J., G.T.B., T.K.F.W., C.A., M.C.P. Project data coordination: P.S., C.C., S.D. Manuscript preparation: W.T.T., T.K.W., S.D., L.S.J., G.T.B., P.S., M.F., K.H.J.G., C.C. All authors gave final approval for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tay, W., Walsh, T., Downes, S. et al. Mitochondrial DNA and trade data support multiple origins of Helicoverpa armigera (Lepidoptera, Noctuidae) in Brazil. Sci Rep 7, 45302 (2017). https://doi.org/10.1038/srep45302
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep45302
This article is cited by
-
Complex multiple introductions drive fall armyworm invasions into Asia and Australia
Scientific Reports (2023)
-
Has insecticidal pressure influenced Spodoptera litura (Fabricius, 1775) population genetic structure and genetic diversity in India?
Biologia (2022)
-
Intra species diversity of Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae) in relation to geography and host plants affiliation in Uttarakhand Himalayan population, India
Phytoparasitica (2022)
-
Helicoverpa armigera Harm 1 Haplotype Predominates in the Heliothinae (Lepidoptera: Noctuidae) Complex Infesting Tomato Crops in Brazil
Neotropical Entomology (2021)
-
Highly diverse and rapidly spreading: Melanagromyza sojae threatens the soybean belt of South America
Biological Invasions (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
